
Abstract
Nanoporous gold (NPG) electrodes were fabricated in film and wire array formats by selectively dealloying Ag from Au0.18Ag0.82. The ammonia borane (AB) oxidation reaction was studied by cyclic voltammetry at the NPG electrodes. The onset potential for the oxidation at NPG in a wire array format shifted to more negative potentials than that observed at a Au disc and higher currents were realised. An onset potential of −1.30 V vs. SCE was recorded which is 0.28 V lower than that at a Au disc. The oxidation current for 20 mM AB in 1 M NaOH increased from 2.65 mA cm−2 at a Au disc to 13.1 mA cm−2 at a NPG wire array. NPG is a viable candidate as an anode catalyst for a direct ammonia borane fuel cell.
Export citation and abstract BibTeX RIS
Ammonia borane (AB) is a chemically stable, non-toxic, environmentally benign solid with a high hydrogen content (19.5 wt %) which can be easily transported. It is a stable solid at room temperature and soluble in relatively polar solvents such as water and methanol. It can be safely handled at room temperature. It has been shown that hydrogen can be released from AB via catalytic hydrolysis 1–10 or thermal decomposition. 11–14 The rate of hydrolysis of AB (Eq. 1) is appreciable in the presence of a suitable catalyst
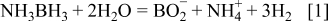
While Pt-based catalysts exhibit the highest activity for AB hydrolysis 8 it was shown that lower cost catalysts such as Ni, Co, Cu and Fe 9 have good catalytic activities for the reaction. Au-Co core-shell nanoparticles were shown to have excellent catalytic activity coupled to long term stability for AB hydrolytic dehydrogentation. 10 AB can thermally decompose in the temperature range 340–410 K to liberate 2.2 mol H2 per mole of AB which is equivalent to a hydrogen storage density of 14.3 wt %. 11 The high energy density coupled with the moderate decomposition temperature makes AB an attractive hydrogen source for fuel cells. However it has been acknowledged that low temperature fuel cells such as those that power electronic devices would benefit from replacing hydrogen with liquid fuels in terms of safety and design simplicity. Liquid fuels such as methanol and ethanol have been used as a substitute to hydrogen for such fuel cells although their oxidations are sluggish. Direct oxidation of AB is a viable alternative that merits attention given its high capacity (5.2 Ah g−1) coupled to its high energy density (8.4 Wh g−1) which compares favourably with that of methanol (6.20 Wh g−1) and ethanol (8.0 Wh g−1).
The direct electro-oxidation of AB (Eq. 2) has been investigated at a Au microdisc 15 and Au disc electrodes in alkaline media. 16, 17 High catalytic activity was demonstrated for AB oxidation at magnetically recyclable Fe-Pt core-shell nanoparticles with amorphous Fe cores. 18 It has recently been proposed that if the direct electrochemical oxidation of AB occurs in a fuel cell a more negative potential and greater power can be obtained than indirectly releasing hydrogen as the fuel source. 16–21 The standard-state potential of Reaction 2 has been calculated as − 1.216 V vs. SHE, which is 0.388 V more negative than the hydrogen electrode in an alkaline medium. A novel direct AB fuel cell (DABFC) can be proposed by combining the anodic oxidation of AB given by Eq. 2 and cathodic reduction of oxygen given by Eq. 3 to give an equilibrium voltage, of 1.616 V, Eq. 4
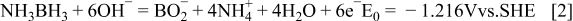
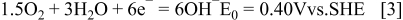
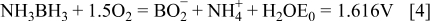
The equilibrium voltage is almost as high that for a direct borohydride fuel cell (DBFC) (1.64 V). AB is stable in aqueous solution at pH ≥ 6.5 unlike borohydride which undergoes self-hydrolysis in alkaline solutions of pH < 9. 22 Kreevoy and Jacobson 23 reported that the hydrolysis of NaBH4 depends on pH and temperature;
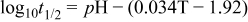
where t1/2 is the half-life for the self-hydrolysis of the NaBH4 solution (min) and T the storage temperature (K). The half life at pH 12 at 298 K is 4.3 days. Hence, it is more effective for the aqueous NaBH4 solution to exist in an alkaline state with pH > 12 in order to prevent its self-hydrolysis. This is an advantage of a DABFC over a DBFC. The BO2 − byproduct can be converted into BH4 − upon reaction with MgH which can then be converted into AB through reaction with diethyl ether. 16 AB spontaneously yields BH3(OH)− in alkaline solution. Careful selection of the anode catalyst in a DABFC coupled to a high OH−:BH3(OH)− ratio is crucial in minimising unwanted competing heterogeneous hydrolysis of BH3(OH)− given by Eq. 5 which results in non-faradaic hydrogen evolution thereby decreasing fuel utilisation and lowering cell performance
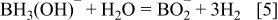
Au is an effective catalyst for oxidation of BH3(OH)− and shows low catalytic activity with respect to its hydrolysis over a certain concentration range. The electrooxidation of AB at Au is believed to involve a CE type reaction; the chemical dissociation of BH3 from AB which reacts with OH- to form BH3(OH)− (Eq. 6) followed by electrochemical oxidation to B(OH)4 −. The complete oxidation of BH3(OH)− to B(OH)4 − follows a stepwise mechanism in which intermediates can either be hydrolysed to H2 or electrochemically oxidised (Eq. 7) such that n can range from 3 to 6, depending on how many electrons are lost to H2 formation 24
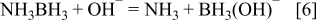
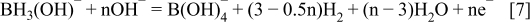
In strongly alkaline solutions an n value of 6 is favoured for the electrooxidation of BH3(OH)−. 15, 24 AB was shown to oxidize at a Au microdisc in 1 M NaOH with maximum coulombic efficiency of 6 when the ratio of OH−:AB is sufficiently high to minimize competing hydrolysis at a 10 μm microdisc in 1 M NaOH. 15 The oxidation commenced at a potential of −1.06 V vs. Ag/AgCl (− 1.10 V vs. SCE) to produce two 3-electron steady-state mass transport controlled at − 0.65 V and − 0.15 V vs. Ag/AgCl. Finkelstein et al. 25 and Zhang et al. 17 showed that Au promotes the partial heterogeneous hydrolysis of BH3(OH)− especially at low potential. Establishing a technology for a DABFC hinges on understanding the reaction mechanism and kinetics and the associated catalytic species at the anode.
We have identified nanoporous Au (NPG) as a potential anode catalyst for a DABFC. NPG can be formed by dealloying which involves selective metal dissolution. This has an ancient history, Incan civilisations dealloyed Cu from the surface of Cu-Au alloys to create an illusion of a pure Au artefact known as depletion gilding. Forty 26 showed that depletion gilding of a less-noble metal from Au alloys results in an open, continuous nanoporous Au structure. When the Ag component in an AgxAuy alloy is dissolved in acid the remaining Au atoms gather together in clusters to create a rough surface that causes Au to evolve into a porous material. The sponge-like 3D structure is a system of interconnecting pores/tunnels in a skeleton of filaments of the metal. The filaments can range from 5 to 50 nm in size with surface areas as high as 20 m2g−1 having a porosity of 70% or higher. It was highlighted that NPG brings together two seemingly conflicting properties - high porosity and high strength. 27 NPG was shown to be as strong as bulk Au, despite being a highly porous material and that its ligaments approach the theoretical yield strength of Au. Structurally NPG resembles naturally occurring zeolites 28 (filament size 1–2 nm and surface areas 100 m2 g−1). This useful but relatively unstudied form of Au most likely contains an intrinsically high step density.
Given that NPG has an interconnected, bicontinuous ligament network containing regions of both negative and positive curvature, a high step density is topologically required. This characteristic makes NPG attractive for catalysis studies; it is even more attractive because it can be formed into thin, high-conductivity foils that are easily adapted to electrocatalytic measurements. The bicontinuous open porous structure allows unlimited transport of molecules through the material. In terms of catalytic applications, NPG offers at least two advantages over other catalysts or supported Au nanoparticles. Firstly, NPG remains active at low temperature (room temperature or lower), unlike Pt or Pd catalysts. 29, 30 Secondly, NPG exhibits good thermal stability and resistance to oxidation 31 and thus can overcome the aggregation or sintering limitations which Au nanoparticles encounter at elevated temperatures or in an oxidative environment. 32 Zeis et al. 33 demonstrated that NPG is an effective catalyst for reduction of hydrogen peroxide to water. Their results are consistent with their overall hypothesis that the central difference between NPG and bulk Au is due to the increased density of step edges in NPG over bulk Au. It was shown that NPG shows much higher electrocatalytic activity towards methanol than polycrystalline Au. 34 Wittstock et al. 35 demonstrated selectivity levels in excess of 97% for gas-phase oxidative coupling of methanol to methyl formate at temperatures below 80°C with high turnover frequencies. High electrocatalytic activity in the oxidation of glucose at NPG was shown by Li et al. 36 We have recently investigated borohydride oxidation reaction at NPG electrodes. The onset potential for borohydride oxidation at NPG in a wire array format shifted to more negative potentials than that observed at a Au disc and higher currents were realized. 37 An onset potential of − 1.07 V vs. SCE was recorded for 20 mM borohydride in 1 M NaOH which is 0.207 V lower than that at a Au disc and the corresponding oxidation current increased to 73.6 mA cm−2 from 3.2 mA cm−2. An n value of 7.49 was determined for the oxidation peak at high potential (− 0.49 V) while a value of 4.26 was determined at low potential for the oxidation plateau centered at − 0.05 V. NPG presents an attractive alternative to Au nanoparticle-based catalysts for use in direct borohydride fuel cells. It is important to note that the pace of discovery of reactions for which NPG is a good catalyst is growing rapidly thereby prompting the investigation of many different systems in which NPG may be the superior catalyst. In this paper we report on the exploitation of NPG with superior electrocatalytic activity and high specific surface area to develop an anode catalyst for a DABFC.
Experimental
Ammonia borane (minimum purity 97%), sodium carbonate (minimum purity 99%) and sodium hydroxide (minimum purity 99%) were purchased from Sigma Aldrich and used as received. KAg(CN)2 (minimum purity 99%) and KAu(CN)2 (minimum purity 99%) were purchased from Johnson Matthey and used as received. Deionized water of resistivity 18 MΩ cm was used to prepare all solutions. Anodisc alumina circular membranes (Anodisc 25) 2.5 cm in diameter, 60 μm thick, 200 nm pore size and 109 pore openings per centimeter square of membrane were supplied by Whatman. These show compatibility with a wide range of solvents and acids and can be dissolved in sodium hydroxide and ammonium hydroxide solutions. The maximum temperature at which they are stable is 400°C. E-beam evaporation was used to deposit a conducting backing layer of Au (350 nm in thickness) on the rear side of the alumina membranes prior to the templated-electrodeposition of Au and AuxAg1–x wire arrays. The working electrode was a 5 mm Au disc (Princeton Applied Research) supplied by Advanced Measurement Technology, U.K. This was polished with 0.5 µm alumina powder obtained from Struers on a Buehler polishing cloth for 2 min and rinsed in deionized water. A commercial gold bath (Puramet 402) was purchased from AMI DODUCO. A 1 mm diameter Au wire 50 mm in length was used as counter electrode. Cyclic voltammograms (CV) were recorded with respect to a standard calomel electrode (SCE). The potential of the working electrode was controlled using a CH Instruments potentiostat model 660C with picoamp booster. All solutions were purged with nitrogen for 20 min prior to experiments in order to remove oxygen, and the experiments were performed at 20°C. Current densities for the electrodes are based on the geometric surface areas of the electrodes. The geometric surface area of the 5 mm diameter Au disc is 0.196 cm2. The geometric surface area of the NPG and Au wire array electrodes supported on the Au-backed 2.5 cm diameter Anodisc is 3.928 cm2. TEM images were recorded using JEOL 2000FX at an accelerating voltage of 200 kV. SEM images were recorded using Nova Nanosem 630 at an accelerating voltage of 15 kV.
Results and Discussion
Fabrication of NPG electrodes
NPG on a planar Au film
It was shown by Ji and Searson 38 that Au0.18Ag0.82 alloy can be deposited from a solution of 100 mM KAg(CN)2 and 20 mM KAu(CN)2 in 250 mM Na2CO3, pH 13, at a constant potential of − 1.2 V. XPS was used to confirm the alloy composition. Herein a film of Au0.18Ag0.82 was deposited on a 200 nm Au film on a pyrex wafer for 5000 s according to the method of Ji and Searson. 38 It was demonstrated that the morphology and porous structure of NPG depends on the composition of the AuxAg1–x alloy for the range 0.18 ≤ x ≥ 0.32. 38 The alloy composition Au0.18Ag0.82 gives the shortest ligament size of 20–30 nm and the highest surface area NPG (6.9 m2 g−1) when dealloyed in nitric acid. The CV response recorded for the alloy in 1 M NaOH from − 0.9 to 0.6 V vs SCE at 10 mV s−1 (not shown here) indicated the presence of Au and Ag from the metal oxide reduction peaks seen at − 0.10 and 0.40 V, respectively. Upon immersing Au0.18Ag0.82 in 30% nitric acid for 2 h a high surface area form of Au referred to as NPG is obtained. The NPG film was delaminated from the underlying Au film and sandwiched in a holey Cu folding TEM grid in order to obtain the TEM image shown in Fig. 1a. The pore and ligament size are 20 and 30 nm, respectively.

Figure 1. (a) TEM image of NPG (b) SEM image of NPG wire array (c) SEM image of NPG wires grown on Au wire array and (d) SEM image showing top-down view of NPG-coated Au wire array.
NPG in a wire array
The fabrication of NPG in a 3D wire array format was achieved by dealloying Au0.18Ag0.82 nanowires that were deposited in Anodisc alumina membranes with 200 nm pore size using the conditions given for the fabrication of NPG on a planar Au film. The alumina was dissolved by soaking it in 0.5 M NaOH for 2 h to release the NPG wires. The SEM image shown in Fig. 1b was recorded for the resulting NPG wires which were 2 μm in length.
It was attempted to add structural rigidity to the NPG wires by depositing (i) a coating of NPG on a Au wire array and (ii) NPG wires onto the ends of a Au wire array, as outlined below. This invariably leads to an increase in the amount of Au used in the electrode fabrication, however.
NPG-coated Au wire array
A Au wire array was deposited in Anodisc alumina membranes with a 350 nm Au conducting backing layer from a commercial Puramet 402 bath. The Au wires were deposited to a length of 1 μm and 200 nm in diameter. The alumina was dissolved by soaking it in 0.5 M NaOH for 2 h to release the Au wires. The Au wire array was then coated with NPG. This was achieved by dealloying depositing Au0.18Ag0.82 on the on the Au wires nanowires which was subsequently dealloyed using the conditions given above for the fabrication of NPG on a planar Au film. The SEM image shown in Fig. 1c was recorded for the resulting NPG-coated Au wires. The NPG coating was 285 nm in thickness.
NPG-Au segmented wire array
A Au wire array was deposited in Anodisc alumina membranes with a 350 nm Au conducting backing layer from a commercial Puramet bath. The Au wires were deposited to a length of 1 μm and 200 nm in diameter. The fabrication of NPG onto the Au wires was achieved by dealloying AgxAuy wires that were then deposited on the Au wires in the Anodisc alumina membranes using the conditions given for the fabrication of NPG on a planar Au film. The alumina was dissolved by soaking it in 0.5 M NaOH for 2 h to release the segmented NPG-Au wires. The SEM image shown in Fig. 1d was recorded for the resulting wire array. The length of the NPG wire segment was 1.5 μm.
The electrochemistry of NPG in 1 M NaOH
Typical behaviour for Au in 1 M NaOH is seen in the CV recorded for NPG deposited on a Au disc shown in Fig. 2. The CV shows that negligible current flows over the potential range − 1.0 to + 0.05 V. The onset for monolayer oxide formation is shown to occur above + 0.05 V with the corresponding oxide reduction peak on the reverse sweep.
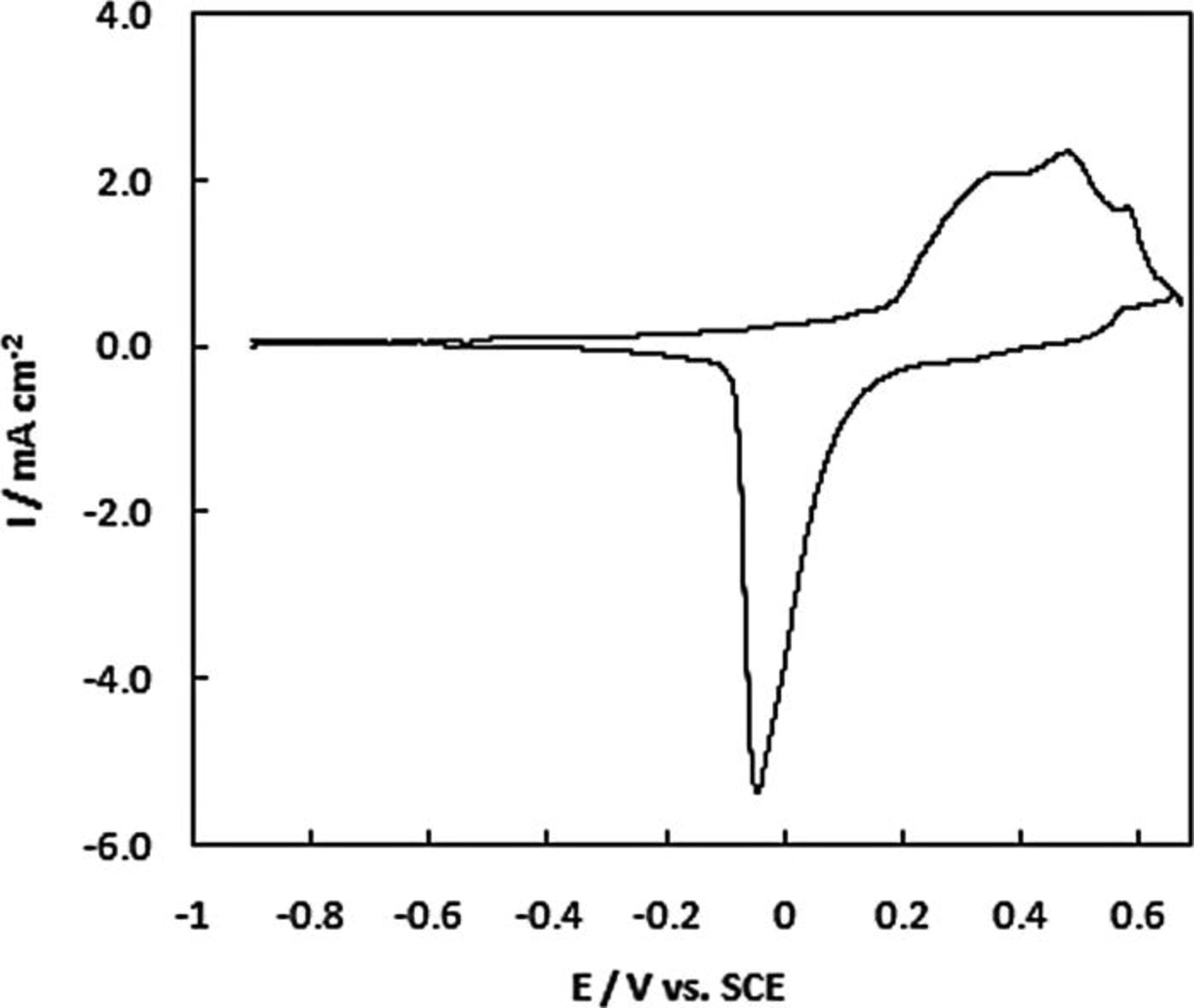
Figure 2. Cyclic voltammogram for NPG on a Au disc electrode in 1 M NaOH recorded at 10 mV s−1.
Electrooxidation of AB at Au disc
The oxidation response for AB at a clean Au disc is shown in Fig. 3. The upper scan potential limit was restricted to 0.10 V in the forward sweep to avoid the formation of Au oxide as BH3(OH)− does not show oxidation activity on that material. The oxidation of AB commences at −1.02 V which is 0.08 V more positive than that at a Au microdisc. 15 In the forward sweep an oxidation peak a1 was recorded at − 0.84 V followed by a broad oxidation wave a2 from –0.49 to –0.10 V. The location of the anodic features a1 and a2 agree with those reported in our microdisc studies of AB and DMAB oxidation at Au 15, 21 and with those reported by Finkelstein et al. 25 They identified two regions of mass transport limited current for BH3(OH)− from − 0.72 to − 0.46 V and from − 0.29 to 0.28 V vs. Ag/AgCl (or − 0.77 to − 0.51 V and − 0.34 to 0.24 V vs. SCE) in their rotating disk electrode (RDE) analysis of DMAB oxidation in 1 M NaOH. 25 Both waves were attributed to CE-type reactions from rotation rate normalised current profiles. The electrochemical step in the CE peak at lower potential was assigned to oxidation of both H2 and BH3(OH)− demonstrating that Au actually has activity for H2 oxidation despite claims to the contrary by Krishnan et al. 39 Recent studies have shown that the faradaic efficiency of Au for BH3(OH)− oxidation is lower than was assumed previously as it can catalyse BH3(OH)− hydrolysis to produce H2. 40 This was demonstrated in their online electrochemical mass spectrometry study of the oxidation of borohydride and its BH3(OH)− intermediate at Au. The peak a1 at − 0.84 V was also reported 17 for the oxidation of 20 mM AB in 2 M NaOH at 100 mV s−1 at a Au disc. They assigned this to electrooxidation of H2 generated from the hydrolysis of AB which they confirmed by its disappearance upon the addition of thiourea which inhibits the recombination of surface hydrogen radicals. The second broad oxidation wave a2 seen at − 0.30 V in Fig. 3 can be attributed to the direct oxidation of AB 15, 16, 19 according to Eq. 7. The current decreases as the potential is swept into the Au monolayer oxide region. Sadik et al. 41 showed that BH3(OH)− has intermediates that can only be oxidised at very high potentials.
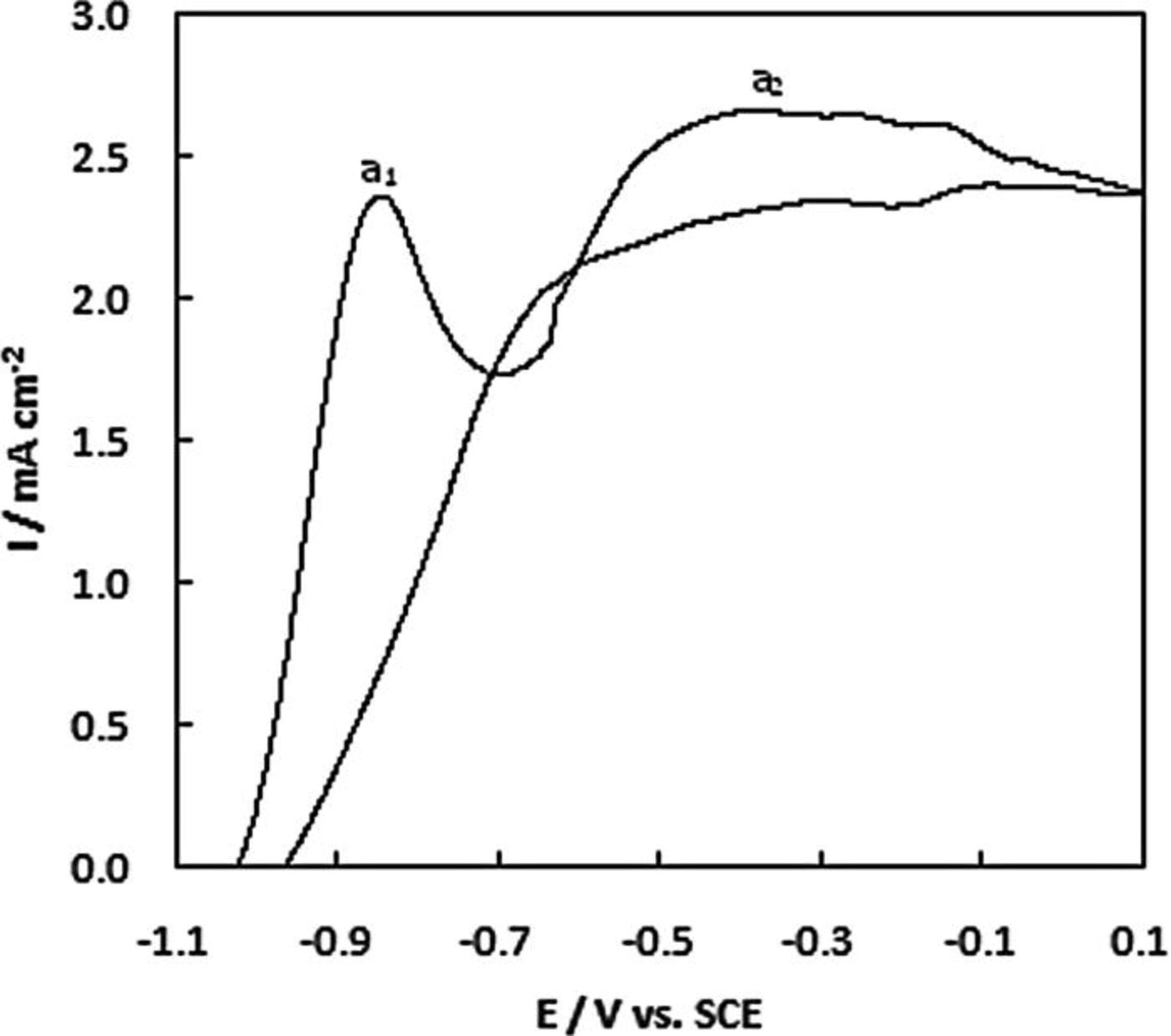
Figure 3. Cyclic voltammogram for NPG on a Au disc in 1 M NaOH containing 20 mM AB recorded at 10 mV s−1.
Electrooxidation of AB at NPG electrodes
An open circuit potential (OCP) of − 1.27 V was recorded for the NPG wire array which cannot entirely be linked to hydrogen oxidation as Eo(H2/H+) = −0.818 V vs. SHE (i.e. − 1.060 V SCE). This provides evidence for the direct electrooxidation of BH3(OH)−. The linear scan voltammetric responses (LSV) for 20 mM AB in 1 M NaOH at a Au disc and at a Au wire array are shown in Figs. 4a and 4b, respectively and the comparable responses at the fabricated NPG electrodes are shown in Figs. 4c, 4d and 4e. The current densities for the electrodes are based on their geometric surface areas. At the NPG wire array the onset potential for the oxidation of AB has shifted to − 1.30 V, 0.28 V more negative than that at the Au disc. The peak a1 * located at − 0.95 V may be assigned partially to the electrooxidation of BH3(OH)− and partially to the oxidation of hydrogen generated from its heterogeneous hydrolysis. This is located at a potential 0.11 V more negative than peak a1 for a Au disc shown in Fig. 4a. The broad anodic wave from − 0.49 to − 0.10 V associated with direct electrooxidation of BH3(OH)− observed at the Au disc has been shifted to lower potential at the NPG wire array and resolved into peaks a2 * at − 0.50 V and a3 * at − 0.15 V. The oxidation current recorded for peak a2 * of 13.1 mA cm−2 is 10.5 mA cm−2 higher than that recorded for peak a2 located at − 0.30 V at a Au disc electrode, this represents more than a fivefold increase in current density.
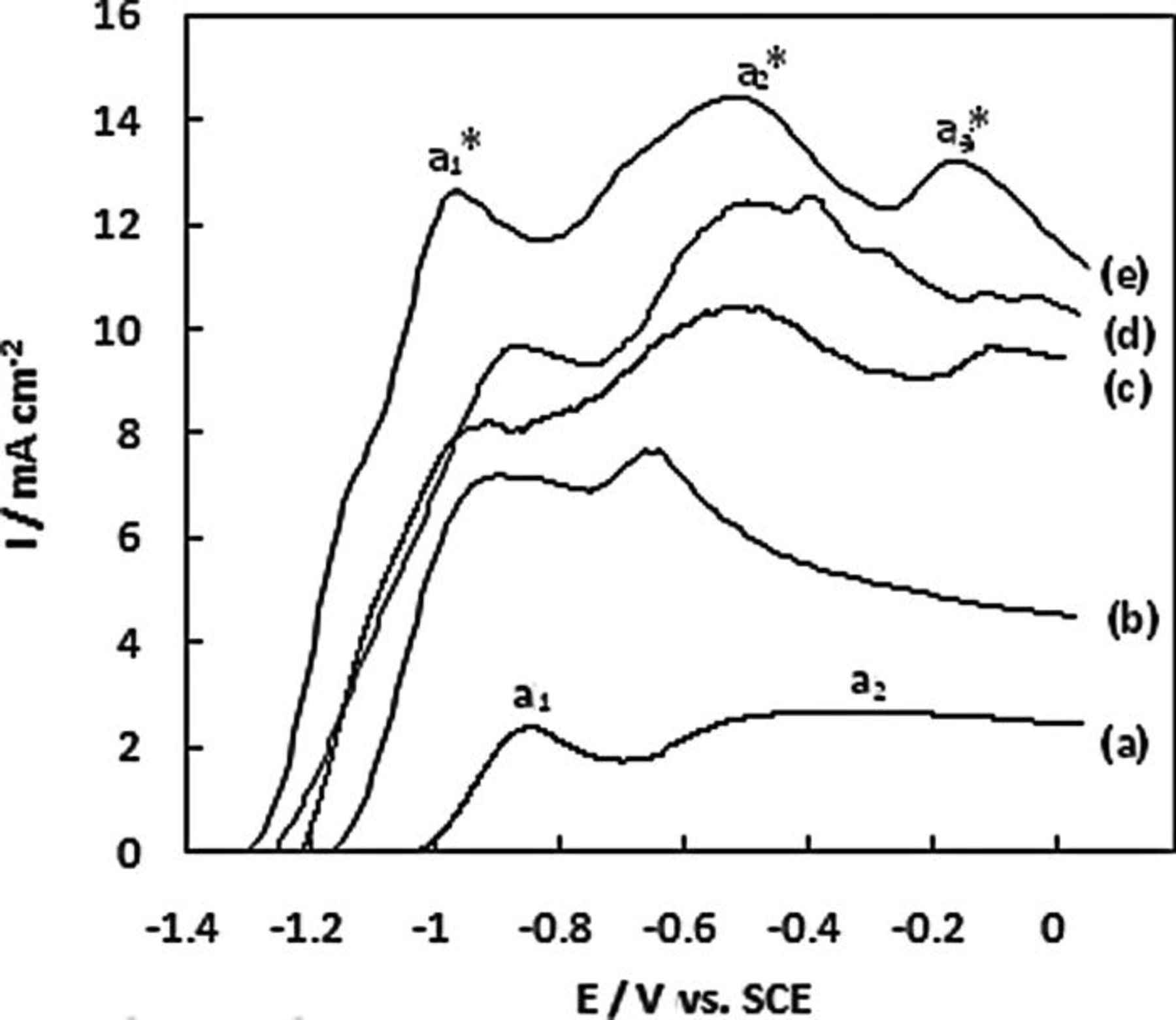
Figure 4. Linear scan voltammogram recorded at 10 mV s−1 for (a) Au disc (b) Au wire array (c) NPG-coated Au wire array (d) NPG-Au segmented wire array and (e) NPG wire array in 1 M NaOH containing 20 mM AB.
At the NPG-coated Au wire array the onset potential for the oxidation of AB has shifted to −1.21 V which is 0.19 V more negative than that at the Au disc. The anodic peaks located at − 0.90, − 0.49 and − 0.08 V may be tentatively assigned in the same manner as peaks a1 *, a2 *, a3 *, respectively, observed at the NPG wire array shown in Fig. 4e. The anodic peak at − 0.49 V has an oxidation current of 10.5 mA cm−2 which is 9 mA cm−2 greater than that for peak a2 at the Au disc electrode. At the NPG-Au segmented wire array the onset potential for the oxidation of AB has shifted to − 1.28 V, also more negative than that at a Au disc by 0.26 V. The anodic peaks located at − 0.90, − 0.49 and − 0.08 V may be tentatively assigned in the same manner as peaks a1 *, a2 *, a3 *, respectively, observed at the NPG wire array.The three anodic peaks observed at − 0.85, − 0.47 and − 0.09 V are also assigned in a similar manner as peaks a1 *, a2 *, a3 *, respectively observed at the NPG wire array. The oxidation current recorded for the anodic peak at − 0.47 V of 12.7 mA cm−2 is 10 mA cm−2 higher than that recorded for peak a2 at a Au disc electrode. Contrastingly, it was shown that the onset potential for AB oxidation at a non-porous high surface area Au wire array with 2.5 μm wire length and 200 nm diameter Au was − 1.13 V and the oxidation current at − 0.64 V was 5.2 mA cm−2. This demonstrates that the enhanced AB oxidation response recorded at NPG is not attributable to simply an increase in surface area.
RDE linear scan voltammograms recorded at NPG-coated Au disc in 20 mM AB in 1 M NaOH deposited on a Au disc are shown in Fig. 5 for a range of rotation rates. Two regions of mass-transport-limited current were observed for a low potential wave from − 0.90 to − 0.85 V and for a high potential wave from − 0.10 to 0.22 V. These waves were observed at more negative potentials than the similar waves that were recorded at a Au disc for DMAB (which is a source of BH3(OH)−) at − 0.72 to − 0.46 V and − 0.29 to 0.28 V vs. Ag/AgCl by Finkelstein et al. 25 Levich plot analysis (using Eq. 8) shown in Figs. 6 and 7 for the high- and low potential waves, respectively, revealed a diffusion-controlled reaction
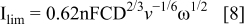
where ν is the kinematic viscosity and D is the diffusion coefficient.

Figure 5. Linear scan voltammetric response for NPGcoated Au disc at rotation rates (a) 200, (b) 400, (c) 1500, (d) 2500 and (e) 3500 rpm in 1 M NaOH containing 20 mM AB at 5 mV s−1.
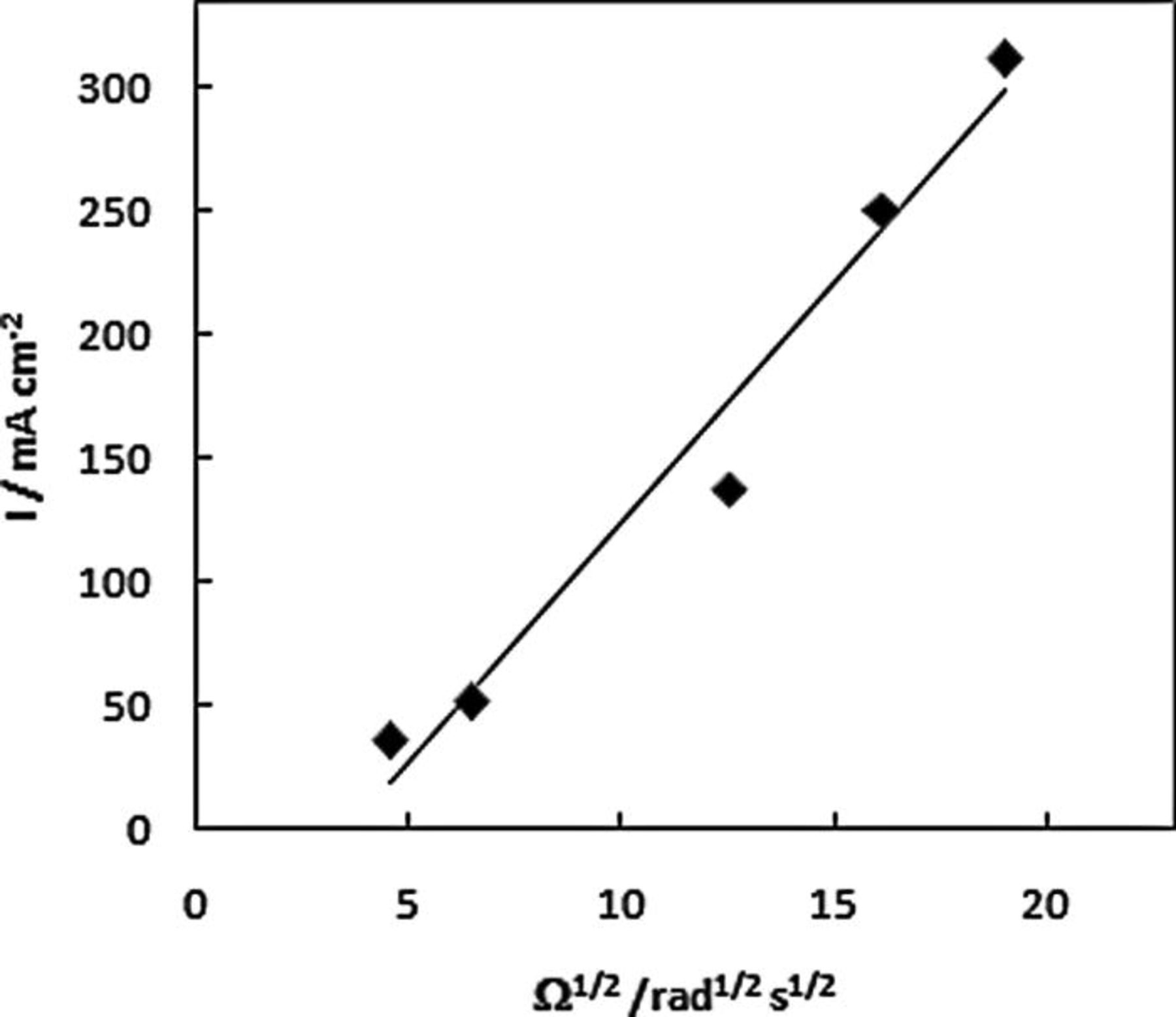
Figure 6. Levich plot for low potential wave for NPG-coated Au disc in 1 M NaOH containing 20 mM AB at 10 mV s−1.
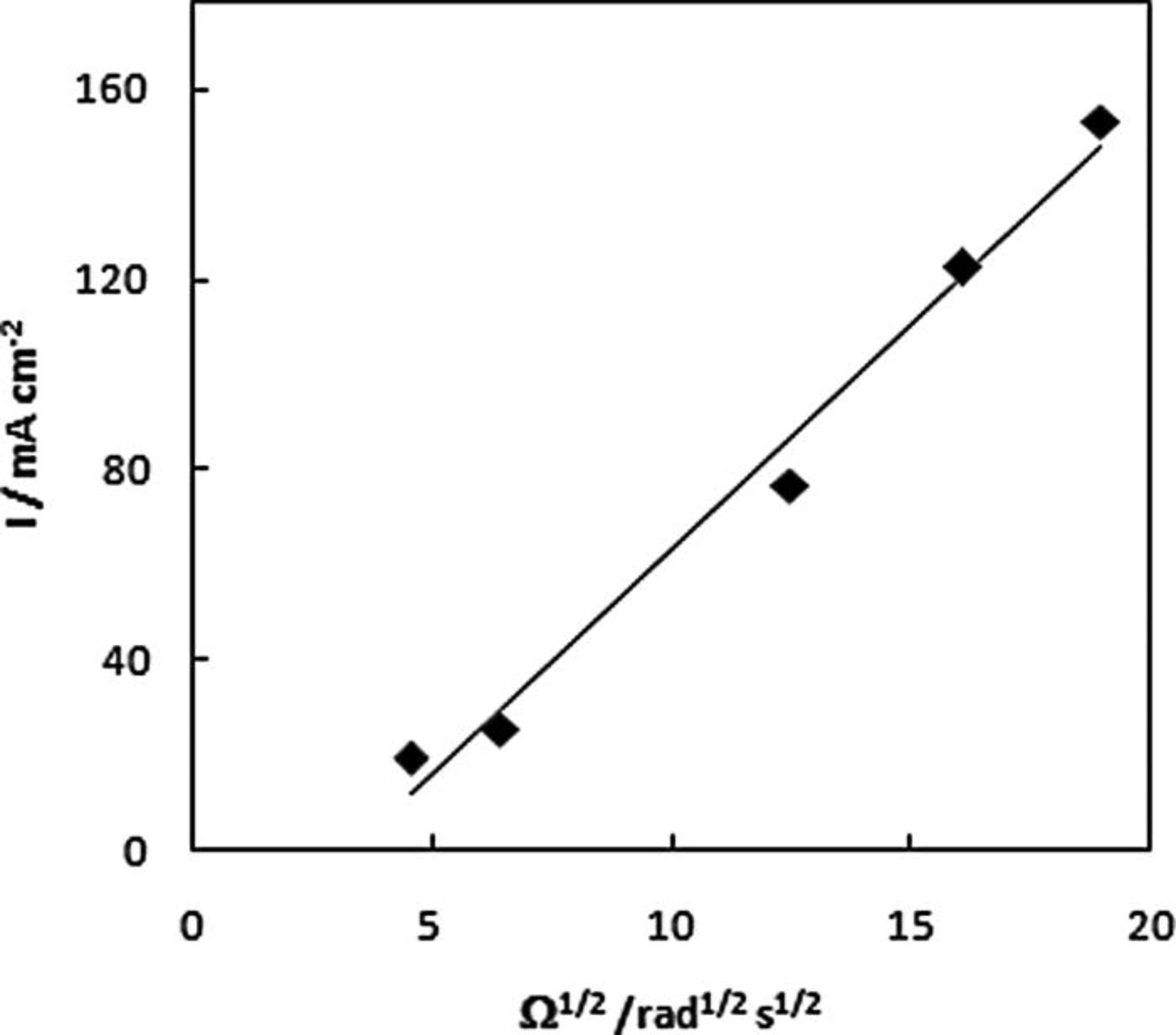
Figure 7. Levich plot for high potential wave for NPG-coated Au disc in 1 M NaOH containing 20 mM AB at 10 mV s−1.
The influence of scan rate on the oxidation response for AB was studied at the NPG-coated Au disc. The peak current for the low potential wave increases linearly with the square root of scan rate as shown in Fig. 8, suggesting that it can be attributed to the oxidation of a species in solution. The positive shift of the peak potential for this wave with scan rate is indicative of a sluggish reaction. The peak current for the high potential wave increases linearly with scan rate as shown in Fig. 9, this behavior is typical of the oxidation of an adsorbed species. A positive shift of the peak potential was also observed for the high potential wave with increasing scan rate. The increase in peak potential with scan rate is indicative of a CE mechanism which was earlier proposed by Finkelstein et al. 25 from a rotation-rate normalized study of BH3(OH)−) oxidation at Au.
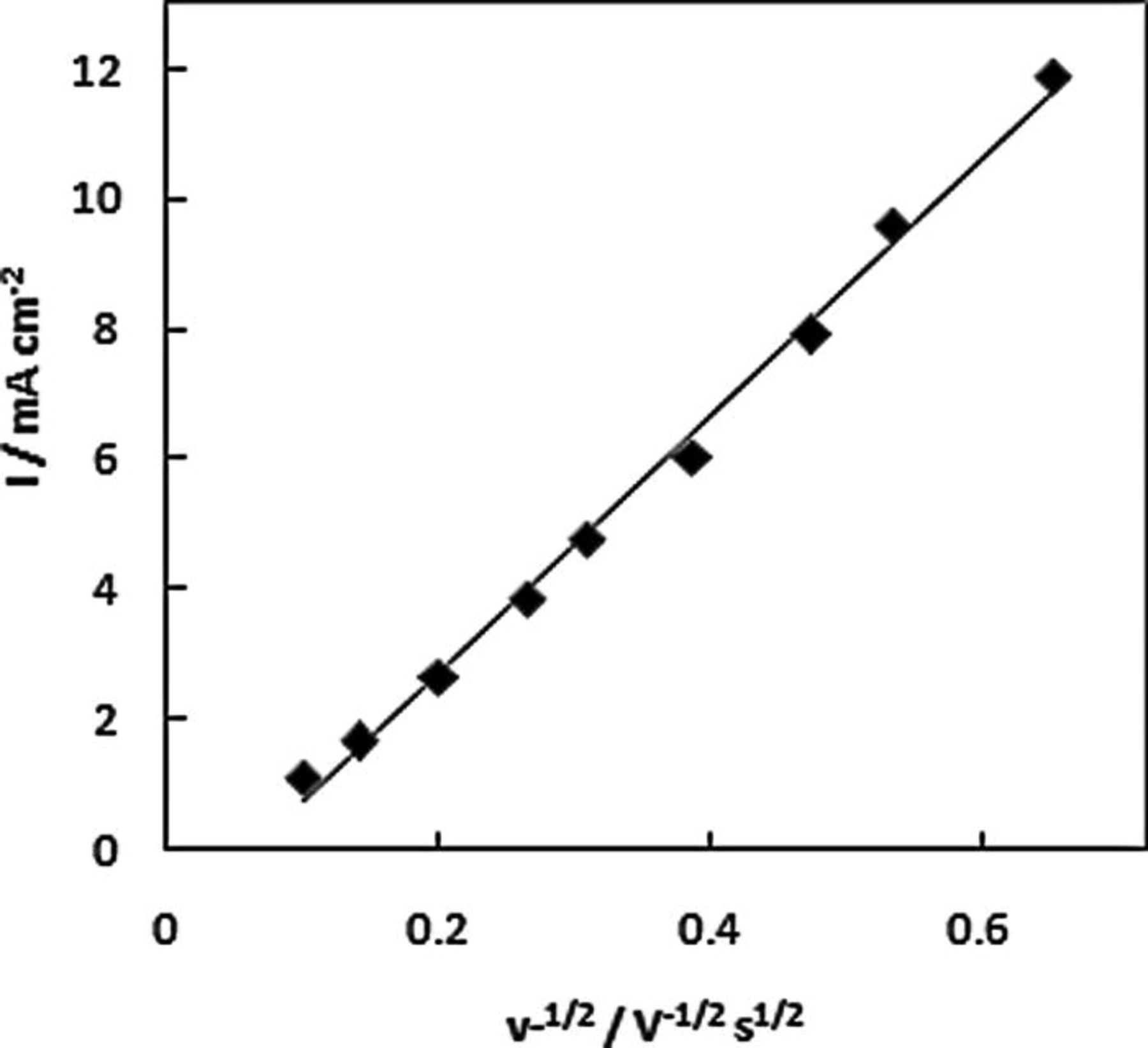
Figure 8. Peak current for low potential wave at NPG-coated Au disc in 1 M NaOH containing 20 mM AB as a function of the square root of scan rate.
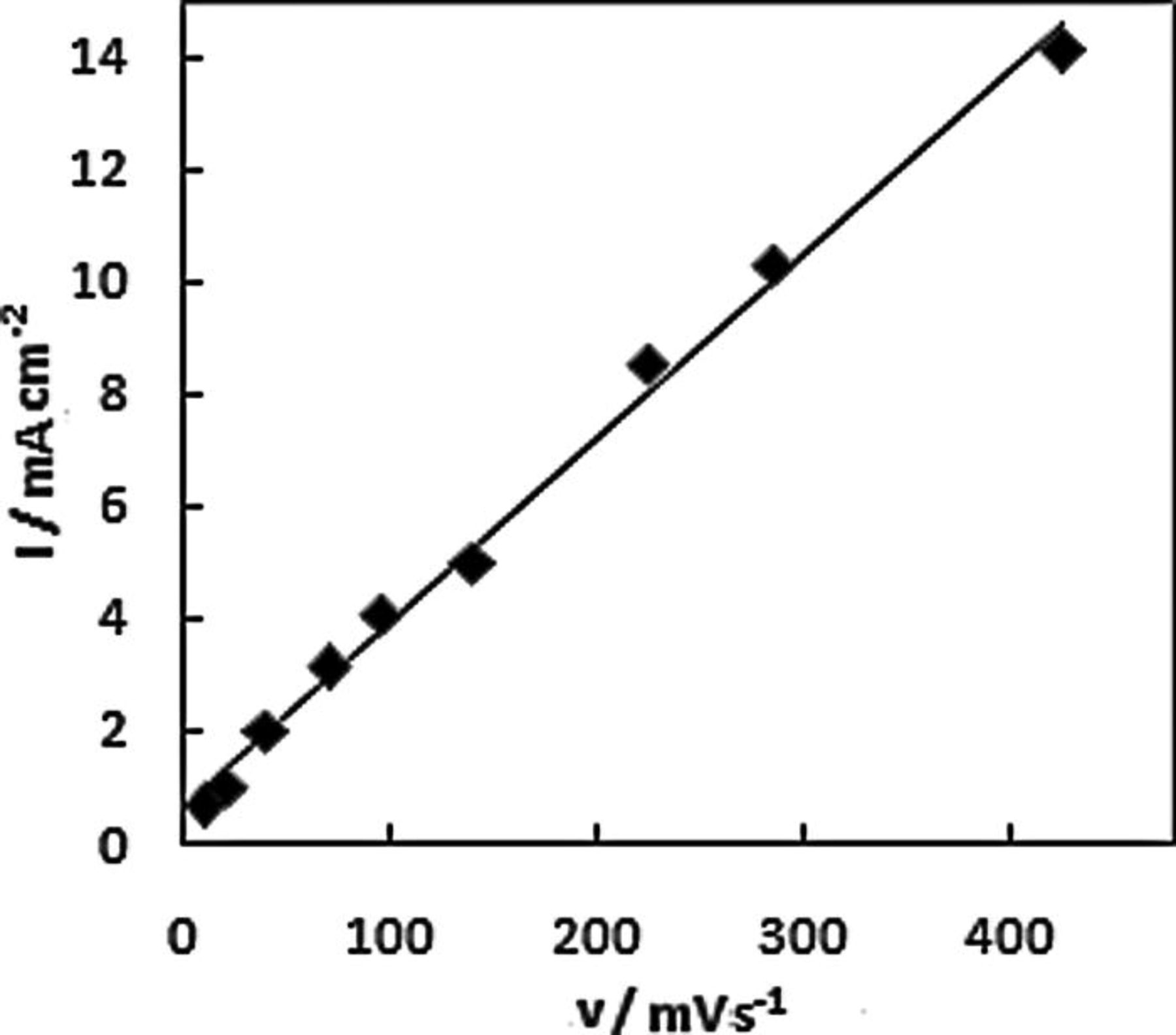
Figure 9. Peak current for high potential wave at NPG-coated Au disc in 1 M NaOH containing 20 mM AB as a function of scan rate.
Chronoamperometry was used to further test the catalytic activity of NPG and to complement the voltammetry studies on static and rotating electrodes. The potential was stepped from − 1.30 V s at a NPG-coated Au disc in 20 mM AB in 1 M NaOH to − 0.10 V and the current was recorded (Fig. 10). From the resulting current transient the number of electrons involved in the oxidation (n) was determined from the Cottrell equation (Eq. 9) as 5.97
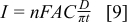
where A is electrode are and D is diffusion coefficient which was estimated as 8.45 × 10−6 cm2 s−1 from a study of AB oxidation at a Au microdisc. 15 The value for n is in close agreement with the value of 6 which was found for the AB oxidation wave at − 0.15 V vs. Ag/AgCl at a Au microdisc 15 and represents high coulombic efficiency for the reaction at NPG.
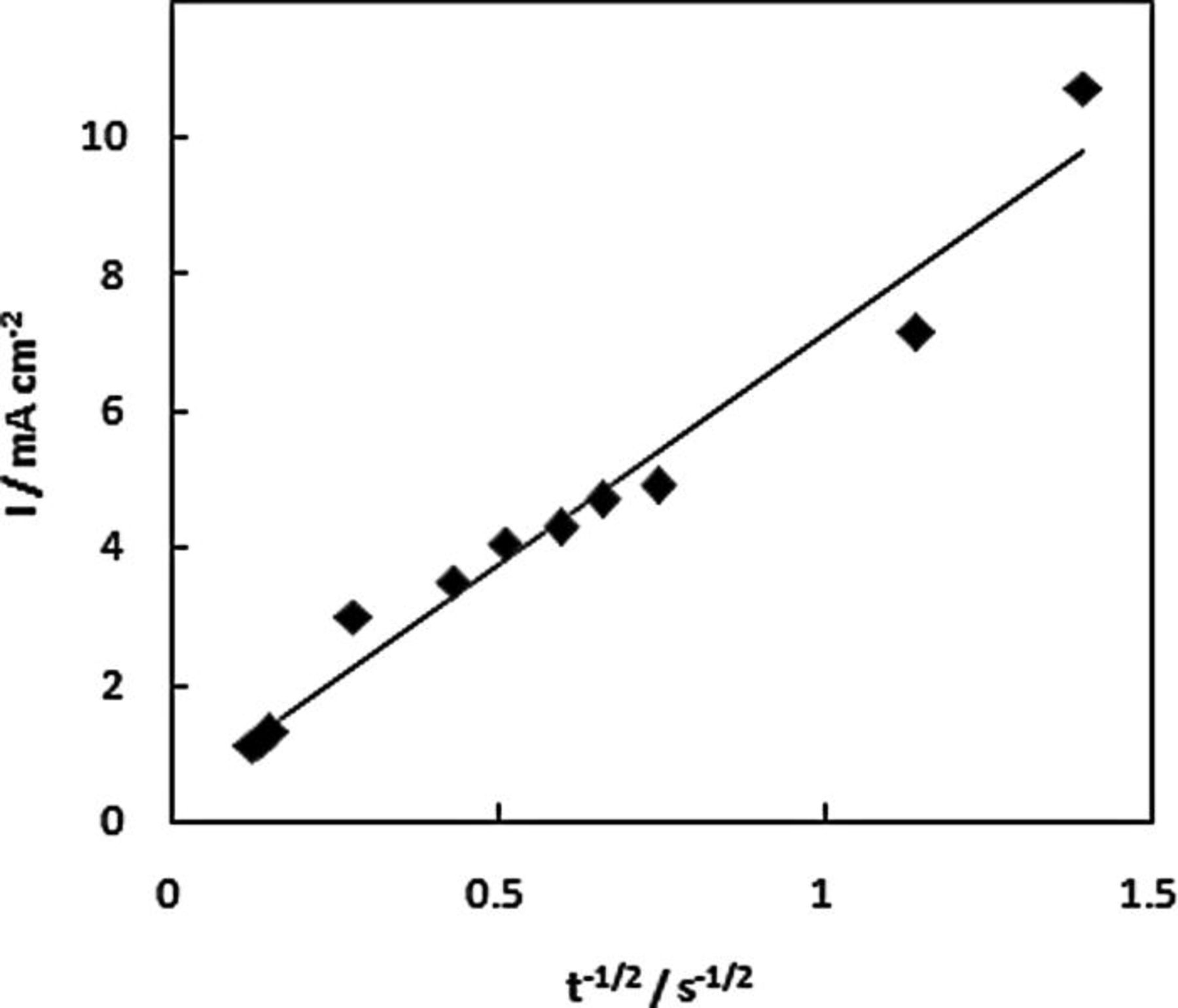
Figure 10. Plot of I vs. t−1/2 for 20 mM AB in 1 M NaOH at NPG-coated Au disc over a time scale of 60 s in response to a potential step from − 1.30 to − 0.10 V.
The characteristics of NPG for AB oxidation for the series of electrode formats that were studied are summarised in Table I. The onset potential for AB oxidation is lowest at the NPG wire array and the oxidation current is highest.
Table I. Variation of onset potential for AB oxidation and current density at a range of Au substrates.
| Substrate | AB onset potential/V vs SCE | I/mA cm−2 |
|---|---|---|
| Planar Au | − 1.02 | 2.65 |
| Au wire array | − 1.13 | 5.2 |
| NPG on planar Au | − 1.20 | 3.1 |
| NPG wire array | − 1.30 | 13.1 |
| NPG-coated Au wire array | − 1.21 | 10.5 |
| NPG wires supported on Au wire array | − 1.28 | 12.7 |
Our results highlight that NPG shows higher catalytic activity for AB oxidation than is observed at bulk planar Au which was shown to be catalytically active for both AB oxidation and hydrogen evolution and oxidation1 at low potential; AB oxidation is more favoured at low potentials than hydrogen evolution at NPG. Different sites co-exist on Au, some catalyse AB oxidation while others catalyse hydrogen evolution indicating the structural sensitivity of the reactions. It is possible that the density of sites at which AB undergoes oxidation is increased at NPG over that at bulk Au. It is also possible that the NPG active layer structure promotes an increase in the residence time of reaction intermediates at the surface leaving more time for complete oxidation to borate. Such behaviour was recently reported by Molina Concha et al. for Pt and Pt/C electrodes 6, 42 and for the multi-step oxygen reduction reaction at Pt nanostructure arrays at planar glassy carbon electrodes. 43 A consensus has not been reached as to what particular property or properties of NPG lead(s) to its catalytic properties despite a growing body of literature evidence. A single definitive mechanism does not exist which can explain all aspects of catalysis for nanoporous or nanostructured Au. The only overarching hypothesis is that the catalytic activity results from some special site, the density of which is highly sensitive to material properties, and the effect of which is amplified in materials of very high specific surface areas where the special sites have a particular electronic configuration. 44
An intrinsically higher step density and hence greater percentage of low coordination number active site atoms are believed to be present at NPG relative to bulk Au. Such sites may assist in the adsorption of reactants via steric or electronic interactions. Indirect evidence exists for the presence of low coordinated surface atoms at NPG which show unusually high catalytic activity. Zhang et al. 34 showed that by galvanically displacing Au by Pt from the surface of NPG the thermal stability of NPG is greatly increased; the most likely sites for galvanic displacement are low coordination step edges. It was suggested that the high curvature ligaments present in NPG may expose various facet orientations 33 and it is possible that a particular facet may be responsible for a particular reaction.
The presence of residual silver was detected in NPG by EDX to concentration levels of less than 1 and 4% (Refs. 33, 35 ) and by XPS to concentration levels of 2.0 and 4.4 atom % Ag. 30, 31 Wittstock et al. 35 showed that residual Ag in NPG participates in the activation of molecular oxygen in the oxidation of methanol. They showed that the oxidation power of NPG may be tuned by adjusting the Ag concentration. It was suggested that residual Ag buried in NPG may contribute to its high catalytic activity for CO oxidation. 30, 31 It is possible that residual Ag present in NPG may assist in promoting the direct oxidation of AB and shifting the OCP to more negative values than at bulk Au. Support for this hypothesis is provided in a study of the direct oxidation of AB at Ag/C by Zhuang et al. 20 The oxidation of 0.1 M AB in 2 M KOH was shown to commence at − 1.10 V vs. Hg/HgO (or − 1.20 V vs. SCE) which is more negative than that of hydrogen evolution (− 0.93 V vs. Hg/HgO) indicating that the anodic current is not entirely attributable to the oxidation of hydrogen produced from AB hydrolysis at this potential at Ag. Chatenet et al. 45 also showed that Ag appears to be a better catalyst for BH3(OH)− oxidation than Au with an onset for oxidation 0.15 V lower at Ag than at Au. More detailed investigation into the catalytic activity of NPG in terms of the role of low-coordinated surface Au atoms and the presence of residual Ag is needed to further elucidate the underlying reaction mechanism for AB oxidation at NPG.
Conclusions
The application of NPG as a fuel cell electrode heralds exciting new avenues for catalyst design. NPG presents an attractive alternative to Au nanoparticle-based catalysts for fuel cells as it eliminates the need for a carbon support thereby removing the associated stability issues. To the best of our knowledge there have been no reports in the literature of the investigation of the oxidation of AB at Au nanoparticles which can be compared with NPG. However the following comparisons serve to highlight the superior activity of NPG over other nanostructured Au-based electrodes for borohydride oxidation at similar concentrations. Ponce-de-Leon et al. 46 reported an oxidation current for 20 mM borohydride in 3 M NaOH at Au nanoparticles supported on titanate nanotubes of 10 mA cm−2 while the onset potential for oxidation was − 0.9 V vs. SCE. Wei et al. 47 investigated the oxidation of 100 mM borohydride in 1 M NaOH at carbon-supported Au hollow nanospheres and reported an oxidation current of 57 mA cm−2 and an onset potential of − 0.80 V vs Ag/AgCl (− 0.845 V vs. SCE) for the oxidation. An onset potential for oxidation of 10 mM borohydride in 1 M NaOH at carbon-supported Au nanoparticles of − 0.57 V vs NHE (− 0.812 V vs. SCE) was reported. 45 An onset potential of − 1.07 V vs. SCE and an oxidation current of 73.6 mA cm−2 was recorded for 20 mM borohydride in 1 M NaOH. 37
NPG can provide a solution to the poor long-term stability and sintering problems that plague supported Au nanoparticle catalysts which limit their use in industrial heterogeneous catalysis. Furthermore, the diffusion of an electroactive species to carbon supported-Au nanoparticles is limited by the low degree of porosity of the support. NPG permits more intimate contact with an electrical substrate to be established. Its porous structure promotes mass transport of the reactant to the active sites and release of gaseous by-products. NPG may be incorporated in thin foil format as a porous catalyst electrode as it is shapeable and has high mechanical, thermal and chemical stability coupled to high catalytic activity. It has a dual functionality in that it can act as a current collector and as a catalyst. From an economic viewpoint the viability of NPG as a catalyst can be raised by crushing it or coating the precursor AgAu alloy on a template backbone prior to dealloying. 35 An advantage of incorporating NPG in place of, or in combination with, Pt in fuel cells would be the enhanced electrical conductivity that could be derived due to the lower electrical resistivity of Au. NPG may be integrated into nafion-based MEAs in conventional PEM fuel cells. An example of a PEM fuel cell using Pt modified-NPG was demonstrated by Zeis et al. 48 They reported a method for integrating Pt-modified NPG electrodes into PEM fuel cells which involved stamping NPG onto pre-swelled, wet nafion in the construction of the MEA. NPG in the form of a membrane was floated on water baths and lifted on mica disks which serve as stamps.
We have shown that the characteristics of AB oxidation at Au electrodes are strongly influenced by electrode morphology. The data summarized in Table I indicates that the oxidation current for AB increases from 2.5 mA cm−2 for a planar Au film to 13.1 mA cm−2 for a NPG wire array, while the onset of oxidation shifts from − 1.02 to − 1.30 V vs. SCE. Attempts were made to enhance the stability of NPG wires by either reinforcing the NPG wires with a Au wire core or by anchoring NPG wires onto Au wires. NPG wires supported on a Au wire array showed comparable electrocatalytic activity to a NPG-only wire array while reinforcing NPG with a Au core led to a slight decay in electrocatalytic activity. Our findings will provide useful information in identifying a viable anode catalyst for the oxidation of AB as an environmentally friendly, high energy density alternative fuel.
Acknowledgments
This work is supported by Irish Environmental Protection Agency funding; it is part of a STRIVE research fellowship entitled "Zero Carbon Emission Micro Fuel Cell Design"; Contract No. 2007-FS-ET-6-M5. We thank Mr. Vince Lodge for recording SEM images. The SEM was funded by "INSPIRE" Higher Education Authority via PRTLI4.