
Abstract
The unusual low-potential Li reactivity toward simple 3d-metal oxides can be accounted for by classical thermodynamic predictions and simple acido-basic considerations. The Smith's scale, defined in solids for acido-basic reactions involving  species exchange, is successfully used to check that, among the numerous simple oxides, the basic ones such as MnO, FeO, CoO, NiO, and CuO should reversibly react with lithium. Besides the basicity criteria, we stressed that the nanometric character of the reduced composite electrode (e.g., metallic nanoparticles immersed in a highly divided
species exchange, is successfully used to check that, among the numerous simple oxides, the basic ones such as MnO, FeO, CoO, NiO, and CuO should reversibly react with lithium. Besides the basicity criteria, we stressed that the nanometric character of the reduced composite electrode (e.g., metallic nanoparticles immersed in a highly divided  media) is a must to enable the reversible reactivity of metal oxides toward Li. Such a simple approach was finally implemented to other compounds (sulfides, nitrides, vanadates...) and the predictions confronted with experimental data. © 2002 The Electrochemical Society. All rights reserved.
media) is a must to enable the reversible reactivity of metal oxides toward Li. Such a simple approach was finally implemented to other compounds (sulfides, nitrides, vanadates...) and the predictions confronted with experimental data. © 2002 The Electrochemical Society. All rights reserved.
Export citation and abstract BibTeX RIS
Simple 3d-metal oxides (MO with  Co, Ni, Cu,...), because of the absence of interstitial sites within their crystallographic structure for Li insertion/deinsertion and of the nonexistence of Li-M alloys,1
2
3 have long been disregarded as possible reversible electrode materials for rechargeable Li-based batteries. In contrast, one of these oxides (CuO) is used in commercial primary cells.4 Recently, our group showed that these materials could reversibly react with large amounts of Li, leading to sustained capacities as high as 700 mAh/g of CoO upon cycling.5
6
7
8 The Li reactivity process in these oxides mainly consists of two steps. The first one, clearly visualized during the first reduction by a voltage plateau in the 1 V range corresponding to a capacity of 2 lithium ions, unambiguously entails a Li-driven decomposition of the metal oxide into a composite matrix made of metallic nanograins
Co, Ni, Cu,...), because of the absence of interstitial sites within their crystallographic structure for Li insertion/deinsertion and of the nonexistence of Li-M alloys,1
2
3 have long been disregarded as possible reversible electrode materials for rechargeable Li-based batteries. In contrast, one of these oxides (CuO) is used in commercial primary cells.4 Recently, our group showed that these materials could reversibly react with large amounts of Li, leading to sustained capacities as high as 700 mAh/g of CoO upon cycling.5
6
7
8 The Li reactivity process in these oxides mainly consists of two steps. The first one, clearly visualized during the first reduction by a voltage plateau in the 1 V range corresponding to a capacity of 2 lithium ions, unambiguously entails a Li-driven decomposition of the metal oxide into a composite matrix made of metallic nanograins  dispersed into
dispersed into  Such a scenario was established from an arsenal of experimental techniques [electrochemical, magnetic, in situ X-ray diffraction (XRD), high-resolution electron microscopy (HREM) and X-ray absorption spectroscopy (XAS)]. The origin of the second step, visualized by the sloping part of the voltage-composition discharge curve down to 0 V, involves an organic-type layer growth that disappears upon the subsequent charge. However, this phenomenon, which results from enhanced electrolyte reactivity promoted by the formation of metal nanoparticles, is beyond the scope of this paper.
Such a scenario was established from an arsenal of experimental techniques [electrochemical, magnetic, in situ X-ray diffraction (XRD), high-resolution electron microscopy (HREM) and X-ray absorption spectroscopy (XAS)]. The origin of the second step, visualized by the sloping part of the voltage-composition discharge curve down to 0 V, involves an organic-type layer growth that disappears upon the subsequent charge. However, this phenomenon, which results from enhanced electrolyte reactivity promoted by the formation of metal nanoparticles, is beyond the scope of this paper.
Based on a compilation of many results, we demonstrate that the unusual electrochemical reaction 
 experimentally observed, can be accounted for using a classical thermodynamic approach assisted by a relatively favorable kinetic. Finally, based on this concept, this unusual Li reaction can be extended to various kinds of inorganic compounds (chalcogenides, nitrides...).
experimentally observed, can be accounted for using a classical thermodynamic approach assisted by a relatively favorable kinetic. Finally, based on this concept, this unusual Li reaction can be extended to various kinds of inorganic compounds (chalcogenides, nitrides...).
Experimental
Electrochemical tests were done on Swagelok-type cells, with an  -based material as positive electrode
-based material as positive electrode  lithium metal as negative electrode (Aldrich), and a borosilicate glass fiber separator soaked with a molar
lithium metal as negative electrode (Aldrich), and a borosilicate glass fiber separator soaked with a molar  solution (in EC:DMC/1:1 in mass ratio) as electrolyte. The used materials were either homemade or provided by Aldrich or by UM (Belgium) for CoO powders. Plastic composite positive electrodes were made according to Bellcore's plastic Li-ion technology (PLiON™), in which the studied powder, poly(vinylidene fluoride)-hexafluoropropylene (PVDF-HFP), SP carbon, and dibutyl phthalate (DBP) weight ratios were 56, 12, 10, and 22%, respectively. 1 cm2 and 75 μm thick disks containing between 7 and 10 mg of the active material were punched out of the plastic laminate and extracted for their DBP by diethyl ether prior to being used as electrode.
solution (in EC:DMC/1:1 in mass ratio) as electrolyte. The used materials were either homemade or provided by Aldrich or by UM (Belgium) for CoO powders. Plastic composite positive electrodes were made according to Bellcore's plastic Li-ion technology (PLiON™), in which the studied powder, poly(vinylidene fluoride)-hexafluoropropylene (PVDF-HFP), SP carbon, and dibutyl phthalate (DBP) weight ratios were 56, 12, 10, and 22%, respectively. 1 cm2 and 75 μm thick disks containing between 7 and 10 mg of the active material were punched out of the plastic laminate and extracted for their DBP by diethyl ether prior to being used as electrode.
The assembled cells were electrochemically tested using a MacPile automatic cycling/data recording system (Biologic S.A., Claix, France) that can be operated in both galvanostatic and potentiostastic modes. Transmission electron microscopy (TEM) observations were made by means of a Philips transmission electron microscope CM12 instrument equipped with a home-designed sample holder for moisture-sensitive samples.
Results and Discussion
A survey of the reactivity of numerous binary compounds  toward Li at low voltage was carried out. In order to simplify, metal elements (M) recognized to electrochemically form Li alloys2
3 as well as compounds giving insertion reactions with lithium such as
toward Li at low voltage was carried out. In order to simplify, metal elements (M) recognized to electrochemically form Li alloys2
3 as well as compounds giving insertion reactions with lithium such as  9
9  10
10  11
11  12
12 
 13
14
15 or
13
14
15 or  were excluded from such studies. Finally,
were excluded from such studies. Finally,  (rutile), MnO, FeO, CoO, NiO,
(rutile), MnO, FeO, CoO, NiO,  and CuO were the only oxides tested down to 0.02 V vs. Li. We only noted an electrochemical activity for the 3d metals situated on the right side of the periodic table starting from Cu, Ni, Co, Fe, Mn.5
6
7
8 The voltage-composition curves, reported in Fig. 1 for a few MO, exhibited similar features with a well-defined plateau during the first discharge followed by an additional capacity and a hysteresis-type profile upon cycling. For all these metal oxides, the end of this plateau was shown, as deduced by TEM, to be associated to the formation of metal nanoparticles,
and CuO were the only oxides tested down to 0.02 V vs. Li. We only noted an electrochemical activity for the 3d metals situated on the right side of the periodic table starting from Cu, Ni, Co, Fe, Mn.5
6
7
8 The voltage-composition curves, reported in Fig. 1 for a few MO, exhibited similar features with a well-defined plateau during the first discharge followed by an additional capacity and a hysteresis-type profile upon cycling. For all these metal oxides, the end of this plateau was shown, as deduced by TEM, to be associated to the formation of metal nanoparticles,  embedded in a
embedded in a  matrix. Note that the electrochemical behavior of CuO is slightly different due to the Cu(I) stability, as reported elsewhere.7
16 In contrast,
matrix. Note that the electrochemical behavior of CuO is slightly different due to the Cu(I) stability, as reported elsewhere.7
16 In contrast,  did not react, and we experienced a limited capacity for MnO when cycled in galvanostatic mode down to 0.02 V (1 Li per 5 h). Finally, besides the oxides associated to the ones on the right side of the 3d-metal series, we found that the corresponding sulfides such as
did not react, and we experienced a limited capacity for MnO when cycled in galvanostatic mode down to 0.02 V (1 Li per 5 h). Finally, besides the oxides associated to the ones on the right side of the 3d-metal series, we found that the corresponding sulfides such as  8 and
8 and  17 (in collaboration with Telcordia) exhibited the same electrochemical behavior (e.g., plateau followed by a sloping voltage). Such observations are explained by an extended description of the unusual oxides reactivity vs. Li according to Reaction 1
17 (in collaboration with Telcordia) exhibited the same electrochemical behavior (e.g., plateau followed by a sloping voltage). Such observations are explained by an extended description of the unusual oxides reactivity vs. Li according to Reaction 1
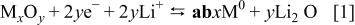
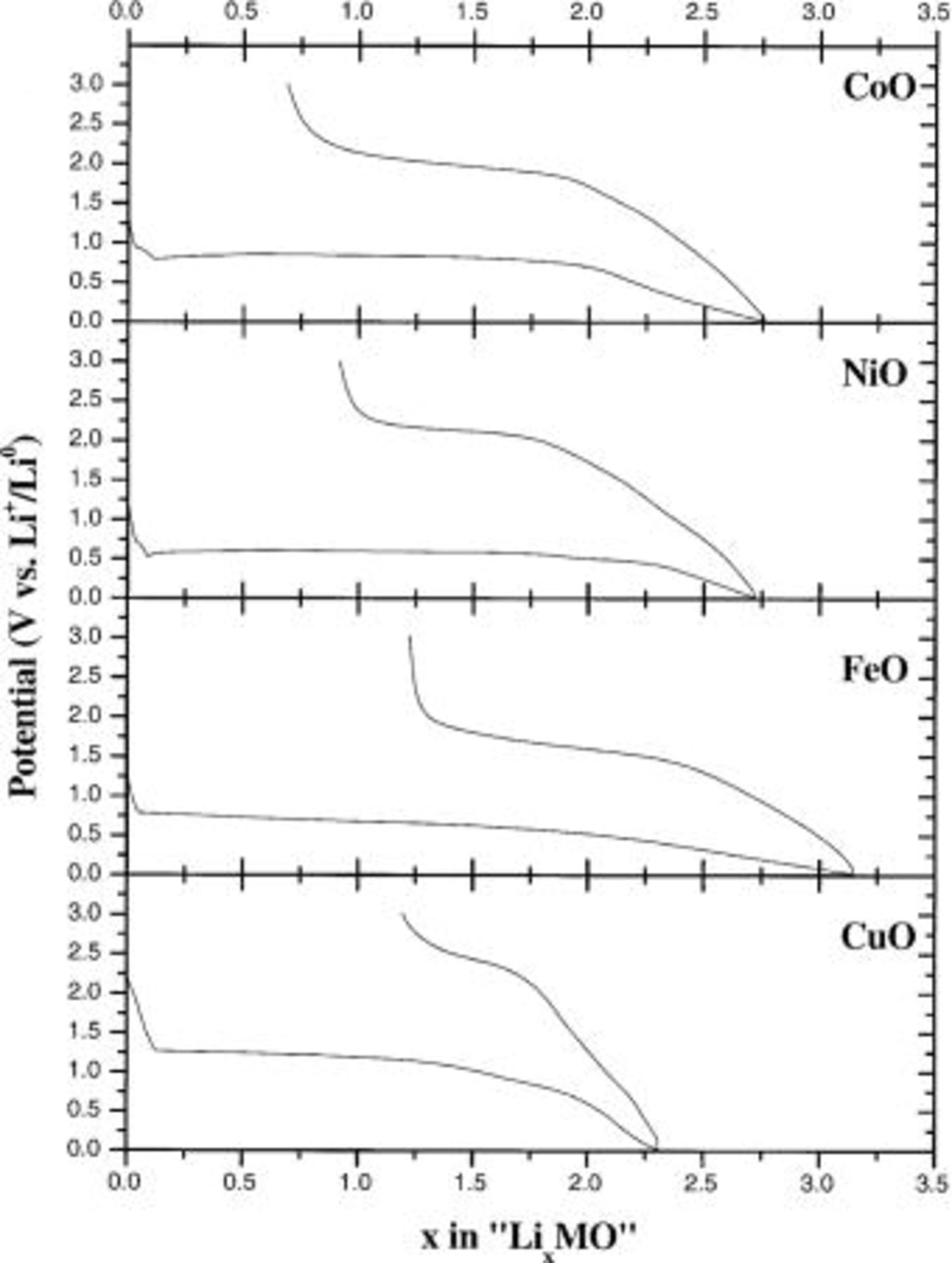
Figure 1. Voltage-composition profiles for various MO/Li cells (with  Ni, Fe, and Cu) cycled at a C/5 rate between 0.02 and 3 V, with the voltage plateau corresponding to the reduction of MO into
Ni, Fe, and Cu) cycled at a C/5 rate between 0.02 and 3 V, with the voltage plateau corresponding to the reduction of MO into  nanoparticles and the sloping voltage consisting in an electrocatalysis decomposition of the electrolyte.
nanoparticles and the sloping voltage consisting in an electrocatalysis decomposition of the electrolyte.
Although this electrode reaction clearly differs from classical insertion or Li-alloying processes, such reversible systems can exist as previously reported.6 8 We just have to check if Reaction 1 is thermodynamically spontaneously feasible in the a side (reduction process) for all the tested oxides. That is to say, one must calculate the standard Gibbs free energy change by simply considering a cell as conventionally described18
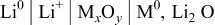
The overall cell reaction is
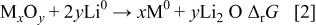
Under reference pressure  and
and  (experimental conditions in our Swagelok-cells), we can easily calculate the Gibbs free energy change
(experimental conditions in our Swagelok-cells), we can easily calculate the Gibbs free energy change  involved in Reaction 2 (equal in our case to
involved in Reaction 2 (equal in our case to  ) and finally the electromotive force (emf) value E from the well-known relation, where F is Faraday's constant
) and finally the electromotive force (emf) value E from the well-known relation, where F is Faraday's constant
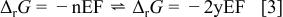
For the oxides considered in this study, we calculated  (and E) using the thermodynamic data given in the literature for massive compounds (Table I).18
19 The Gibbs free energy values are negative, proving unambiguously that such oxides are, under these conditions, spontaneously reducible (a side). The standard potential
(and E) using the thermodynamic data given in the literature for massive compounds (Table I).18
19 The Gibbs free energy values are negative, proving unambiguously that such oxides are, under these conditions, spontaneously reducible (a side). The standard potential  of the electrode reaction, Reaction 1, can be easily deduced by the Hess law according to the following relation
of the electrode reaction, Reaction 1, can be easily deduced by the Hess law according to the following relation
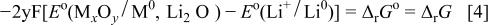
whereas the equilibrium potential  of Reaction 1 is deduced from the Nernst law. Note that the
of Reaction 1 is deduced from the Nernst law. Note that the  redox system being our reference electrode,
redox system being our reference electrode,  and with
and with  we have the final relation
we have the final relation 
 Consequently, since Reaction 1 is thermodynamically spontaneous in the a side, as we have shown, it should proceed in the b side if an external emf greater than E is applied. Thus, by considering a thermodynamic reversible process (at equilibrium), we expect Reaction 1 to proceed on both sides with the onset being a well-defined plateau at an equilibrium potential
Consequently, since Reaction 1 is thermodynamically spontaneous in the a side, as we have shown, it should proceed in the b side if an external emf greater than E is applied. Thus, by considering a thermodynamic reversible process (at equilibrium), we expect Reaction 1 to proceed on both sides with the onset being a well-defined plateau at an equilibrium potential  with a capacity of
with a capacity of 
 For instance, with CoO
For instance, with CoO  this plateau should be at
this plateau should be at  vs.
vs. 
Table I.
Calculated Gibbs free energy change 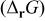 and emf and emf 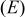 values for the reaction values for the reaction 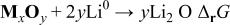 . . | ||
|---|---|---|
| Compound |
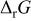 (kJ mol−1) (kJ mol−1) |
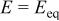 (V) (V) |
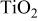 | −233.53 | 0.605 |
| MnO | −199.17 | 1.032 |
| FeO | −317.10 | 1.643 |
| NiO | −346.24 | 1.794 |
| CoO | −347.01 | 1.798 |
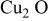 | −414.18 | 2.146 |
| CuO | −428.26 | 2.219 |
Nevertheless, Fig. 1 shows that the reduction potential of MO/Li cells (especially during the first discharge) is quite low compared with the theoretical predictions. For instance, the cobalt monoxide reduction takes place at about 0.8 V. Such deviation is naturally of kinetic nature and represents the important overpotential needed to initiate and pursue the decomposition reaction. As expected, a relaxation step in the middle of the plateau [open-circuit voltage (OCV) measurement] increases the cell voltage (Fig. 2). Note that the OCV value after 100 h is only about 1.3 V by comparison with the theoretical value  indicating a slow system. This is consistent with Novak's results on CuO cells claiming that a 2 month period was necessary for a CuO electrode to go back to its steady equilibrium potential after a current pulse.20 Assuming that the 1 V overpotential needed for reducing CoO is of the same amplitude as for most of the other 3d oxides considered, one can, according to Table I, anticipate some difficulties in reducing MnO and a fortiori
indicating a slow system. This is consistent with Novak's results on CuO cells claiming that a 2 month period was necessary for a CuO electrode to go back to its steady equilibrium potential after a current pulse.20 Assuming that the 1 V overpotential needed for reducing CoO is of the same amplitude as for most of the other 3d oxides considered, one can, according to Table I, anticipate some difficulties in reducing MnO and a fortiori  However, using potentiostatic intermittent titration technique (PITT) measurements to minimize the needed overpotential value,21 the overall MnO reduction was observed at just about 100 mV above the
However, using potentiostatic intermittent titration technique (PITT) measurements to minimize the needed overpotential value,21 the overall MnO reduction was observed at just about 100 mV above the  system (Fig. 3). As expected, no reactivity was measured for
system (Fig. 3). As expected, no reactivity was measured for  under similar conditions.
under similar conditions.
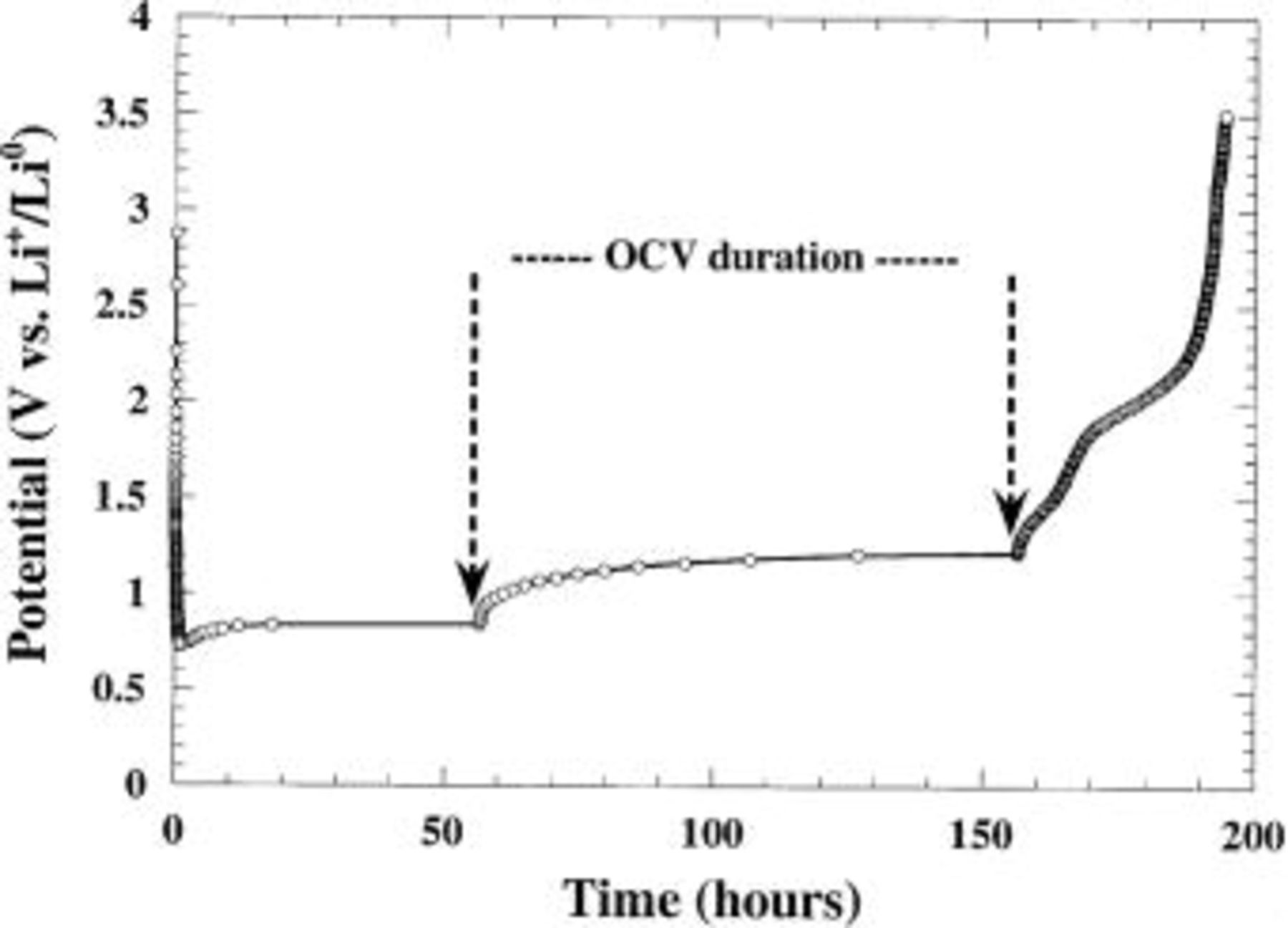
Figure 2. Chronopotentiogram for CoO/Li cell reduced at a C/5 rate with an OCV period in the middle of the plateau  in order to reach the equilibrium potential (OCV duration: 100 h).
in order to reach the equilibrium potential (OCV duration: 100 h).
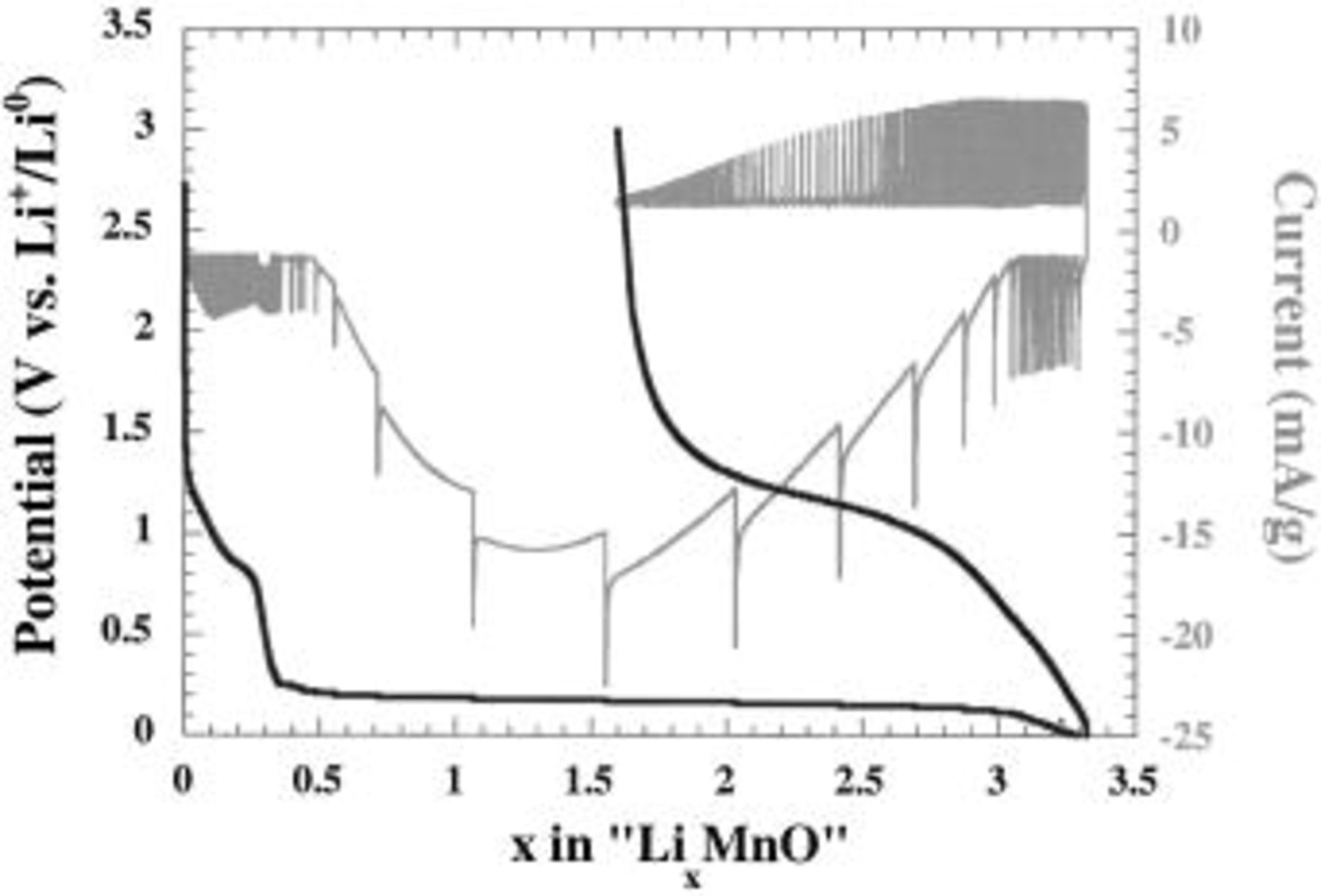
Figure 3. The voltage composition curves as deduced from PITT measurements for MnO/Li cell cycled between 0.02 and 3 V. The voltage steps were of 10 mV with a current limit corresponding to C/300 (e.g., 1 Li in 300 h).
Returning to Reaction 1, the electrochemical reversibility "especially the back side reaction (b)," although thermodynamically possible, was kinetically unexpected due to the difficult task to reconvert  (solid-state phases) into metal oxide
(solid-state phases) into metal oxide  We experienced this difficulty by forcing the reoxidation
We experienced this difficulty by forcing the reoxidation  of stoichiometric amounts of metallic cobalt and
of stoichiometric amounts of metallic cobalt and  powders mechanically milled by means of a SPEX 800 apparatus that operates in a shock mode. Whatever the milling time tried, we never succeeded in recharging a Li cell using such a positive mixture at the positive electrode. Besides, note that the well-defined plateau observed during the first discharge (Fig. 1) is vanishing on subsequent discharges, leading to "sloppy voltage plateaus." This is mainly due to the nanosized character of the electroproduced particles, which generates many new indefinite interfaces. Consequently, we believed that the reversibility experienced with various
powders mechanically milled by means of a SPEX 800 apparatus that operates in a shock mode. Whatever the milling time tried, we never succeeded in recharging a Li cell using such a positive mixture at the positive electrode. Besides, note that the well-defined plateau observed during the first discharge (Fig. 1) is vanishing on subsequent discharges, leading to "sloppy voltage plateaus." This is mainly due to the nanosized character of the electroproduced particles, which generates many new indefinite interfaces. Consequently, we believed that the reversibility experienced with various  cells is mainly nested in the nanoparticle character of the positive electrode after it has been reduced once. Indeed, the specific
cells is mainly nested in the nanoparticle character of the positive electrode after it has been reduced once. Indeed, the specific  particles confinement on a nanoscale decreases considerably the activation energy by comparison with massive solid-state compounds. Nevertheless, electrochemically speaking, Reaction 1 remains slow, as seen by the large difference observed between the discharge and the charge-voltage composition curves in cycling (Fig. 1).
particles confinement on a nanoscale decreases considerably the activation energy by comparison with massive solid-state compounds. Nevertheless, electrochemically speaking, Reaction 1 remains slow, as seen by the large difference observed between the discharge and the charge-voltage composition curves in cycling (Fig. 1).
Although unconventional for the battery community because quite unlike the classical Li-insertion or Li-alloying mechanism, the suggested electrochemical reaction, Reaction 1, bears some striking similarity with the "second-kind" type of electrode, well known in conventional electrochemistry. To support such a statement, one must further analyze what is behind Reaction 1, especially in terms of mechanism.
A "second-kind"-type electrode has, by definition, two solid phases in contact with the electrolyte as well as an inert electrical lead. One of the phases is typically a metal; the second one is a nonsoluble salt or compound of the metal.22 Examples of such electrode type can be oxide-based and, finally, Reaction 1 can be compared with the well known and commercialized aqueous  reference electrode that operates according to Reaction 5, with
reference electrode that operates according to Reaction 5, with 
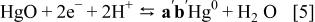
This reaction is basically a classical precipitation reaction that involves the  ion (super basic), which immediately reacts either with the protons of the medium to form a water molecule or with
ion (super basic), which immediately reacts either with the protons of the medium to form a water molecule or with  (acidic species) to form HgO. More specifically, the underlying mechanism, as shown in Table II, can be described as two main steps: (i) a charge transfer (CT) between
(acidic species) to form HgO. More specifically, the underlying mechanism, as shown in Table II, can be described as two main steps: (i) a charge transfer (CT) between  and
and  with (ii) an acido-basic consecutive reaction (CR) during which the oxygen of mercury oxide (basic species) is captured by protons (acid species) to form a water molecule. The overall reversibility of Reaction 5 implies the reversibility of the two CT and CR reactions together with the basicity of the oxygen-based compounds (e.g., HgO and
with (ii) an acido-basic consecutive reaction (CR) during which the oxygen of mercury oxide (basic species) is captured by protons (acid species) to form a water molecule. The overall reversibility of Reaction 5 implies the reversibility of the two CT and CR reactions together with the basicity of the oxygen-based compounds (e.g., HgO and  ). Thus, as Reactions 1 and 5 look alike, it is quite tempting to also describe Reaction 1 as a CT between
). Thus, as Reactions 1 and 5 look alike, it is quite tempting to also describe Reaction 1 as a CT between  and
and  with the oxygen being taken by
with the oxygen being taken by  to form
to form  instead of
instead of  via an acido-basic reaction.
via an acido-basic reaction.
Table II.
Detailed electrode reaction for 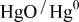 system showing the charge transfer (CT) and the acido-basic consecutive reaction (CR). system showing the charge transfer (CT) and the acido-basic consecutive reaction (CR). | |||
|---|---|---|---|
| Discharge | Charge | ||
| Steps | Reactions | Steps | Reactions |
| CT |
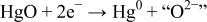 | CT |
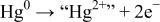 |
| CR |
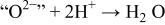 | CR |
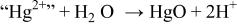
|
Nevertheless, there is an important physicochemical difference between 
 and
and 
 for instance. In fact, within the mercury electrode, water that constitutes the electrolyte medium (solvent) is in excess. Furthermore, its liquid state enables a large surface contact with the redox couple
for instance. In fact, within the mercury electrode, water that constitutes the electrolyte medium (solvent) is in excess. Furthermore, its liquid state enables a large surface contact with the redox couple  favoring the electrochemical reaction. On the contrary,
favoring the electrochemical reaction. On the contrary,  is present in the solid state and in limited amount. In addition, the occurrence of Reaction 1, that simultaneously brings in three solid phases
is present in the solid state and in limited amount. In addition, the occurrence of Reaction 1, that simultaneously brings in three solid phases 
 and
and  ), obviously implies a mass transport through intergranular diffusion. However, as explained previously, we should recall that we are dealing with a heterogeneous nanometric system so that the reactivity diffusion length is approaching that of a liquid medium, thereby lifting the kinetic limitation.
), obviously implies a mass transport through intergranular diffusion. However, as explained previously, we should recall that we are dealing with a heterogeneous nanometric system so that the reactivity diffusion length is approaching that of a liquid medium, thereby lifting the kinetic limitation.
At this point a legitimate question is whether our proposed electrochemically assisted acido-basic reaction favored by nanoparticles (see the summary scheme in Fig. 4) could be a satisfactory approach. As an attempt to answer such a question, among the 3d-metal oxides that should thermodynamically react with Li, one must compare the acido-basic character of metal oxides, and therefore go back to acidity concepts. Different definitions exist depending on the considered species and the medium. For instance, we have the well-known Lewis approach on the electronic doublet (⋅ ⋅), the Brønsted definition dealing with proton  exchange, and the Lux-Flood23 concept dealing with
exchange, and the Lux-Flood23 concept dealing with  anion transfer. Since
anion transfer. Since  ions are directly involved in our process, we chose the latter. In order to compare the acidity/basicity of oxides, Smith defined a scale in which the oxide basicity is defined by the a constant fixed to 0 for
ions are directly involved in our process, we chose the latter. In order to compare the acidity/basicity of oxides, Smith defined a scale in which the oxide basicity is defined by the a constant fixed to 0 for  and that will be positive or negative for acidic and basic oxides, respectively.24 The more negative a parameter will be, the more basic the oxide will be. The a values for the investigated oxides are reported in Table III. Finally, all oxides that reacted reversibly with Li are basic as
and that will be positive or negative for acidic and basic oxides, respectively.24 The more negative a parameter will be, the more basic the oxide will be. The a values for the investigated oxides are reported in Table III. Finally, all oxides that reacted reversibly with Li are basic as  itself. Based on this acido-basicity complex, an explanation was given to a recent report25 claiming the misunderstood feasibility of reoxidizing
itself. Based on this acido-basicity complex, an explanation was given to a recent report25 claiming the misunderstood feasibility of reoxidizing  into SnO basic and not into
into SnO basic and not into  acid when cycling a
acid when cycling a  cell.
cell.
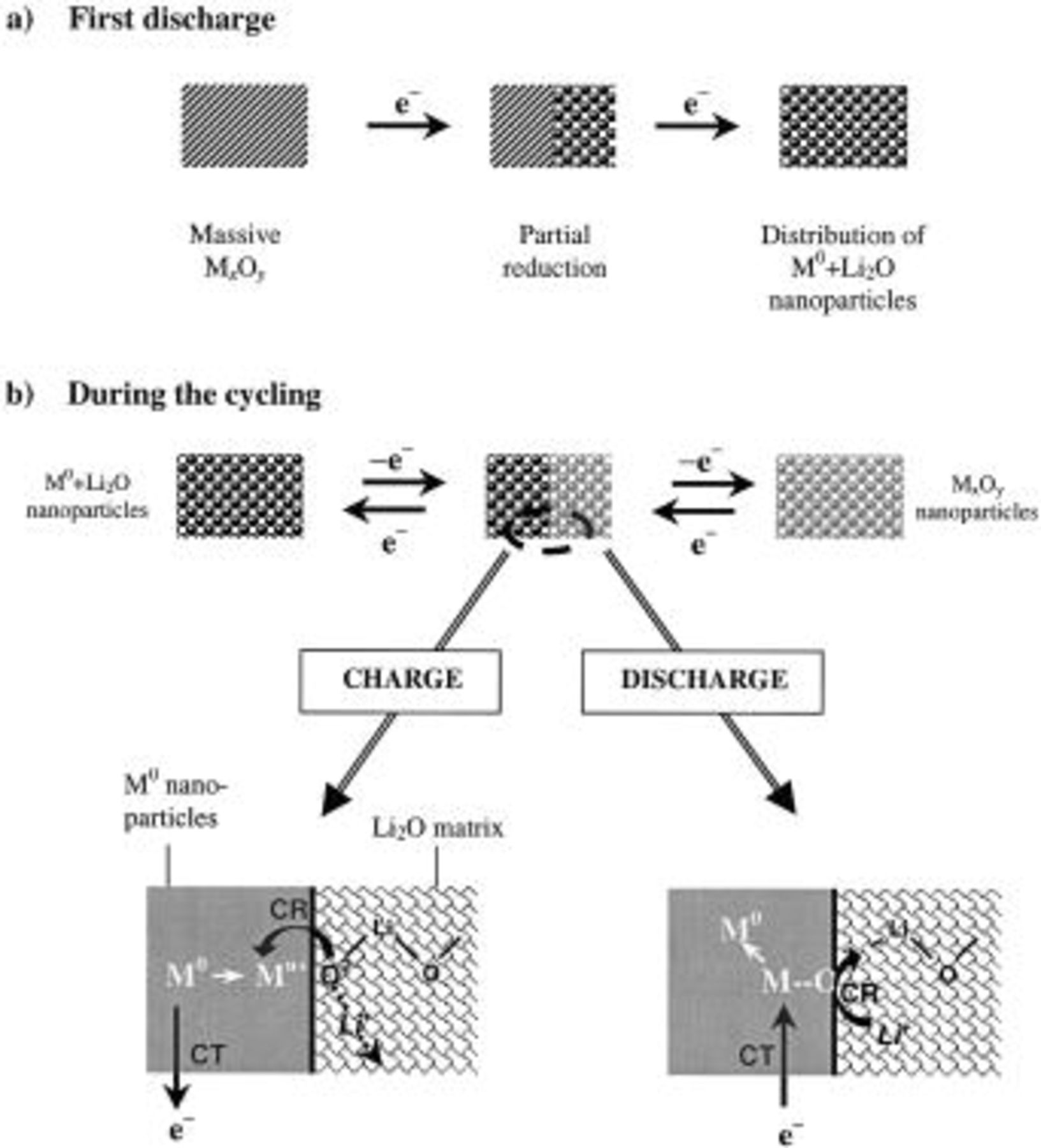
Figure 4. A schematic of the overall reaction mechanism is shown within (a) the textural evolution of the electrode material upon the first discharge leading to a nanometric composite. The evolution of this latter on subsequent cycles are given in (b) together with a zoom of the local reaction occurring at the nanoparticle size.
Table III.
Values of parameter 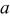 for binary oxides, relative to for binary oxides, relative to 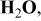 given by Smith's scale.24 given by Smith's scale.24 | |
|---|---|
| Compound | Parameter a |
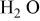 | 0 |
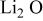 | −9.2 |
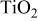 | 0.7 |
| MnO | −4.8 |
| FeO | −3.4 |
| NiO | −2.4 |
| CoO | −3.8 |
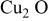 | −1.0 |
| CuO | −2.5 |
At this point, it is worth stressing that in the 1980s, the reversible reactivity of metal oxides toward Li according to Reaction 1 was put forward by Thackeray et al. for high-temperature molten salts  KCl/LiAl at 420°C.9
10 At that time, the proposed mechanism consisted of a "displacement" reaction of either iron or Li from the initial
KCl/LiAl at 420°C.9
10 At that time, the proposed mechanism consisted of a "displacement" reaction of either iron or Li from the initial  structure, thus leaving the initial oxygen skeleton untouched. Such a mechanism, which implies the quantitative iron diffusion (relatively heavy cation), is hardly believable in our case where the temperature does not exceed 25°C. Although the work of these authors regarding the molten salts medium is well accepted, it is worth recalling that Trémillon26 explained through the oxidation of ilmenite
structure, thus leaving the initial oxygen skeleton untouched. Such a mechanism, which implies the quantitative iron diffusion (relatively heavy cation), is hardly believable in our case where the temperature does not exceed 25°C. Although the work of these authors regarding the molten salts medium is well accepted, it is worth recalling that Trémillon26 explained through the oxidation of ilmenite  in the presence of
in the presence of  (Reaction 6) that
(Reaction 6) that  species (free and stable in molten salt media) play a role in redox reactions involving oxides
species (free and stable in molten salt media) play a role in redox reactions involving oxides
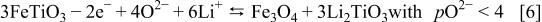
Thus, in light of such a report, our electrochemically assisted acido-basic reaction could satisfactorily explain Thackeray's reaction as well.
In light of these encouraging results it was tempting to generalize our electrochemically assisted acido-basic mechanism to other compounds besides oxides when it is thermodynamically possible to reduce them. As  is not the only anion showing basicity, one can go further by studying the electrochemical reactivity of other simple materials having the same transition cations as the studied oxides but different anions (
is not the only anion showing basicity, one can go further by studying the electrochemical reactivity of other simple materials having the same transition cations as the studied oxides but different anions ( and
and  for instance) so that the envisaged reaction can be written as follows
for instance) so that the envisaged reaction can be written as follows
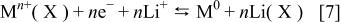
Such a generalization boils down to verify both the thermodynamic feasibility of Reaction 7 and the exchange facility of the anion X (e.g., its basicity). Thermodynamically wise Reaction 7 is feasible for 
 In contrast, while
In contrast, while  can be electrochemically reduced by Li, no
can be electrochemically reduced by Li, no  estimate could be calculated due to the lack of existing thermodynamic data in the literature. Regarding acido-basicity, one needs a scale quantifying the intrinsic basicity of various anions, which have no solvating effect since Reaction 7 deals with solid-state couples. Such a classification does exist and consists in measuring the strength of gaseous acids and bases [e.g., measuring the intrinsic proton affinity (PA) for X, which is taken positive by convention]. Waddington27 indirectly determined from the Born-Harber cycle the term PA used for chalcogenides and the nitride anions. The values are reported in Table IV.
estimate could be calculated due to the lack of existing thermodynamic data in the literature. Regarding acido-basicity, one needs a scale quantifying the intrinsic basicity of various anions, which have no solvating effect since Reaction 7 deals with solid-state couples. Such a classification does exist and consists in measuring the strength of gaseous acids and bases [e.g., measuring the intrinsic proton affinity (PA) for X, which is taken positive by convention]. Waddington27 indirectly determined from the Born-Harber cycle the term PA used for chalcogenides and the nitride anions. The values are reported in Table IV.
Table IV.
| Values of the intrinsic PA for chalcogenide and nitride ions given by Waddington.27 | |
|---|---|
| Anion | PA in the gas phase (kJ/mol) |
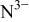 | 3084 |
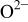 | 2318 |
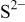 | 2300 |
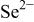 | 2200 |
The nitride ion shows a 33% higher PA (e.g., large basicity) as compared to  consistent with the large electrostatic attraction of the three negative charges.
consistent with the large electrostatic attraction of the three negative charges.  basicity is in the same size order as for the oxide, whereas the selenium ion is slightly more acid. Nevertheless, in our case
basicity is in the same size order as for the oxide, whereas the selenium ion is slightly more acid. Nevertheless, in our case  is not suitable because its metal nature makes it electroactive above 2 V
is not suitable because its metal nature makes it electroactive above 2 V  just like tellurium.28
29 Consequently, simple sulfides and 3d-transition metal nitrides can react along the same principle as Reaction 1. Here, we only show the electrochemical reaction of a sulfite, namely,
just like tellurium.28
29 Consequently, simple sulfides and 3d-transition metal nitrides can react along the same principle as Reaction 1. Here, we only show the electrochemical reaction of a sulfite, namely,  (from Aldrich). Figure 5 shows the potential-composition curve obtained when in classical galvanostatic mode and for 1 Li/5 h. Like for the oxides, a well-defined plateau is noticed during the first discharge, with a capacity close to the expected theoretical value if we refer to the following reaction
(from Aldrich). Figure 5 shows the potential-composition curve obtained when in classical galvanostatic mode and for 1 Li/5 h. Like for the oxides, a well-defined plateau is noticed during the first discharge, with a capacity close to the expected theoretical value if we refer to the following reaction
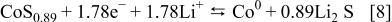
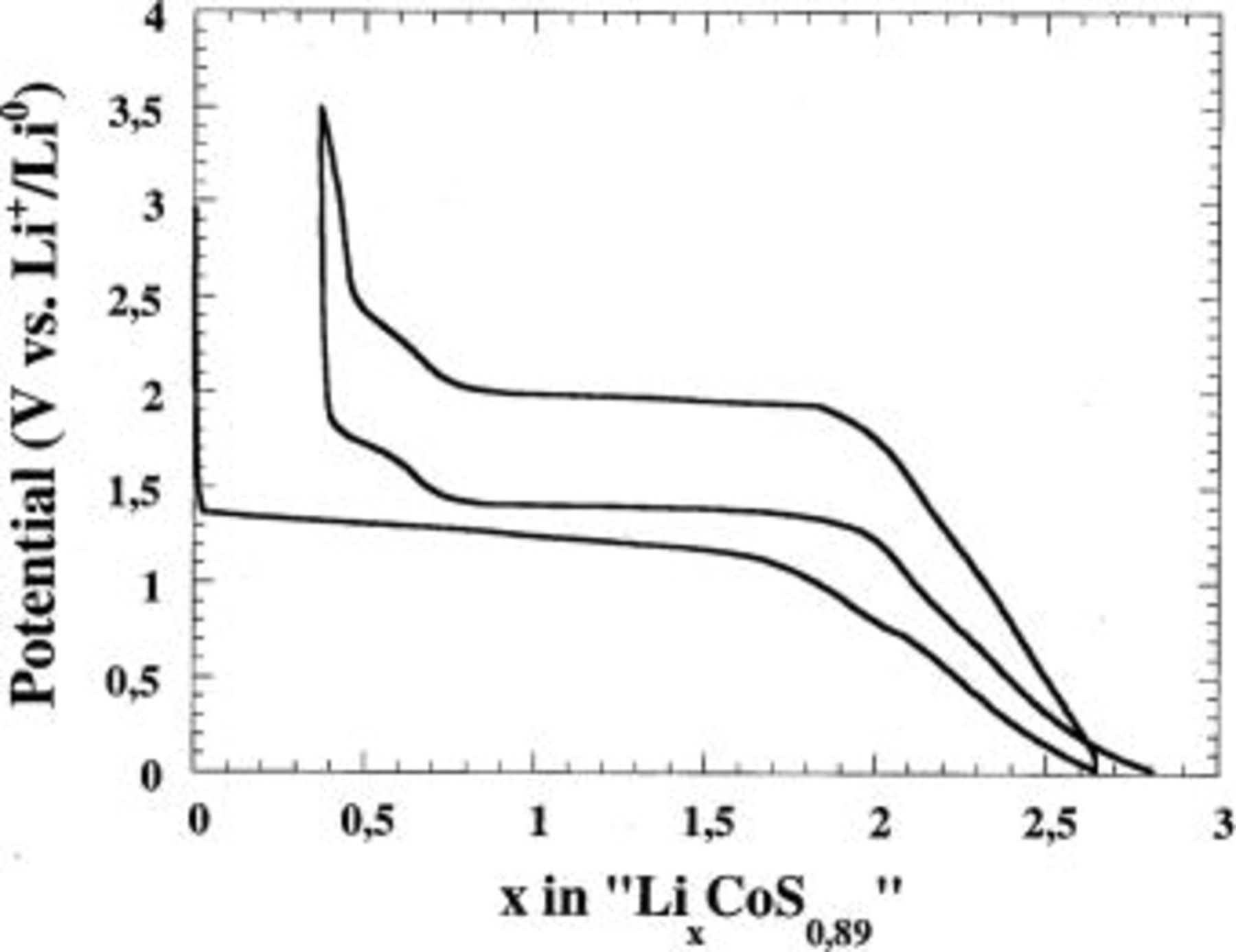
Figure 5. The voltage-composition profile for  cell, cycled between 0.02 and 3.5 V at a C/5 rate.
cell, cycled between 0.02 and 3.5 V at a C/5 rate.
In contrast, the reaction overpotential is clearly smaller than for the oxides (0.3 instead of 1 V), and therefore the plateau average potential is closer to the theoretical equilibrium potential  as determined using similar testing parameters as for the oxides. The recharge evolution is nearly the same as that of the oxides except for the plateau at 2.2 V, which is less ill defined than for the oxides. TEM studies were carried out to further validate the oxide mechanism similarity. Figure 6a shows active material particles after a reduction carried out down to 0.02 V. Again, there is nanoparticle formation (∼50 Å) together with the growth of an inorganic/organic layer around the initial particle of the active material that preserves its shape. From scanning area electron diffraction (SAED) the presence of
as determined using similar testing parameters as for the oxides. The recharge evolution is nearly the same as that of the oxides except for the plateau at 2.2 V, which is less ill defined than for the oxides. TEM studies were carried out to further validate the oxide mechanism similarity. Figure 6a shows active material particles after a reduction carried out down to 0.02 V. Again, there is nanoparticle formation (∼50 Å) together with the growth of an inorganic/organic layer around the initial particle of the active material that preserves its shape. From scanning area electron diffraction (SAED) the presence of  and
and  is clearly defined for the discharged electrode (Fig. 6b). Upon subsequent oxidation,
is clearly defined for the discharged electrode (Fig. 6b). Upon subsequent oxidation,  vanishes to the expense of
vanishes to the expense of  in a similar way as that observed for the oxides.
in a similar way as that observed for the oxides.

Figure 6. TEM micrograph (a) of a  powder recovered from a
powder recovered from a  cell stopped after the first discharge
cell stopped after the first discharge  and the corresponding SAED pattern (b) showing the presence of
and the corresponding SAED pattern (b) showing the presence of  and
and  inside the agglomerate.
inside the agglomerate.
These results clearly indicate that the reactivity mechanism of Li toward the oxide defined by Reaction 1 can be extended to chalcogenides and even nitrides, as recently demonstrated by our Telcordia colleagues (U.S.), for  17 When M does not alloy with Li and X is a base, the reversible reaction, Reaction 7, can be observed as long as nanometric particles are produced. Using this electrochemical process, Li is absolutely not inserted but acts as an acid to trap and fix the base X.
17 When M does not alloy with Li and X is a base, the reversible reaction, Reaction 7, can be observed as long as nanometric particles are produced. Using this electrochemical process, Li is absolutely not inserted but acts as an acid to trap and fix the base X.
In light of this mechanism we can explain our early work dealing with the large reversible capacity of the unusual reversible reactivity of 3d-metal vanadates  Ni, Fe,...) toward Li, which until now was a mystery to us, in spite of the fact that we had most of the pieces to solve the puzzle. Indeed, by studying the
Ni, Fe,...) toward Li, which until now was a mystery to us, in spite of the fact that we had most of the pieces to solve the puzzle. Indeed, by studying the 
 we established a correlation between the hydrated vanadates electrochemical capacity and their oxygen content,30 and furthermore, we witnessed, through in situ X-ray absorption near-edge spectroscopy (XANES) and
we established a correlation between the hydrated vanadates electrochemical capacity and their oxygen content,30 and furthermore, we witnessed, through in situ X-ray absorption near-edge spectroscopy (XANES) and  Mössbauer measurements,31
32 the reduction of
Mössbauer measurements,31
32 the reduction of  to
to  and its reoxidation upon cycling but neglected the sample morphology evolution upon cycling. A fully reduced iron vanadate phase,
and its reoxidation upon cycling but neglected the sample morphology evolution upon cycling. A fully reduced iron vanadate phase,  was recently investigated by performing TEM measurements.8 We evidenced the nanometric character of the sample together with, as determined by SAED, the reversible reduction of
was recently investigated by performing TEM measurements.8 We evidenced the nanometric character of the sample together with, as determined by SAED, the reversible reduction of  in
in  (e.g., Fe-O/metallic iron), thus giving some meaning to our early oxygen correlation.
(e.g., Fe-O/metallic iron), thus giving some meaning to our early oxygen correlation.
Since our first report, a wide variety of 3d-metal-based inorganic materials including oxides (

 ), borates
), borates 
 ), or phosphates have been investigated by others and by our group for their reactivity toward Li at low potential. For instance,
), or phosphates have been investigated by others and by our group for their reactivity toward Li at low potential. For instance,  was shown to present the same electrochemical characteristics as oxides (extra capacity, important polarization, amorphization of the initial material). Although no indication was given by the authors regarding the implied process, we checked the fully reduced material by performing TEM and evidenced the nanometric character of the sample together with the formation/decomposition of
was shown to present the same electrochemical characteristics as oxides (extra capacity, important polarization, amorphization of the initial material). Although no indication was given by the authors regarding the implied process, we checked the fully reduced material by performing TEM and evidenced the nanometric character of the sample together with the formation/decomposition of  as determined by SAED. The same mechanism holds as well for the twenty Co-, Ni-, or Fe-based binary or ternary oxides that we have already studied.
as determined by SAED. The same mechanism holds as well for the twenty Co-, Ni-, or Fe-based binary or ternary oxides that we have already studied.  with a sustained reversible capacity as large as 1100 mAh/g is among the most attractive.
with a sustained reversible capacity as large as 1100 mAh/g is among the most attractive.
Conclusion
We have shown that the reversible electrochemical reactivity of metal oxides toward Li recently reported, leading to the formation of metallic nanoparticles, can be predicted from a classical thermodynamic approach. Formally, the considered reaction can be compared to a simple "second-kind"-type electrode well known in conventional solution electrochemistry where an acido-basic type reaction is involved (" " exchange). To support such a statement, among the 3d-metal oxides that thermodynamically reacted with Li, we have successfully checked their basic character using the Smith's scale. Nevertheless, the unexpected reversibility of the
" exchange). To support such a statement, among the 3d-metal oxides that thermodynamically reacted with Li, we have successfully checked their basic character using the Smith's scale. Nevertheless, the unexpected reversibility of the  reaction is kinetically promoted in solid state by the nanoparticle character of the active material. Finally, this simple description was extended to other classes of materials
reaction is kinetically promoted in solid state by the nanoparticle character of the active material. Finally, this simple description was extended to other classes of materials  where X is basic too. Such oversimplified generalization appears to satisfactorily describe most of the examples reported so far, dealing with the low potential Li reactivity of 3d-metal-based inorganic compounds. In short, through this work we clearly showed that the reversible reactivity of metal oxides toward Li, at first totally unexpected, could simply be explained from well-known and founded concepts.
where X is basic too. Such oversimplified generalization appears to satisfactorily describe most of the examples reported so far, dealing with the low potential Li reactivity of 3d-metal-based inorganic compounds. In short, through this work we clearly showed that the reversible reactivity of metal oxides toward Li, at first totally unexpected, could simply be explained from well-known and founded concepts.
Acknowledgments
The authors give special thanks to D. Larcher, L. Dupont, G. Amatucci, N. Pereira, and M. Dollé.
Université de Picardie Jules Verne assisted in meeting the publication costs of this article.