
Abstract
Tartaric acid is added to sulfuric acid anodizing baths to generate porous anodic film that provides corrosion resistance to practical aerospace alloys and reduces the environmental impact of the traditional chromic acid anodizing process. Here, a fundamental study on the effects of the addition of tartaric acid to the sulfuric acid anodizing electrolyte has been undertaken. During anodizing, it was evident that tartaric acid does not significantly affect the mechanism of porous film growth, but it reduces the growth rate of the porous anodic film. After anodizing, in acidic environments, it may reduce the dissolution rate of a previously formed oxide. Furthermore, it was found that in a nearly neutral, chloride-rich environment, tartaric acid limits the anodic reaction of aluminum dissolution at concentrations in the hundreds of ppm range. The previous suggests that the good anticorrosion performance of alloys anodized in the presence of tartaric acid is due to residues of tartaric acid in the pore solution.
Export citation and abstract BibTeX RIS
AA 2024 T3 is a relatively high-strength, damage-tolerant, aluminum alloy that is widely used in the aerospace industry. The presence of copper and magnesium as alloying elements is responsible for the high mechanical performance, but specific measures are required to ensure adequate corrosion protection. Traditionally, the anticorrosion performance is achieved by anodizing in chromic acid,1 followed by painting. However, the increasing environmental issues and stringent legislation related to the use of chromic acid have necessitated the development of anodizing electrolytes with reduced environmental impact and associated disposal costs. In order to achieve this goal, a full understanding of the anodizing behavior of the alloy is required to implement anodizing cycles based on knowledge of the resultant performance.
Although the contribution of tartaric acid in improving the corrosion resistance of aluminum alloys after anodizing in sulfuric acid is generally recognized,2, 3 the precise mechanism is the subject of discussion. Specifically, two main aspects are unclear, namely, the nature of the interaction of tartaric acid with the newly forming oxide during anodizing and the manner in which tartaric acid (or its derivatives) acts to reduce corrosion susceptibility after anodizing (i.e., when the workpiece is exposed to service conditions).
Experimental
Anodizing was undertaken on aluminum  and AA 2024 T3 aluminum alloy. Prior to anodizing, the individual specimens were degreased in acetone, rinsed in deionized water, and dried in a cold airstream. Degreasing was followed by etching in
and AA 2024 T3 aluminum alloy. Prior to anodizing, the individual specimens were degreased in acetone, rinsed in deionized water, and dried in a cold airstream. Degreasing was followed by etching in  sodium hydroxide solution at
sodium hydroxide solution at  for 60 and
for 60 and  for aluminum and for the AA 2024 T3 alloy, respectively. After etching, smut present on the specimen surface was removed by immersion in 30% nitric acid for
for aluminum and for the AA 2024 T3 alloy, respectively. After etching, smut present on the specimen surface was removed by immersion in 30% nitric acid for  at room temperature. Anodizing was undertaken in
at room temperature. Anodizing was undertaken in  sulfuric acid and
sulfuric acid and  ammonium pentaborate solutions at room temperature. Anodizing was generally performed in a three-electrode cell, with the specimen as the working electrode, a graphite bar counter electrode, and a saturated calomel electrode (SCE) as the reference electrode; the potentiostat employed was a Solartron SI 1287. For reanodizing studies, the relatively high voltages achieved during anodizing at constant current densities required the use of a power supply and a two electrode cell con figuration. During anodizing,
ammonium pentaborate solutions at room temperature. Anodizing was generally performed in a three-electrode cell, with the specimen as the working electrode, a graphite bar counter electrode, and a saturated calomel electrode (SCE) as the reference electrode; the potentiostat employed was a Solartron SI 1287. For reanodizing studies, the relatively high voltages achieved during anodizing at constant current densities required the use of a power supply and a two electrode cell con figuration. During anodizing, 
 or
or 
 tartaric acid were added to the sulfuric acid solution. After anodizing, electrochemical impedance spectroscopy was employed to determine the response of the anodized substrate during immersion in
tartaric acid were added to the sulfuric acid solution. After anodizing, electrochemical impedance spectroscopy was employed to determine the response of the anodized substrate during immersion in  sulfuric acid or in
sulfuric acid or in  sulfuric acid with the addition of
sulfuric acid with the addition of  tartaric acid.
tartaric acid.
The effects of tartaric acid on the corrosion behavior of the AA 2024 T3 aluminum alloy exposed to  NaCl were evaluated in split-cell experiments.4 In the split cell, two identical specimens were immersed in
NaCl were evaluated in split-cell experiments.4 In the split cell, two identical specimens were immersed in  NaCl in separate compartments, divided by a porous glass frit. After an appropriate holding time, nitrogen was passed through one side and air through the other side. The nitrogen purge compartment becomes the net anode because of the reduced concentration of oxygen in the electrolyte, and the specimen in the air purged side becomes the net cathode.
NaCl in separate compartments, divided by a porous glass frit. After an appropriate holding time, nitrogen was passed through one side and air through the other side. The nitrogen purge compartment becomes the net anode because of the reduced concentration of oxygen in the electrolyte, and the specimen in the air purged side becomes the net cathode.
The anodic films attached to the alloy substrates were observed by transmission electron microscopy (TEM) of ultramicrotomed sections using a JEOL JEM-2000FXII instrument. For ultramicrotomy, the specimens were trimmed initially with a glass knife, and final sections, of nominal thickness of  , were generated with a diamond knife in the usual manner.5
, were generated with a diamond knife in the usual manner.5
Results
Anodizing under linear polarization conditions
Linear anodic polarization of individual specimens of aluminum and the AA 2024 T3 alloy was performed from the open-circuit potential (OCP) to  SCE in
SCE in  sulfuric acid, with and without addition of
sulfuric acid, with and without addition of  tartaric acid (Fig. 1 and 2), at a sweep rate of
tartaric acid (Fig. 1 and 2), at a sweep rate of  . Concerning aluminum in the absence of tartaric acid (Fig. 1), from the commencement of anodizing, at an OCP of
. Concerning aluminum in the absence of tartaric acid (Fig. 1), from the commencement of anodizing, at an OCP of  SCE, the current increased rapidly until
SCE, the current increased rapidly until  SCE and, subsequently, reached a plateau, related to initial nonuniform film growth and thickening of the barrier layer.6 After the plateau, a transition region in which the current density increased significantly with applied potential, associated with the initial development of a porous film morphology, was observed. Finally, above
SCE and, subsequently, reached a plateau, related to initial nonuniform film growth and thickening of the barrier layer.6 After the plateau, a transition region in which the current density increased significantly with applied potential, associated with the initial development of a porous film morphology, was observed. Finally, above  SCE, the expected increase of the current with the applied potential was evident. This region is associated with development of the usual porous morphology.6–8 With the addition of
SCE, the expected increase of the current with the applied potential was evident. This region is associated with development of the usual porous morphology.6–8 With the addition of  tartaric acid to the previous solution, a similar overall behavior was observed. However, a reduction in the current density by
tartaric acid to the previous solution, a similar overall behavior was observed. However, a reduction in the current density by  in the plateau region and an extension of the potential axis of the current plateau were disclosed. At the potential of
in the plateau region and an extension of the potential axis of the current plateau were disclosed. At the potential of  SCE, in the region associated with the initial development of the porous morphology, the difference in the values of current density was more significant, revealing a current reduction of
SCE, in the region associated with the initial development of the porous morphology, the difference in the values of current density was more significant, revealing a current reduction of  . At potentials of
. At potentials of  , where the porous morphology develops, the current reduction associated with the presence of tartaric acid was less marked with increase of applied potential (e.g., from 30% at
, where the porous morphology develops, the current reduction associated with the presence of tartaric acid was less marked with increase of applied potential (e.g., from 30% at  to 20% at
to 20% at  ).
).
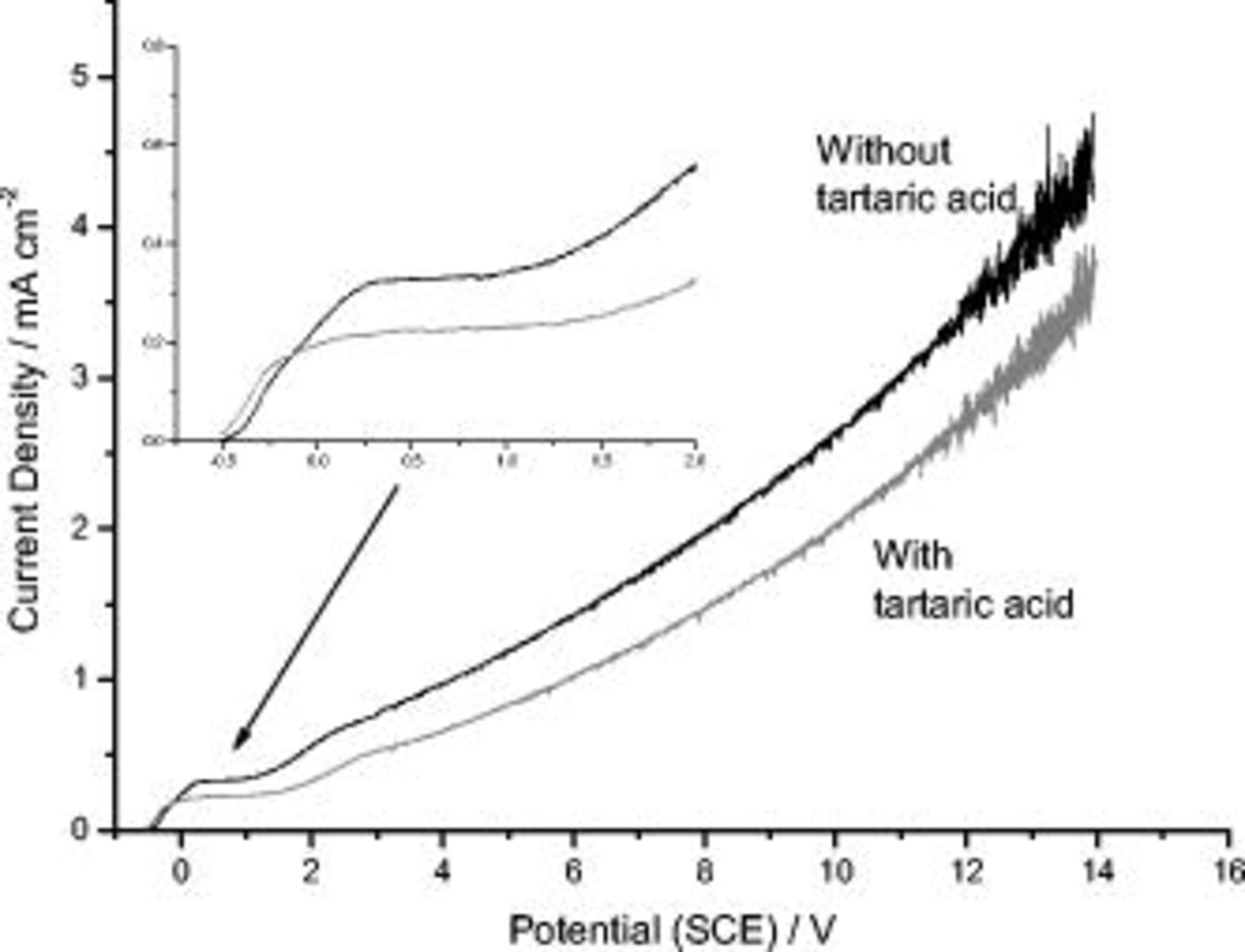
Figure 1.
 behavior for 99.99% aluminum in
behavior for 99.99% aluminum in  sulfuric acid, in the presence and absence of
sulfuric acid, in the presence and absence of  tartaric acid; each line is the average of three curves. In the inset the region between
tartaric acid; each line is the average of three curves. In the inset the region between  and
and  is enlarged.
is enlarged.
For the AA 2024 T3 aluminum alloy (Fig. 2), in the absence of tartaric acid, the usual behavior was observed;2 at  SCE, a first current density peak, associated with the dissolution of S
SCE, a first current density peak, associated with the dissolution of S  phase particles, was revealed. This was followed by the expected plateau region, associated with the thickening of the anodic oxide, and by the second current peak, between 2 and
phase particles, was revealed. This was followed by the expected plateau region, associated with the thickening of the anodic oxide, and by the second current peak, between 2 and  , associated with the oxidation of other constituents [i.e., Al–Cu and Al–Cu–Fe–(Mn) containing particles].2 The previous do not contain magnesium in their composition. After the second peak, at
, associated with the oxidation of other constituents [i.e., Al–Cu and Al–Cu–Fe–(Mn) containing particles].2 The previous do not contain magnesium in their composition. After the second peak, at  SCE, a region of increase of current density with applied potential was observed.
SCE, a region of increase of current density with applied potential was observed.
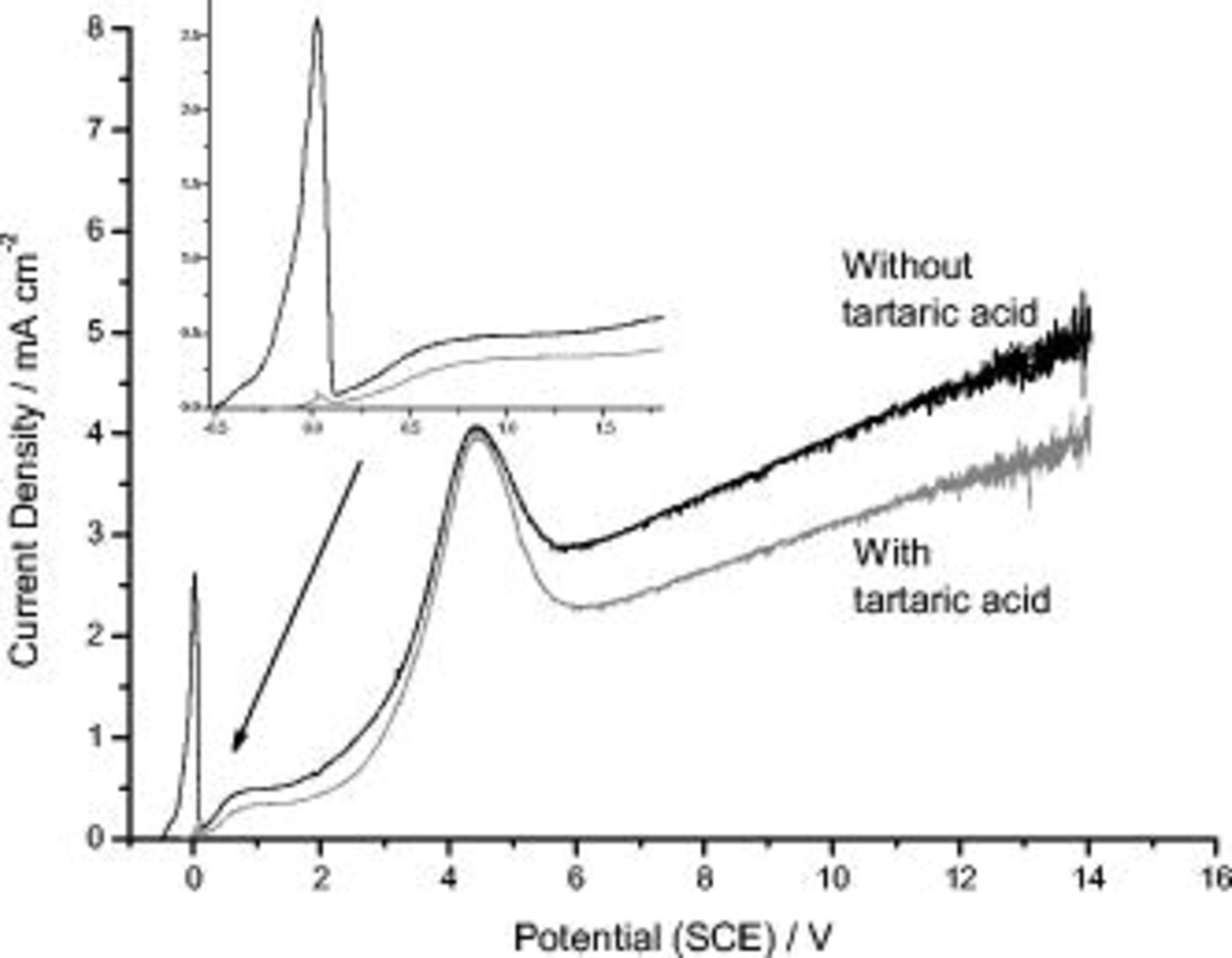
Figure 2.
 behavior for AA 2024 T3 aluminum alloy in
behavior for AA 2024 T3 aluminum alloy in  sulfuric acid, in the presence and absence of
sulfuric acid, in the presence and absence of  tartaric acid; each line is the average of three curves. In the inset the region between
tartaric acid; each line is the average of three curves. In the inset the region between  and
and  is enlarged.
is enlarged.
With the addition of  tartaric acid the OCP increased from
tartaric acid the OCP increased from  SCE and the first peak was suppressed. The anodizing current was generally reduced by 15–25%, particularly at potentials of
SCE and the first peak was suppressed. The anodizing current was generally reduced by 15–25%, particularly at potentials of  . Interestingly, between 2 and
. Interestingly, between 2 and  , when the oxidation of the Al–Cu– and Al–Cu–Fe–(Mn)–containing particles proceeds, the current reduction associated with the presence of the tartaric acid was
, when the oxidation of the Al–Cu– and Al–Cu–Fe–(Mn)–containing particles proceeds, the current reduction associated with the presence of the tartaric acid was  .
.
The difference between high-purity aluminum and AA 2024 T3 aluminum in the overall behavior at  (quasi-exponential and quasi-linear increase of current with potential in the case of high-purity aluminum and of the AA 2024 T3 aluminum alloy, respectively) is presently under investigation. However, preliminary results suggest that such a difference may be attributed to the process of oxygen evolution during anodizing, associated with the presence of copper in the practical aluminum alloy and its oxidation at the alloy∕film interface.
(quasi-exponential and quasi-linear increase of current with potential in the case of high-purity aluminum and of the AA 2024 T3 aluminum alloy, respectively) is presently under investigation. However, preliminary results suggest that such a difference may be attributed to the process of oxygen evolution during anodizing, associated with the presence of copper in the practical aluminum alloy and its oxidation at the alloy∕film interface.
Reanodizing under galvanostatic conditions
Reanodizing was performed in order to determine if the presence of tartaric acid in the anodizing solution results in incorporation of electrolyte-derived species with consequent modification of the electrical properties of the anodic film. The anodizing conditions were selected to form a relatively uniform, nonporous film, on high-purity aluminum. Specifically, anodizing was undertaken under galvanostatic conditions at  in
in  sulfuric acid and
sulfuric acid and  sulfuric acid with
sulfuric acid with  tartaric acid electrolyte, respectively. Subsequently, the specimens were reanodized in
tartaric acid electrolyte, respectively. Subsequently, the specimens were reanodized in  ammonium pentaborate at
ammonium pentaborate at  . In this condition, film growth proceeds at 100% efficiency, with film formation at the metal-film and film-solution interfaces. Consequently, the film formed during the first anodizing step can be considered to be located within the anodic oxide formed during the second anodizing step (Fig. 3).
. In this condition, film growth proceeds at 100% efficiency, with film formation at the metal-film and film-solution interfaces. Consequently, the film formed during the first anodizing step can be considered to be located within the anodic oxide formed during the second anodizing step (Fig. 3).
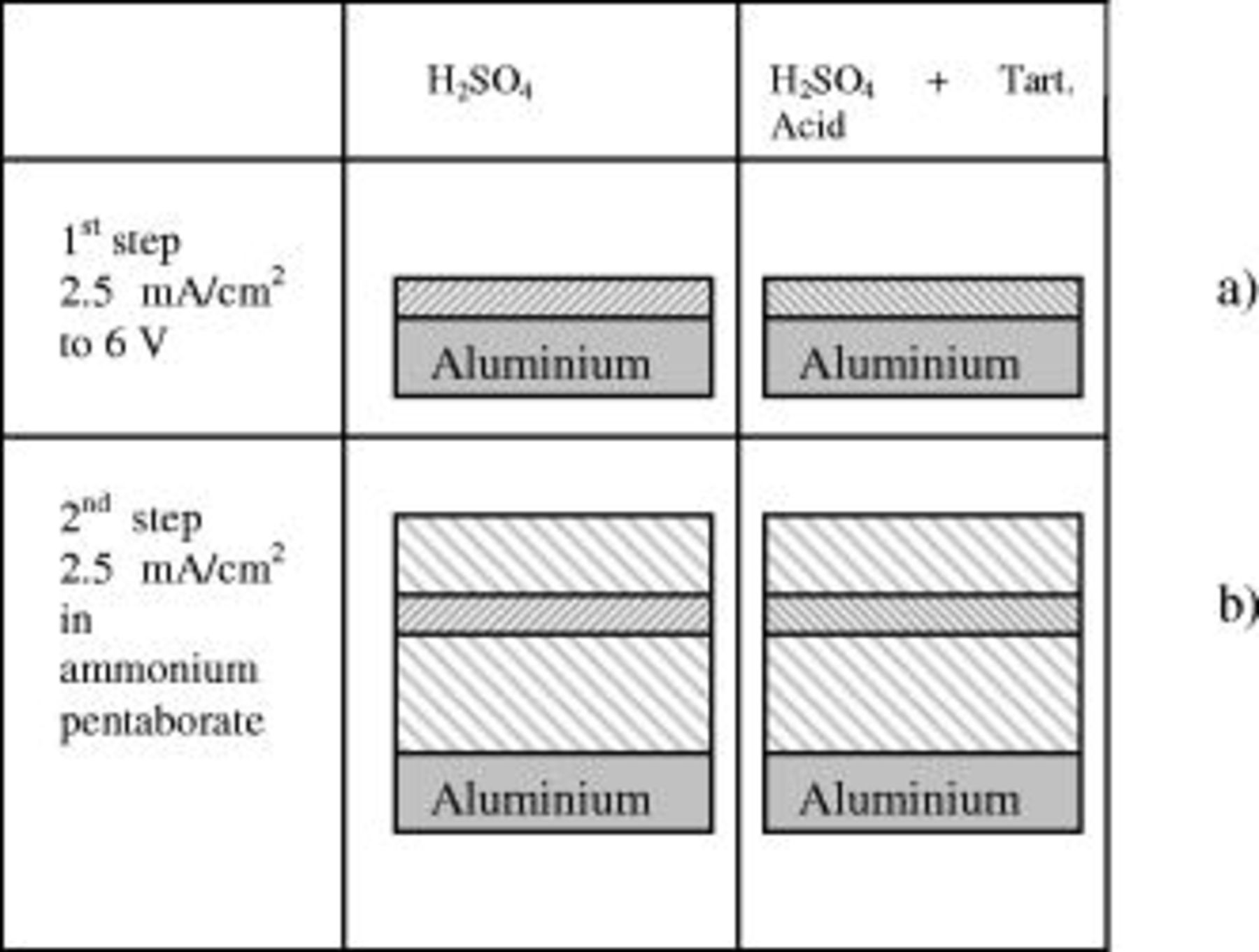
Figure 3. Schematic diagram showing the location of film generated after anodizing and reanodizing: (a) Film formed during the first anodizing step; (b) initial location of the film formed during the first anodizing step in the film formed during the second anodizing step. The effect of ionic migration induced by the second anodizing step is neglected.
During the first anodizing step (Fig. 4), the usual voltage rise of  , due to the pre-existing air-formed film, was observed; subsequently, the voltage increased linearly with time to the final potential of
, due to the pre-existing air-formed film, was observed; subsequently, the voltage increased linearly with time to the final potential of  . The linearity of the
. The linearity of the  behavior confirms that a nonporous film was generated in this potential range. The voltage increases were 0.45 and
behavior confirms that a nonporous film was generated in this potential range. The voltage increases were 0.45 and  in the presence and in the absence of tartaric acid, respectively. The
in the presence and in the absence of tartaric acid, respectively. The  response during reanodizing (Fig. 5) displayed an initial voltage surge to
response during reanodizing (Fig. 5) displayed an initial voltage surge to  , associated with the previously formed, nonporous film; this was followed by a region of linear increase of voltage with time. The voltage increases were 1.03 and
, associated with the previously formed, nonporous film; this was followed by a region of linear increase of voltage with time. The voltage increases were 1.03 and  for the specimens supporting the film containing the layer originally generated in the presence and in the absence of tartaric acid, respectively. The closely similar
for the specimens supporting the film containing the layer originally generated in the presence and in the absence of tartaric acid, respectively. The closely similar  responses during the second anodizing step strongly suggest that the properties of the anodic films generated in the first anodizing steps are comparable.
responses during the second anodizing step strongly suggest that the properties of the anodic films generated in the first anodizing steps are comparable.
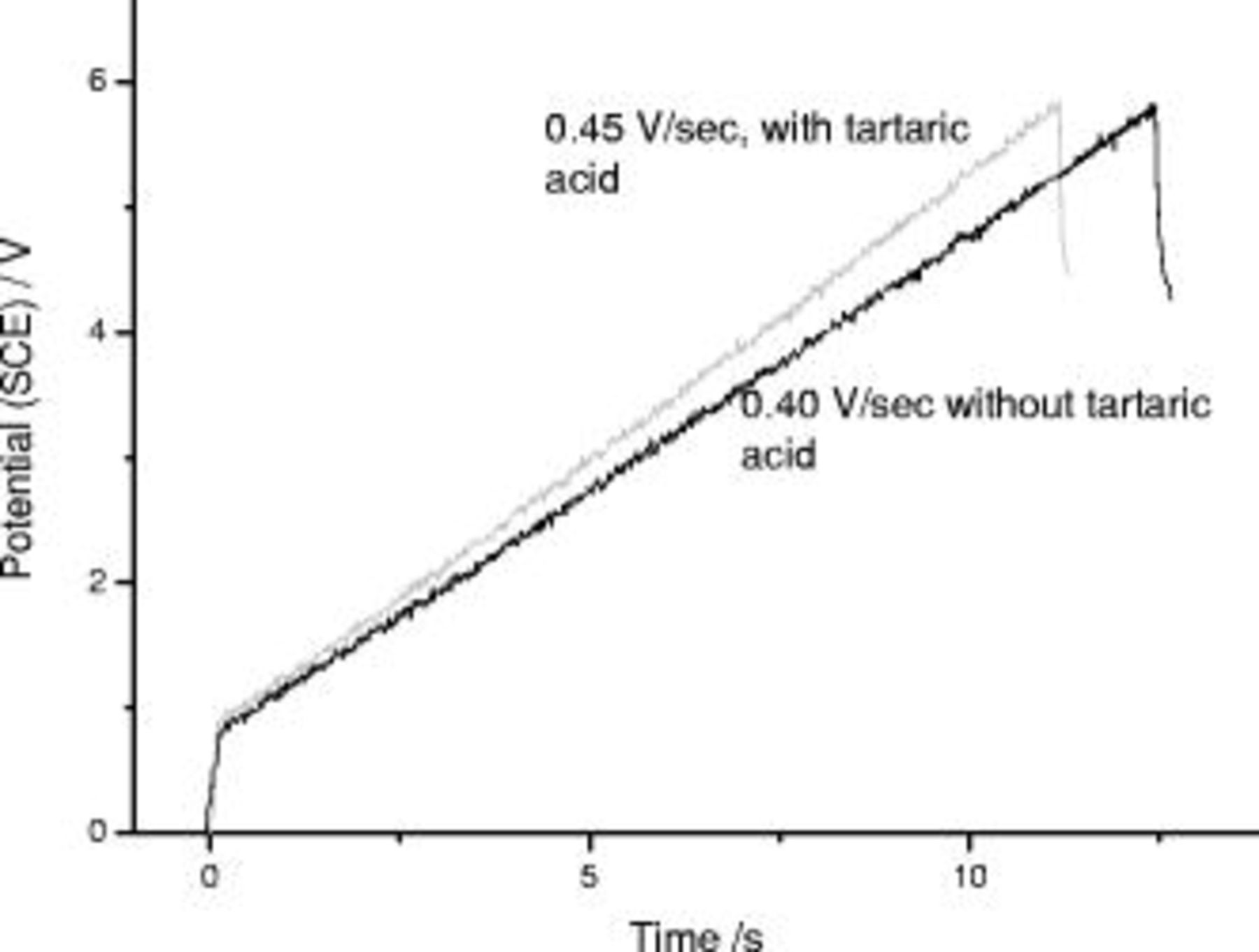
Figure 4.
 behavior for anodizing of 99.99% aluminum sulfuric acid
behavior for anodizing of 99.99% aluminum sulfuric acid  at
at  in the presence and absence of
in the presence and absence of  tartaric acid; average of three curves for each condition.
tartaric acid; average of three curves for each condition.
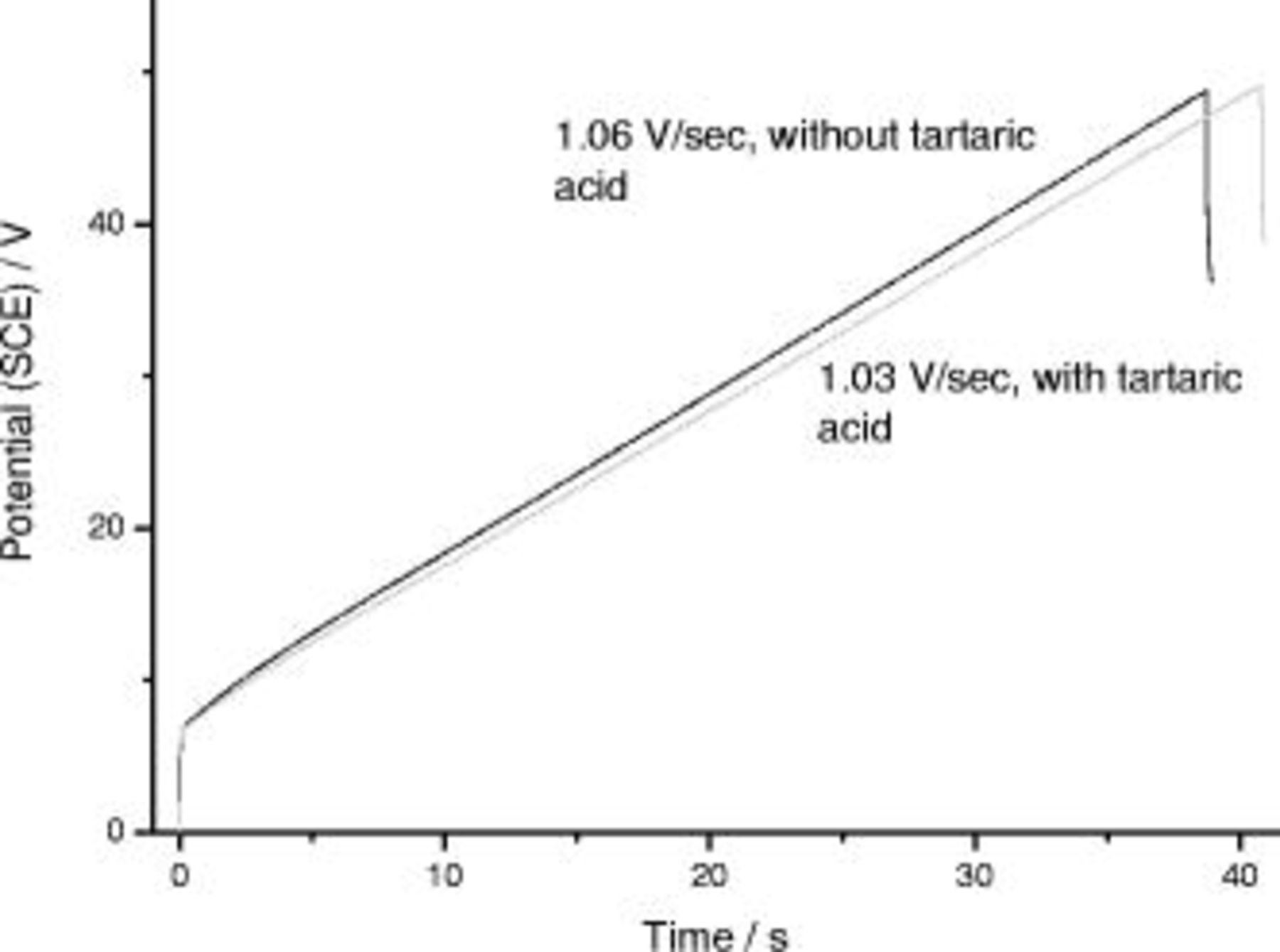
Figure 5.
 behavior for reanodizing of the specimens of Fig. 8 in
behavior for reanodizing of the specimens of Fig. 8 in  ammonium pentaborate at
ammonium pentaborate at  ; average of three curves for each condition.
; average of three curves for each condition.
Layered porous anodic films on AA 2024 T3 alloy in the presence and absence of tartaric acid in  sulfuric acid electrolyte
sulfuric acid electrolyte
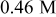
In order to investigate further the possible effect of tartaric acid addition during porous film growth on the practical material, the AA 2024 T3 alloy was anodized under potentiostatic conditions in  sulfuric acid electrolyte in the presence and absence of tartaric acid. Specifically, the alloy specimen was anodized sequentially at 12, 3, and
sulfuric acid electrolyte in the presence and absence of tartaric acid. Specifically, the alloy specimen was anodized sequentially at 12, 3, and  in
in  sulfuric acid, and subsequently at 3, 12, and
sulfuric acid, and subsequently at 3, 12, and  in
in  sulfuric acid with the addition of
sulfuric acid with the addition of  tartaric acid. During each step, the anodizing charge was
tartaric acid. During each step, the anodizing charge was  . This procedure enabled direct comparison of the features and thicknesses of the corresponding film layers by TEM. Concerning the current density-time response (Fig. 6), when the potential was increased, a current spike was observed and, after a relatively short transient, displaying several oscillations, a steady current was achieved. In agreement with previous findings, the steady current in the presence of
. This procedure enabled direct comparison of the features and thicknesses of the corresponding film layers by TEM. Concerning the current density-time response (Fig. 6), when the potential was increased, a current spike was observed and, after a relatively short transient, displaying several oscillations, a steady current was achieved. In agreement with previous findings, the steady current in the presence of  tartaric acid was reduced by 20% compared to the current measured in sulfuric acid only. Conversely, when the potential was reduced from the initial value of
tartaric acid was reduced by 20% compared to the current measured in sulfuric acid only. Conversely, when the potential was reduced from the initial value of  to the value of
to the value of  , the current decreased to
, the current decreased to  . The value of the current remained low for a prolonged period of time, and subsequently, a gradual increase with time was observed, to achieve a final steady value. The duration of this recovery transient had almost doubled in the presence of tartaric acid and the steady current at low potential was reduced by
. The value of the current remained low for a prolonged period of time, and subsequently, a gradual increase with time was observed, to achieve a final steady value. The duration of this recovery transient had almost doubled in the presence of tartaric acid and the steady current at low potential was reduced by  .
.
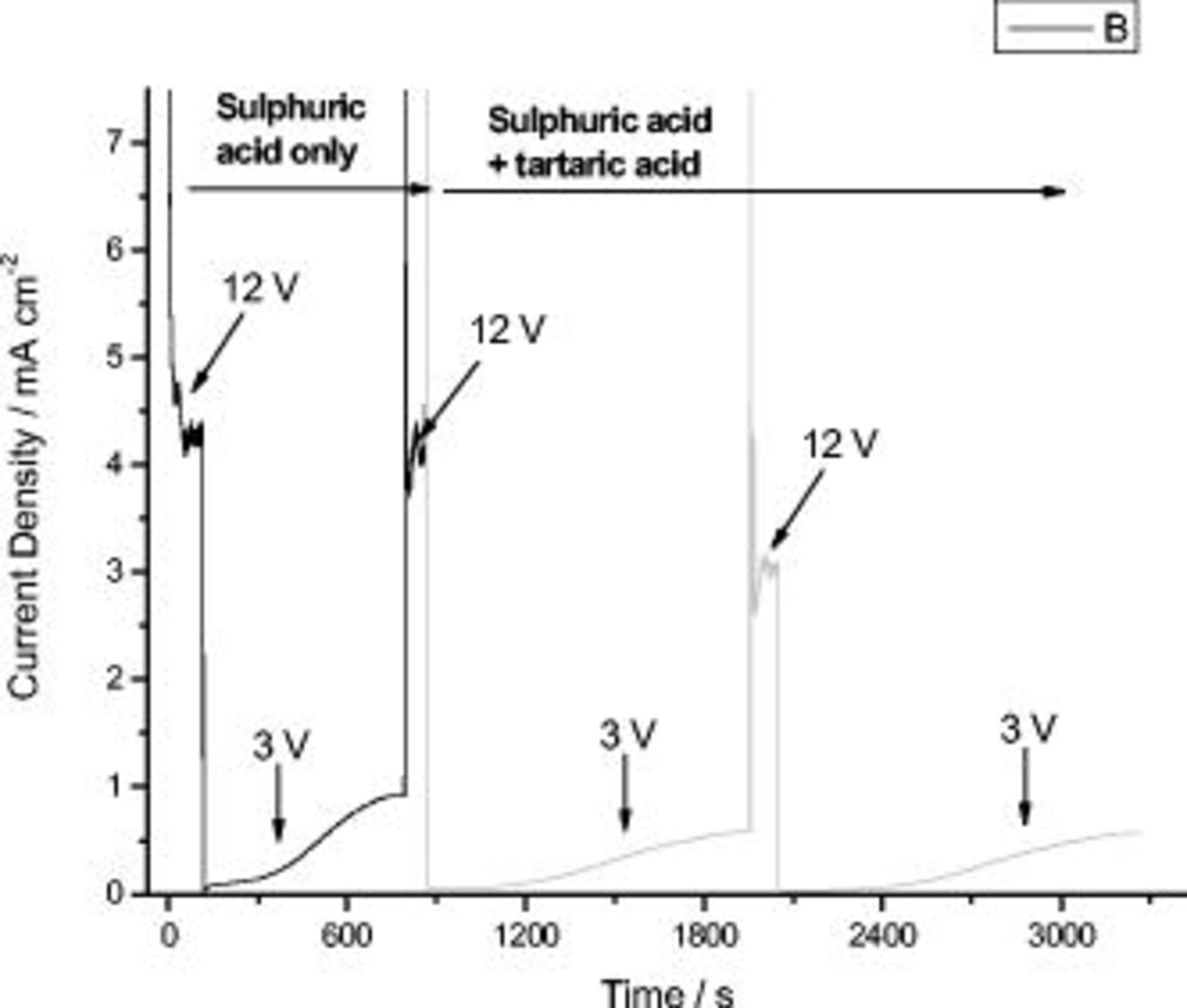
Figure 6.
 behavior for the layered specimen: sulfuric acid
behavior for the layered specimen: sulfuric acid  –sulfuric acid
–sulfuric acid  –sulfuric acid
–sulfuric acid  –sulfuric and tartaric acid
–sulfuric and tartaric acid  , and sulfuric and tartaric acid
, and sulfuric and tartaric acid  –sulfuric and tartaric acid
–sulfuric and tartaric acid  .
.
Figure 7 displays the anodic film attached to the alloy matrix obtained after applying the previously described voltage cycle (i.e., sequential anodizing). As expected, six morphologically differing layers are observed in the section of the anodic film attached to the alloy substrate. Starting from the outer film regions, layers 1  , 2
, 2  , and 3
, and 3  were formed in sulfuric acid, and layers 4
were formed in sulfuric acid, and layers 4  , 5
, 5  , and 6
, and 6  were generated in sulfuric and tartaric acid electrolyte. The reduced thickness of the first layer is due to damage induced by the diamond knife during ultramicrotomy. Layers 2, 4, and 6, formed at the reduced potential of
were generated in sulfuric and tartaric acid electrolyte. The reduced thickness of the first layer is due to damage induced by the diamond knife during ultramicrotomy. Layers 2, 4, and 6, formed at the reduced potential of  , displayed a finer morphology compared to the remaining layers generated at increased potential. Comparing the morphologies and thicknesses of the layers obtained in the presence and in the absence of tartaric acid, no major differences were revealed.
, displayed a finer morphology compared to the remaining layers generated at increased potential. Comparing the morphologies and thicknesses of the layers obtained in the presence and in the absence of tartaric acid, no major differences were revealed.
Figure 7. Layered specimen; from the top: sulfuric acid  –sulfuric acid
–sulfuric acid  , sulfuric acid
, sulfuric acid  –sulfuric and tartaric acid
–sulfuric and tartaric acid  , sulfuric and tartaric acid
, sulfuric and tartaric acid  –sulfuric and tartaric acid
–sulfuric and tartaric acid  .
.
Dissolution studies
In order to investigate the effect of the addition of tartaric acid on the chemical dissolution rate of the anodic film in sulfuric acid, two aluminum specimens were anodized under galvanostatic conditions in  sulfuric acid at
sulfuric acid at  in order to generate a nonporous film. After anodizing, the specimens were immersed in
in order to generate a nonporous film. After anodizing, the specimens were immersed in  sulfuric acid, or
sulfuric acid, or  sulfuric acid with addition of
sulfuric acid with addition of  tartaric acid.
tartaric acid.
Impedance measurements were performed during immersion by applying a sinusoidal waveform of  amplitude over the frequency range from
amplitude over the frequency range from  , with the resulting Bode plots presented in Fig. 8. The general behavior of the two specimens was similar, indicating the presence of one time constant associated with the equivalent circuit of Fig. 9a. In both cases, the modulus of the impedance,
, with the resulting Bode plots presented in Fig. 8. The general behavior of the two specimens was similar, indicating the presence of one time constant associated with the equivalent circuit of Fig. 9a. In both cases, the modulus of the impedance,  , decreased with time, although such a decrease was reduced for the specimen immersed in the solution containing tartaric acid. The phase angle decreased with increasing immersion time, and again, such an effect was less significant for the specimen immersed in the solution containing tartaric acid.
, decreased with time, although such a decrease was reduced for the specimen immersed in the solution containing tartaric acid. The phase angle decreased with increasing immersion time, and again, such an effect was less significant for the specimen immersed in the solution containing tartaric acid.
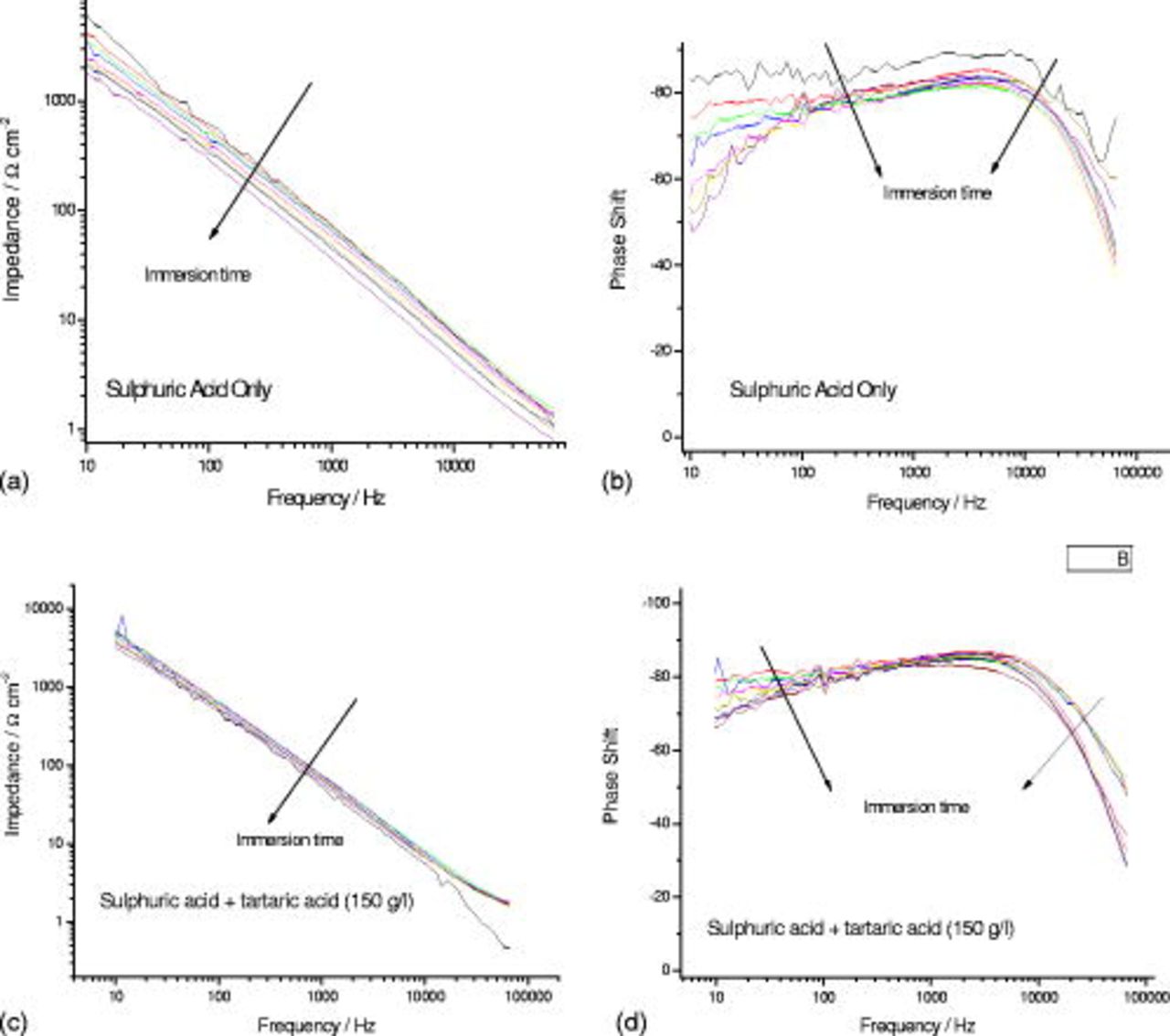
Figure 8. Effect of immersion time on the impedance response and phase shift for a specimen immersed in  sulfuric acid (a,b) and
sulfuric acid (a,b) and  sulfuric acid
sulfuric acid  tartaric acid (c,d).
tartaric acid (c,d).
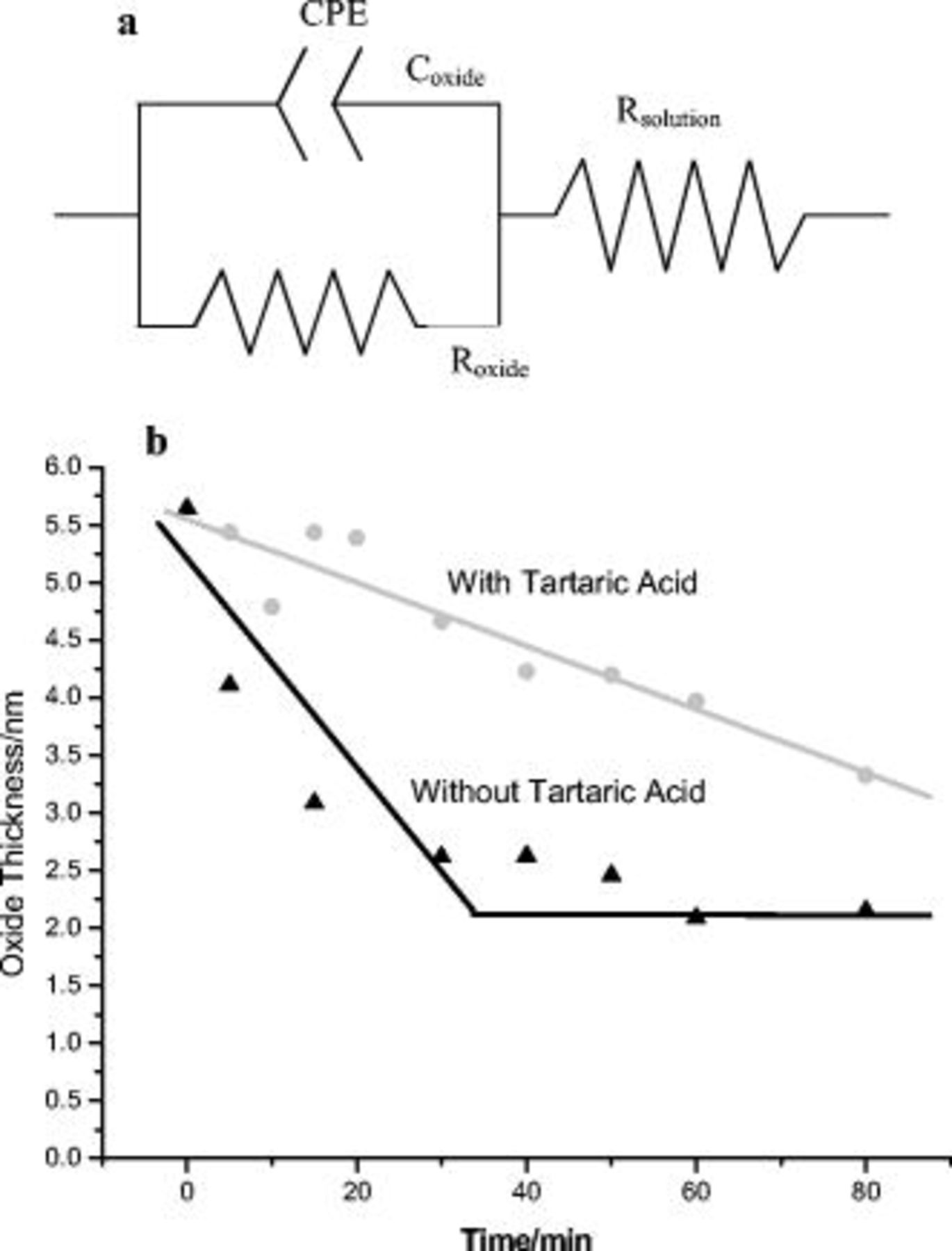
Figure 9. (a) Equivalent circuit used to estimate the anodic film capacitance and resistance, and (b) variation of the thickness of the anodic film with time of immersion in sulfuric acid and sulfuric acid with  tartaric acid.
tartaric acid.
The equivalent circuit of Fig. 9a enabled estimation of the capacitances associated with the presence of the anodic film. The capacitances were then used to estimate the remaining thickness of the oxide film after dissolution in the acidic environment (Fig. 9b). For the calculation, a relative dielectric constant for the aluminum oxide9 of 9.9 was assumed. Interestingly, the thickness of the oxide immersed in sulfuric and tartaric acid decreased linearly with immersion time  , whereas the oxide immersed in sulfuric acid showed an increased rate of dissolution. After immersion for
, whereas the oxide immersed in sulfuric acid showed an increased rate of dissolution. After immersion for  , the thickness of the oxide was close to
, the thickness of the oxide was close to  and did not decrease with increased immersion times. This is due to the growth of the oxide at the metal∕film interface, proceeding at the same rate as its dissolution at the film∕electrolyte interface, thus resulting in a film thickness of
and did not decrease with increased immersion times. This is due to the growth of the oxide at the metal∕film interface, proceeding at the same rate as its dissolution at the film∕electrolyte interface, thus resulting in a film thickness of  .
.
The previous results were confirmed using a specimen supporting a barrier-type film, formed by anodizing at  in
in  ammonium pentaborate, to generate a film thickness of
ammonium pentaborate, to generate a film thickness of  (Fig. 10a). After anodizing, the specimen was cut into four parts: one part was retained for subsequent TEM observation of the as-formed film, and the remaining three parts were immersed in
(Fig. 10a). After anodizing, the specimen was cut into four parts: one part was retained for subsequent TEM observation of the as-formed film, and the remaining three parts were immersed in 
 , with addition of 0, 80, and
, with addition of 0, 80, and  tartaric acid, respectively. In agreement with the impedance measurements, the addition of tartaric acid decreased the dissolution rate of the anodic oxide. The oxide thicknesses remaining after three days of immersion were
tartaric acid, respectively. In agreement with the impedance measurements, the addition of tartaric acid decreased the dissolution rate of the anodic oxide. The oxide thicknesses remaining after three days of immersion were  (average dissolution rate:
(average dissolution rate:  ),
),  (average dissolution rate:
(average dissolution rate:  ), and
), and  (average dissolution rate:
(average dissolution rate:  ) for the specimens immersed in the solution with 150, 80, and
) for the specimens immersed in the solution with 150, 80, and  tartaric acid, respectively (Fig. 10b, 10b, 10c and 10d).
tartaric acid, respectively (Fig. 10b, 10b, 10c and 10d).

Figure 10. TEM images of the anodic film obtained by anodizing at  in ammonium pentaborate at
in ammonium pentaborate at  ; as (a) anodized and (b) after immersion for three days in
; as (a) anodized and (b) after immersion for three days in 
 with the addition of
with the addition of  tartaric acid, (c)
tartaric acid, (c)  tartaric acid, and (d) in the absence of tartaric acid.
tartaric acid, and (d) in the absence of tartaric acid.
Split-cell experiments
In order to gain insight into the effects of tartaric acid on the macroscopic and local corrosion behavior and local activity at flaws in aluminum surfaces supporting an anodic film (i.e., locations where the bare alloy is directly exposed to the electrolyte), split-cell experiments were performed. The split cell comprised two cylindrical cells connected by a tube containing a porous membrane. This cell arrangement allows generation of two environments, with the porous membrane enabling flow of current but limiting the exchange of chemical species between the cell compartments.
The individual cells were filled initially with  of naturally aerated
of naturally aerated  NaCl solution. Individual etched and desmutted AA 2024 T3 specimens (i.e., pretreated but not anodized) were immersed in each compartment and coupled by a zero resistance ammeter (ZRA). The current recorded by the ZRA is displayed in Fig. 11. During the first
NaCl solution. Individual etched and desmutted AA 2024 T3 specimens (i.e., pretreated but not anodized) were immersed in each compartment and coupled by a zero resistance ammeter (ZRA). The current recorded by the ZRA is displayed in Fig. 11. During the first  of immersion, the current oscillated around the zero value, reflecting the random distribution of cathodic and anodic processes on both specimens. After
of immersion, the current oscillated around the zero value, reflecting the random distribution of cathodic and anodic processes on both specimens. After  , high-purity nitrogen was passed through one side of the split cell and air was passed through the other side. This resulted in a progressive increase of the current from the net anode (nitrogen side) to the net cathode (air side). After
, high-purity nitrogen was passed through one side of the split cell and air was passed through the other side. This resulted in a progressive increase of the current from the net anode (nitrogen side) to the net cathode (air side). After  , the net current value was relatively stable around
, the net current value was relatively stable around  . After
. After  from the commencement of the experiment, addition of
from the commencement of the experiment, addition of 
 tartaric acid was made to the anodic or to the cathodic compartment. The addition of
tartaric acid was made to the anodic or to the cathodic compartment. The addition of 
 tartaric acid to the cathodic compartment resulted in a decrease of pH 3 and in a corresponding peak in the current to a maximum value of
tartaric acid to the cathodic compartment resulted in a decrease of pH 3 and in a corresponding peak in the current to a maximum value of  . The peak was followed by a rapid decrease, with several oscillations, prior to reaching a plateau value of
. The peak was followed by a rapid decrease, with several oscillations, prior to reaching a plateau value of  after
after  ; the plateau current density was similar to that prior to the addition. Similar behavior was observed after addition of
; the plateau current density was similar to that prior to the addition. Similar behavior was observed after addition of  of tartaric acid in the anodic side. However, the current peak after the addition was less marked and the plateau value of the current, achieved after
of tartaric acid in the anodic side. However, the current peak after the addition was less marked and the plateau value of the current, achieved after  , was reduced from
, was reduced from  prior to the addition to
prior to the addition to  after the addition.
after the addition.
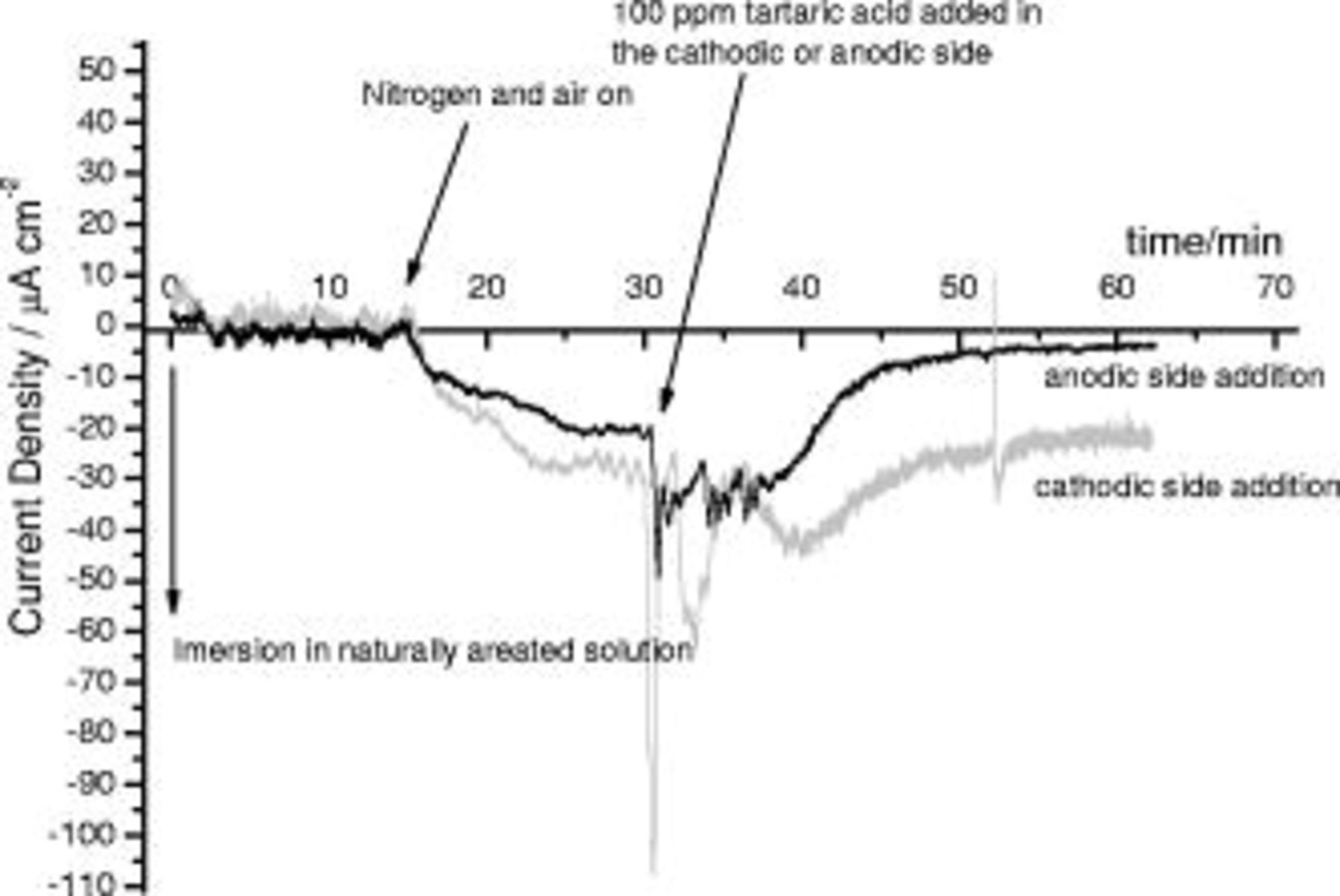
Figure 11. Current∕time behavior recorded in the split-cell experiment,  : immersion in naturally aerated solution;
: immersion in naturally aerated solution;  : air and nitrogen are passed through the cells;
: air and nitrogen are passed through the cells;  : addition of
: addition of  tartaric acid in the anodic or cathodic compartment.
tartaric acid in the anodic or cathodic compartment.
Titrations
In order to gain further insight into the contribution of the tartaric acid and its possible derivatives on the enhanced corrosion performance, titrations on selected solutions containing tartrate species were undertaken using  HCl. The titrated solutions were: solution a,
HCl. The titrated solutions were: solution a,  of
of  NaOH and solution b,
NaOH and solution b,  of
of 
 tartaric acid
tartaric acid  . Solution a was chosen as representative of the behavior of a strong base (NaOH) titrated by a strong acid (HCl), and solution b was used to evaluate the effect of the presence of tartrate species on the titration behavior. The
. Solution a was chosen as representative of the behavior of a strong base (NaOH) titrated by a strong acid (HCl), and solution b was used to evaluate the effect of the presence of tartrate species on the titration behavior. The  NaOH solution (black curve in Fig. 12), displayed an initial pH of
NaOH solution (black curve in Fig. 12), displayed an initial pH of  , which was slightly reduced by the initial addition of
, which was slightly reduced by the initial addition of  HCl. However, after addition of
HCl. However, after addition of  of the titrating solution, the pH decreased more rapidly and, after addition of
of the titrating solution, the pH decreased more rapidly and, after addition of  , it decreased sharply to pH 3. Further addition of HCl solution resulted in minor pH reduction; a final value of 2.5 was measured for addition of
, it decreased sharply to pH 3. Further addition of HCl solution resulted in minor pH reduction; a final value of 2.5 was measured for addition of  HCl solution. The addition of
HCl solution. The addition of  tartaric acid to the
tartaric acid to the  NaOH solution (gray curve in Fig. 12) significantly modified the titration behavior. Specifically, the initial pH of the solution was reduced to
NaOH solution (gray curve in Fig. 12) significantly modified the titration behavior. Specifically, the initial pH of the solution was reduced to  compared to 10.4 in the absence of tartaric acid. In this case, after addition of
compared to 10.4 in the absence of tartaric acid. In this case, after addition of  of
of  HCl, a sharp decrease in the pH was revealed, indicating the neutralization of excess NaOH. However, after the pH dropped from
HCl, a sharp decrease in the pH was revealed, indicating the neutralization of excess NaOH. However, after the pH dropped from  to 4.5, further additions of HCl resulted in a slow decrease in pH with further addition of the titrating solution. The pH decrease from 4.5 to 2.75 required
to 4.5, further additions of HCl resulted in a slow decrease in pH with further addition of the titrating solution. The pH decrease from 4.5 to 2.75 required  of
of  HCl solution, compared to
HCl solution, compared to  during titration of
during titration of  NaOH solution. The buffering capacities of the selected solution
NaOH solution. The buffering capacities of the selected solution  in the pH range from 4.5 to 2.75 are 1.72 and 6.25 for NaOH and
in the pH range from 4.5 to 2.75 are 1.72 and 6.25 for NaOH and  acid, respectively. This indicates that tartrate species at concentration of
acid, respectively. This indicates that tartrate species at concentration of  provide a buffering capacity in the pH range from 4.5 to 2.75.
provide a buffering capacity in the pH range from 4.5 to 2.75.
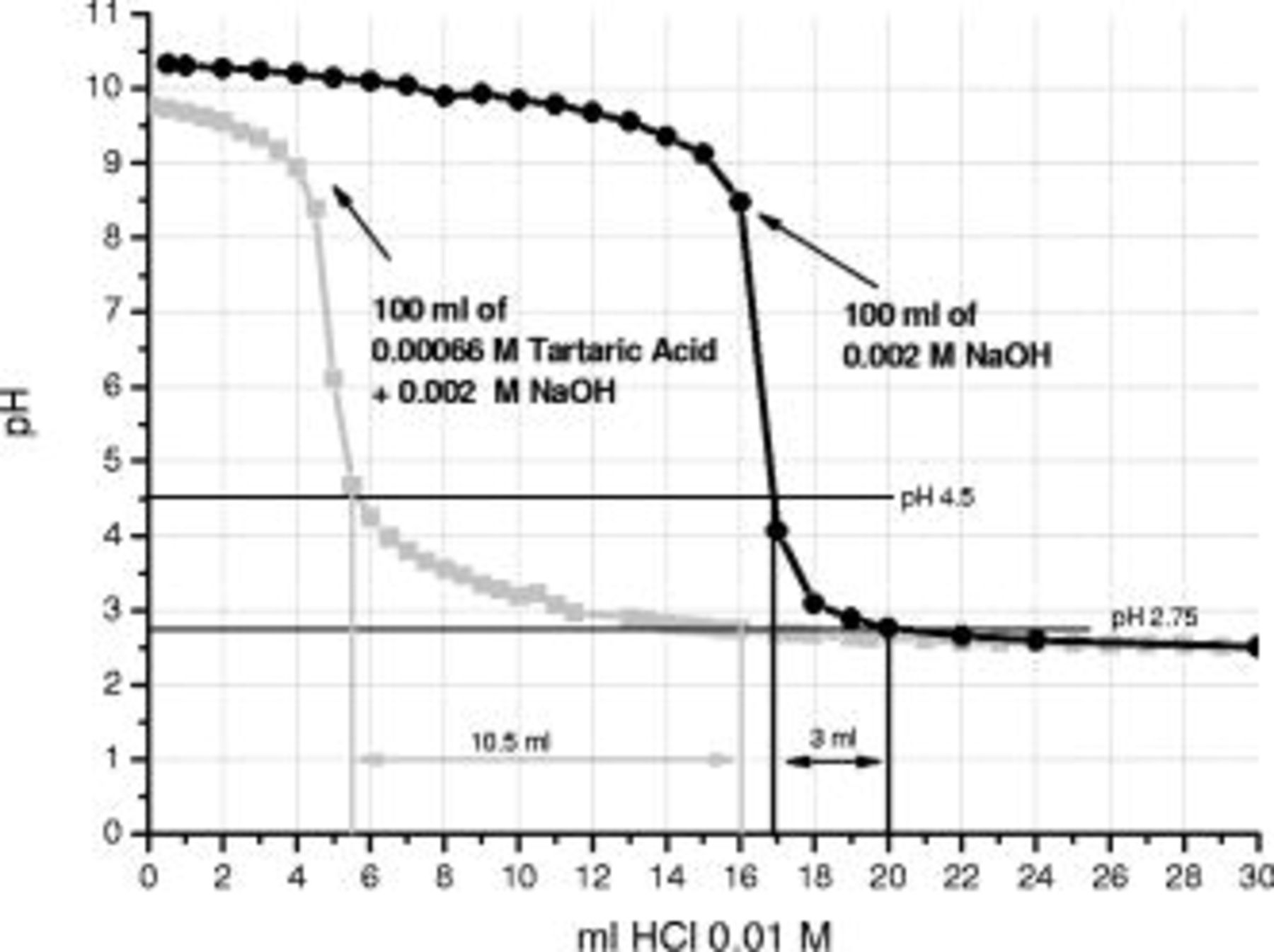
Figure 12. Titration curves for  of
of  NaOH (black curve),
NaOH (black curve), 
 tartaric acid (gray curve).
tartaric acid (gray curve).
Discussion
It is generally recognized that for anodizing under potentiostatic conditions, the presence of tartaric acid in the sulfuric acid bath reduces the anodizing current density.2, 3 The previous was confirmed in the present work by linear polarization studies on high-purity aluminum and AA 2024 T3 aluminum alloy. Scrutiny of the electrical response recorded during anodizing of the practical alloy allows correlation between the peaks observed in the current-voltage ( ) curves and the specific events proceeding on the alloy surface.8 It was found that the addition of tartaric acid results in almost complete suppression of the initial current peak, related to S-phase particle dissolution, suggesting that at potentials close to the OCP, tartaric acid prevents their preferred dissolution. Conversely, at increased potentials, where the oxidation of other second-phase material proceeds along with film disruption through oxygen gas generation, the effect of the tartaric acid addition is negligible. Finally, at high potential, associated with well-established porous growth, the current reduction due to the addition of tartaric acid observed during anodizing of AA 2024 T3 is comparable to the current reduction observed during anodizing of pure aluminum under similar conditions.
) curves and the specific events proceeding on the alloy surface.8 It was found that the addition of tartaric acid results in almost complete suppression of the initial current peak, related to S-phase particle dissolution, suggesting that at potentials close to the OCP, tartaric acid prevents their preferred dissolution. Conversely, at increased potentials, where the oxidation of other second-phase material proceeds along with film disruption through oxygen gas generation, the effect of the tartaric acid addition is negligible. Finally, at high potential, associated with well-established porous growth, the current reduction due to the addition of tartaric acid observed during anodizing of AA 2024 T3 is comparable to the current reduction observed during anodizing of pure aluminum under similar conditions.
Reanodizing provides further insight into the role of the tartaric acid addition. The increased slope in the  curves during the first anodizing step may suggest that (i) tartaric acid is incorporated into the anodic film, increasing the effective electrical resistance or (ii) that the tartaric acid interacts with the oxide only at the film∕solution interface reducing, for a given anodizing potential, the rate of oxygen ion and sulfate ion ingress into the oxide film. However, the behavior revealed during the second anodizing step in ammonium pentaborate allows exclusion of the first suggestion. Thus, if the presence of tartaric acid during the first step had resulted in an increase in the electrical resistance of the anodic film, then the resultant
curves during the first anodizing step may suggest that (i) tartaric acid is incorporated into the anodic film, increasing the effective electrical resistance or (ii) that the tartaric acid interacts with the oxide only at the film∕solution interface reducing, for a given anodizing potential, the rate of oxygen ion and sulfate ion ingress into the oxide film. However, the behavior revealed during the second anodizing step in ammonium pentaborate allows exclusion of the first suggestion. Thus, if the presence of tartaric acid during the first step had resulted in an increase in the electrical resistance of the anodic film, then the resultant  response during the second step of anodizing would display an increased initial voltage rise after the application of the current, or an increased rate of voltage increase. However, the curves recorded during the second step for the anodic films previously formed in the presence and in the absence of tartaric acid are closely similar. Furthermore, the small difference in the curves is contrary to that expected according to the first hypothesis. As a consequence, it can be concluded that tartaric acid interacts with the exposed anodic surface and, for this reason, rinsing of the specimens between the two anodizing steps, results in a very similar reanodizing behavior.
response during the second step of anodizing would display an increased initial voltage rise after the application of the current, or an increased rate of voltage increase. However, the curves recorded during the second step for the anodic films previously formed in the presence and in the absence of tartaric acid are closely similar. Furthermore, the small difference in the curves is contrary to that expected according to the first hypothesis. As a consequence, it can be concluded that tartaric acid interacts with the exposed anodic surface and, for this reason, rinsing of the specimens between the two anodizing steps, results in a very similar reanodizing behavior.
The next step in the characterization of the behavior of tartaric acid was examination of the effect on the porous anodic film morphology generated on the practical AA 2024 T3 aluminum alloy (Fig. 7). In agreement with the previous statements, the anodic film morphology was not influenced by the addition of tartaric acid, for layers formed at relatively high and low voltages. Interestingly, the recovery time subsequent to the potential drop during anodizing of the layered specimen was almost twice that in the presence of tartaric acid. It is generally recognized that when the potential is suddenly dropped during anodizing, high field ionic conduction across the barrier layer beneath the pores is restricted. However, after a sufficient time, the chemical dissolution at the pore base results in thinning of the barrier layer, with consequent increase of the electric field. Eventually, the barrier layer thins sufficiently to allow onset of ionic conduction and porous film growth is reinitiated. The observation that the addition of tartaric acid results in an increase in the recovery time suggests that it decreases the dissolution rate of the oxide in the sulfuric acid electrolyte.
In order to confirm the previous, further dissolution experiments were performed. Thus, a nonporous film was produced in sulfuric acid electrolyte by galvanostatic anodizing to  , and impedance measurements were performed after immersion in sulfuric acid solution with and without the addition of tartaric acid. As expected, the thinning rate was reduced in the presence of tartaric acid. This was confirmed by direct TEM observation of barrier-type anodic film generated in ammonium pentaborate to
, and impedance measurements were performed after immersion in sulfuric acid solution with and without the addition of tartaric acid. As expected, the thinning rate was reduced in the presence of tartaric acid. This was confirmed by direct TEM observation of barrier-type anodic film generated in ammonium pentaborate to  after similar immersion tests. The two different sets of experiments were necessary because, in the first case, a film of chemical composition corresponding to anodizing in sulfuric acid was produced, but its thickness was limited because of the requirement to prevent pore initiation by interrupting the galvanostatic anodizing at the relatively low potential of
after similar immersion tests. The two different sets of experiments were necessary because, in the first case, a film of chemical composition corresponding to anodizing in sulfuric acid was produced, but its thickness was limited because of the requirement to prevent pore initiation by interrupting the galvanostatic anodizing at the relatively low potential of  . As a consequence, the limited thickness of the nonporous film achievable with sulfuric acid electrolyte did not allow precise quantification of the thinning rate by TEM and the indirect method of impedance spectroscopy was employed. Conversely, in the case of the oxide film generated in ammonium pentaborate, the thickness of the barrier-type film was sufficient to evaluate thinning by direct TEM observation but the chemical composition of the oxide film differed from that obtained in sulfuric acid.10–12 This difference may be responsible for the variation in the thinning rate measured in the same solution for the anodic oxide formed in sulfuric acid and the anodic oxide formed in ammonium pentaborate. However, the agreement in the trend of the two results confirmed that the addition of tartaric acid decreases the dissolution rate of the oxide in the acidic environment.
. As a consequence, the limited thickness of the nonporous film achievable with sulfuric acid electrolyte did not allow precise quantification of the thinning rate by TEM and the indirect method of impedance spectroscopy was employed. Conversely, in the case of the oxide film generated in ammonium pentaborate, the thickness of the barrier-type film was sufficient to evaluate thinning by direct TEM observation but the chemical composition of the oxide film differed from that obtained in sulfuric acid.10–12 This difference may be responsible for the variation in the thinning rate measured in the same solution for the anodic oxide formed in sulfuric acid and the anodic oxide formed in ammonium pentaborate. However, the agreement in the trend of the two results confirmed that the addition of tartaric acid decreases the dissolution rate of the oxide in the acidic environment.
Considering the previous as a key point in understanding the role of tartaric acid in enhancing the anticorrosion performances, its behavior was examined during immersion of appropriate specimens in  NaCl solution, using a split cell. Clearly, this approach modifies the fundamental corrosion process with respect to natural immersion, but it provides insight into the role of electrolyte additions.4 In the anodic compartment, the activity of the cathodic sites is significantly limited due to the reduced availability of oxygen; consequently, the current produced by the anodic processes (dissolution of aluminum) is consumed at the cathodic sites on the specimen in the cathodic compartment. Usually, under natural immersion conditions, cathodic and anodic sites are in close proximity and corrosion is generally initiated in the zone of influence of second-phase particles of cathodic nature with respect to the aluminum matrix due to the local alkalinity generated from oxygen reduction. Here, the local increase in pH promotes dissolution of the oxide film present on the adjacent alloy matrix, resulting in localized loss of material. Eventually, a cavity is generated and the cavity may provide the preferred location for subsequent localized attack. In the split-cell experiments, the cathodic activity is localized in the cathodic compartment, and consequently, local alkalinization in the anodic side is largely absent. However, from the split-cell experiments, it was demonstrated that addition of
NaCl solution, using a split cell. Clearly, this approach modifies the fundamental corrosion process with respect to natural immersion, but it provides insight into the role of electrolyte additions.4 In the anodic compartment, the activity of the cathodic sites is significantly limited due to the reduced availability of oxygen; consequently, the current produced by the anodic processes (dissolution of aluminum) is consumed at the cathodic sites on the specimen in the cathodic compartment. Usually, under natural immersion conditions, cathodic and anodic sites are in close proximity and corrosion is generally initiated in the zone of influence of second-phase particles of cathodic nature with respect to the aluminum matrix due to the local alkalinity generated from oxygen reduction. Here, the local increase in pH promotes dissolution of the oxide film present on the adjacent alloy matrix, resulting in localized loss of material. Eventually, a cavity is generated and the cavity may provide the preferred location for subsequent localized attack. In the split-cell experiments, the cathodic activity is localized in the cathodic compartment, and consequently, local alkalinization in the anodic side is largely absent. However, from the split-cell experiments, it was demonstrated that addition of 
 tartaric acid in the anodic side is sufficient to decrease the anodic current by about five times. This results from the reduced overall dissolution rate of the air-formed film covering the macroscopic alloy surface.
tartaric acid in the anodic side is sufficient to decrease the anodic current by about five times. This results from the reduced overall dissolution rate of the air-formed film covering the macroscopic alloy surface.
The current surge revealed after addition is related to the temporary modification of the dynamic equilibrium between anodic and cathodic processes on the single electrodes that may occur after the rapid drop of the pH. In order to understand the mechanism of interaction between tartaric acid and anodic current, it is noted that the chemical dissolution of the oxide covering the aluminum matrix is sensitive to pH. Specifically, alumina reveal a very low dissolution rate at pH values close to neutrality, but the solubility significantly increases in acid or alkaline environments. In the anodic side of the split cell, the local alkalinity associated with cathodic processes should be largely restricted, due to the absence of dissolved oxygen. As a consequence, anodic phenomena and associated local acidification are predominant. In these conditions, the addition of tartaric acid in the presence of high amounts of sodium chloride may result in the generation of a buffer solution, with tartaric acid coexisting in equilibrium with sodium tartrate and hydrogensodium tartrate.
In order to simulate this condition, titrations were performed by HCl solution in NaOH solutions with and without addition of tartaric acid. This allows inspection of the entire pH range (reverse titration). From the titration curves, the addition of relatively small amounts of tartaric acid produces a significant increase in pH for relatively large amounts of HCl added. This suggests that the presence of tartrate anions, partially or totally dissociated, prevent the pH from decreasing to very low values when relatively high concentrations of HCl are present. From the corrosion viewpoint, this effect is beneficial because local acidification corresponding to active anodic sites may be significantly reduced by the presence of tartrate species, reducing the overall anodic activity.
In order to produce this effect on the commercial alloy, significant amounts of tartrate species should be retained in the pores after anodizing. Tartrate species are expected to be adsorbed on the pore wall material, and furthermore, aluminum tartrate may precipitate in the pore during rinsing after anodizing. This may be possible because, during anodizing,  of the aluminum cations produced at the metal∕film interface are directly ejected in the anodizing solution. As a consequence, in the region close to the film∕solution12 interface, a high concentration of aluminum cations is present. Under these specific conditions, it is possible that tartaric acid combines with aluminum cations to produce aluminum tartrate, a compound that is highly soluble in acidic solution but with relatively low solubility in water.13 After anodizing, the pH is rapidly increased during rinsing and relatively large amounts of aluminum tartrate may precipitate at the pore walls and remain located there until the specimen is exposed to the corrosive environment. Here, aluminum tartrate may redissolve, producing a buffer solution that prevents local acidification or alkalinization, thereby limiting the susceptibility to localized corrosion.
of the aluminum cations produced at the metal∕film interface are directly ejected in the anodizing solution. As a consequence, in the region close to the film∕solution12 interface, a high concentration of aluminum cations is present. Under these specific conditions, it is possible that tartaric acid combines with aluminum cations to produce aluminum tartrate, a compound that is highly soluble in acidic solution but with relatively low solubility in water.13 After anodizing, the pH is rapidly increased during rinsing and relatively large amounts of aluminum tartrate may precipitate at the pore walls and remain located there until the specimen is exposed to the corrosive environment. Here, aluminum tartrate may redissolve, producing a buffer solution that prevents local acidification or alkalinization, thereby limiting the susceptibility to localized corrosion.
Conclusions
The effects of tartaric acid addition on the anodizing and postanodizing behavior of AA 2024 T3 aluminum alloy have been examined. It was demonstrated that the addition of tartaric acid to the anodizing bath does not significantly modify the electrical properties of the anodic oxide or the porous anodic film morphology. This suggests that tartaric acid derived species are not significantly incorporated in the oxide during anodizing. Conversely, it was shown that tartaric acid can significantly affect the postanodizing behavior, independent of the chemical composition of the oxide film generated. Specifically, it was revealed that tartaric acid decreases the dissolution rate of the anodic oxide in the acidic environment.
As a consequence of the previous, it is suggested that the beneficial effect on the anticorrosion performance associated with the tartaric acid addition is related to the residual tartaric acid in the pore solution after anodizing. This was confirmed by split-cell studies using "bare" alloy specimens, where addition of tartaric acid in the hundreds of parts-per-million range significantly reduced the rate of the anodic reaction. Finally, it was suggested that the presence of tartaric acid derived species in the pore solution may have a beneficial effect on the pH stability, preventing local alkalinization or acidification associated with corrosion processes.
Acknowledgment
The authors acknowledge the financial support provided by the Engineering and Physical Sciences Research Council Portfolio Award, LATEST.