Article Text

Abstract
Background Individual susceptibility to exacerbations in chronic obstructive pulmonary disease (COPD) is likely influenced by genetic factors; however, most such variance is unexplained. We hypothesised that β2-adrenergic receptor genotypes, Gly16Arg (rs1042713, c.46G>A) and Gln27Glu (rs1042714, c.79C>G) influence risk of severe exacerbations in COPD.
Methods Among 96 762 individuals in the Copenhagen General Population Study, we identified 5262 with COPD (forced expiratory volume in one second divided by forced vital capacity, FEV1/FVC, below 0.7, FEV1 less than 80% of predicted value, age above 40 years and no asthma) who had genotyping performed. Severe exacerbations were defined as acute admissions due to COPD during 5 years of follow-up (mean 3.4 years). 923 individuals with COPD diagnosed similarly in the Copenhagen City Heart Study (CCHS) were used for replication analyses.
Results We recorded 461 severe exacerbations in 5262 subjects. The HRs for severe exacerbations were 1.62 (95% CI 1.30 to 2.03, p=0.00002) for 16Gly/Arg heterozygotes and 1.41 (1.04 to 1.91, p=0.03) for 16Arg homozygotes, compared with 16Gly homozygotes. HRs were 1.35 (1.03 to 1.76, p=0.03) for 27Gln/Glu heterozygotes and 1.49 (1.12 to 1.98, p=0.006) for 27Gln homozygotes, compared with 27Glu homozygotes. Similar trends were observed in the CCHS. Among 27Gln homozygotes only, HRs were 5.20 (1.81 to 14.9, p=0.002) for 16Gly/Arg heterozygotes and 4.03 (1.40 to 11.6, p=0.01) for 16Arg homozygotes, compared with 16Gly homozygotes.
Conclusion Common β2-adrenergic receptor genotypes influence risk of severe exacerbations in COPD, potentially mainly by genetic influence of the 16Arg allele in rs1042713.
- adrenoreceptor polymorphisms
- COPD
- precision medicine
Statistics from Altmetric.com
Key messages
What is the key question?
Genetic susceptibility to risk of severe exacerbations in chronic obstructive pulmonary disease (COPD) has previously been observed. We hypothesised that β2-adrenergic receptor genotypes Gly16Arg (rs1042713, c.46G>A) and Gln27Glu (rs1042714, c.79C>G) influence risk of severe exacerbations in COPD.
What is the bottom line?
Common β2-adrenergic receptor genotypes influence risk of severe exacerbations in COPD, potentially mainly by genetic influence of the 16Arg allele in rs1042713.
Why read on?
This study demonstrates a strong influence of the β2-receptor genotypes 16Gly/Arg and 27Gln/Glu on prospective risk of severe exacerbations in COPD, and since these genotypes are common they could potentially be used as future biomarkers of exacerbations in COPD by inclusion in risk allele scores.
Introduction
Chronic obstructive pulmonary disease (COPD) is a major global health problem ranked as the fourth leading cause of death. COPD often entails acute exacerbations that are important to prognosis and management.1
A twin study has indicated that approximately 60% of individual susceptibility to risk of severe COPD exacerbations requiring hospital admission could be influenced by genetic factors.2 α1-antitrypsin deficiency caused by the Pi Z allele variant (366Glu/Lys) in the SERPINA1 gene on chromosome 14 is acknowledged as the most important genetic risk factor for COPD, and the Z-allele is also associated with increased risk of COPD exacerbations.3 However, this mutation is rare1 and most of the observed genetic variance in susceptibility to COPD exacerbations is unexplained.2 3
In recent post-hoc analyses from a randomised controlled trial it was shown that in COPD patients treated with long-acting β2-agonists,4 the Gly16Arg (rs1042713, c.46G>A) polymorphism was associated with a differential treatment effect on risk of exacerbations.5 Prior to that study, several studies had examined the pharmacogenetic properties of β2-adrenergic receptor polymorphisms, but results have been conflictory.6
In a study of 190 individuals with COPD the authors found no association between 16Gly/Arg or 27Gln/Glu polymorphisms per se and risk of exacerbations.7 A similar lack of association with exacerbations was observed in the placebo arms of two randomised controlled trials when stratified by 16Gly/Arg genotype.8 In contrast, a retrospective study of 92 individuals with COPD observed that 16Arg/Arg homozygotes associated with more use of oral corticosteroids and antibiotics in COPD.9 This association with exacerbations was supported in a retrospective case-control study among 61 individuals with COPD that found an increased risk of having frequent exacerbations among individuals homozygous for 16Arg.10
Nevertheless, there is a paucity of convincing evidence on possible influences of β2-adrenergic receptor genotypes per se on the risk of exacerbations.
We hypothesised that the β2-adrenergic receptor genotypes Gly16Arg and Gln27Glu influence risk of severe exacerbations in COPD.
Methods
Study populations
Copenhagen General Population Study
Data on 96 762 individuals from the prospective Copenhagen General Population Study (CGPS; examined 2003–2013) with lung function measured was used. We defined COPD in concordance with other studies as the ratio between forced expiratory volume in one second divided by forced vital capacity, FEV1/FVC, below 0.7, and FEV1 <80% of the predicted value (% predicted)11 using local reference equations; that is, corresponding to global initiative of obstructive lung disease grades two to four.1 Only prebronchodilator spirometry was performed. We excluded individuals with self-reported asthma,12 and individuals below 40 years of age. This way, we identified a prospective cohort of 5262 individuals with COPD. Of these, 5219 (99.2%) had successful genotyping of rs1042713 (Gly16Arg, c.46G>A) and 5160 (98.1%) had successful genotyping of rs1042714 (Gln27Glu, c.79C>G).
Copenhagen City Heart Study
For replication analyses we used the Copenhagen City Heart Study (CCHS), another prospective cohort study. All individuals with data on lung function and genotyping performed in the third examination (examined 1991–1994), and individuals with lung function and genotyping in the fourth examination13 (examined 2001–2003) who had not previously participated in the third examination, were included. We identified a total of 923 individuals with COPD defined as described above. Of these, 880 (95.3 %) had successful genotyping of Gly16Arg and 879 (95.2 %) had successful genotyping of Gln27Glu.
β2-Adrenergic receptor genotypes
Gly16Arg (rs1042713) and Gln27Glu (rs1042714) polymorphisms were in Hardy-Weinberg equilibrium in the total CGPS (p=0.57 and p=0.40, respectively), as well as in individuals with COPD (p=0.54 and p=0.54). For genotyping in the CGPS, a TaqMan-based assay was used (Applied Biosystems, Foster City, California, USA, details available from authors). Similarly, we observed Hardy-Weinberg equilibrium in the total CCHS (p=0.13 and p=0.10) and in individuals with COPD (p=0.94 and p=0.17). A Nanogen NMW 1000 Nanochip Molecular Biology Workstation (Nanogen, San Diego, California, USA) was used to genotype the two polymorphisms in this cohort.
Severe exacerbations
Severe exacerbations in the CGPS and the CCHS were recorded as acute admissions with a discharge diagnosis of COPD in the national Danish Patient Registry14 (International Classification of Diseases ICD-8 490–492, and ICD-10 J41-44; ICD-9 was never used in Denmark). We used a maximum of 5 years of follow-up (mean: 3.4 years) in the CGPS. In the smaller CCHS, a longer follow-up period was available due to the earlier examination dates, and to increase statistical power we used a maximum of 20 years of follow-up (mean: 9.2 years).
Study approval
The study was approved by Herlev and Gentofte Hospital and a regional ethics committee (H-KF01-144/01) and was conducted according to the Declaration of Helsinki. Written informed consent was obtained from all individuals.
Role of the funding sources
The funding sources had no role in the design and conduct of the study; in the collection, management, analysis and interpretation of the data; or in the preparation, review, or approval of the manuscript.
Statistical analyses
All analyses were done using the statistical software package R.15 Baseline characteristics were analysed by χ2 tests or one-way analysis of variance (ANOVA) as appropriate.
The influence of Gly16Arg and Gln27Glu polymorphisms on future risk of severe exacerbations in the CGPS and the CCHS were studied by cumulative incidence curves and univariable competing risks regression analyses of time to first exacerbation. We used COPD mortality recorded in the national Danish Register of Causes of Death16 as competing risk. Censoring was death (CGPS: No.=362; CCHS: No.=480), emigration (CGPS: No.=2; CCHS: No.=1), or end of follow-up (April 2013, both cohorts). Competing risks regression was applied using the ‘comp.risk’ function of the ‘timereg’ package in R using a proportional model (model=‘prop’) with censoring weights calculated by a Cox model (cens.model=‘cox’).17 Furthermore, Kolmogorov-Smirnov and Cramer von Mises tests and visual examination of observed and simulated test processes under the null were applied to test for possible time-varying effects.
Sensitivity analyses
Although genotypes are expected to be largely unconfounded,18 we did a first sensitivity analysis adjusting for lung function as FEV1% predicted. Furthermore, a recent study has indicated a differential genotype treatment effect of Gly16Arg among COPD patients using long-acting β2-agonists. Therefore, as data on drug utilisation were available from 1995 and forward in the national Danish Prescription Registry,19 in our second sensitivity analysis we tested for possible interactions between genotypes and use of long-acting β2-agonists in 1 year prior to the baseline examination. In our third sensitivity analyses, we excluded all individuals with use of any inhaled medicines (short-acting β2-agonists, long-acting β2-agonist with or without inhaled corticosteroids, or long-acting anticholinergics) in 1 year prior to the baseline examination, to minimise any possible confounding from medication use. Finally, we adjusted our models for baseline characteristics of significant associations with the single nucleotide polymorphisms studied.
Subgroup analyses
Gly16Arg and Gln27Glu polymorphisms are closely located in the ADRB2 gene on chromosome 5. Previous studies have shown that these two polymorphisms are in linkage disequilibrium20 21; that is, their mutual genotypes are inherited in a non-random way. This implies that in a single nucleotide polymorphism analysis, the observed influence of the 16Arg allele in rs1042713 could be affected by a 16Gly homozygote reference group with presence of a non-random combination of 27Gln and 27Glu in rs1042714. Similarly, the observed influence of the 27Gln allele in rs1042714 could be affected by a 27Glu homozygote reference group with a non-random distribution of 16Arg and 16Gly in rs1042713.22 We therefore studied the influence of the 16Arg allele among 27Gln homozygotes only (ie, no presence of 27Glu), and the influence of the 27Gln allele among 16Gly homozygotes only (ie, no presence of 16Arg). We used a competing risks regression model adjusted for FEV1% predicted, as described above.
Results
Characteristics
Table 1 shows baseline characteristics of the two cohorts, the 5262 individuals with COPD in the CGPS and the 923 individuals with COPD in the CCHS; the latter consisting of slightly more males and more current smokers. Online supplementary tables S1-S4 shows detailed characteristics of all individuals in the CGPS and the CCHS by genotypes, as well as detailed characteristics of individuals with COPD by genotypes in the two cohorts.
Supplemental material
Characteristics of individuals with chronic obstructive pulmonary disease (COPD)
Genotypes seemed largely unconfounded in both COPD cohorts. However, in the total CGPS cohort, presence of the 16Arg allele was statistically associated with a clinically very small influence on FEV1% predicted (−0.3% for 16Gly/Arg heterozygotes and −0.6% for 16Arg homozygotes, online supplementary table S1). Furthermore, among individuals with COPD in the CGPS a 16Arg allele influence on gender reached statistical significance (48.5% males for 16Gly homozygotes, 47.7% males for 16Gly/Arg heterozygotes and 52.9% males for 16Arg homozygotes, table 1 and online supplementary table S2).
During follow-up among individuals with COPD in the CGPS we recorded 461 subjects with severe exacerbations. During follow-up in the CCHS we recorded 310 subjects with severe exacerbations.
β2-Adrenergic receptor genotypes and risk of severe exacerbations
In the CGPS, the HRs for severe exacerbations were 1.62 (95% CI 1.30 to 2.03, p=0.00002) for 2488 16Gly/Arg heterozygotes and 1.41 (1.04 to 1.91, p=0.03) for 753 16Arg homozygotes, compared with 1978 16Gly homozygotes (figure 1, upper left part). Corresponding HRs in the CCHS with lower statistical power were 1.24 (0.95 to 1.62, p=0.11) and 1.02 (0.67 to 1.55, p=0.92) (figure 1, upper right part).

Influence of β2-adrenergic receptor Gly16Arg (rs1042713) or Gln27Glu (rs1042714) genotypes on risk of severe exacerbations in individuals with chronic obstructive pulmonary disease (COPD) by cumulative incidence curves obtained from unadjusted competing risks regression analyses based on 5262 individuals with COPD in the Copenhagen General Population Study (left panels) and 923 individuals with COPD in the Copenhagen City Heart Study (right panels). Upper panels represent Gly16Arg (16Gly as reference group) and lower panels represent Gln27Glu (27Glu as reference group). Numbers at risk are shown below panels.
In the CGPS, the HRs for severe exacerbations were 1.35 (1.03 to 1.76, p=0.03) for 2576 27Gln/Glu heterozygotes and 1.49 (1.12 to 1.98, p=0.006) for 1553 27Gln homozygotes compared with 1031 27Glu homozygotes (figure 1, lower left part). Corresponding HRs in the CCHS were 1.73 (1.20 to 2.49, p=0.003) and 1.45 (0.97 to 2.17, p=0.07) (figure 1, lower right part).
No significant associations were observed between 16Gly/Arg or 27Gln/Glu and death from COPD, but the CIs were wide (online supplementary table S5). There were no indications of time-varying effects (p>0.10 for both genotypes and all covariates in Kolmogorov-Smirnov and Cramer von Mises tests).
Sensitivity analyses
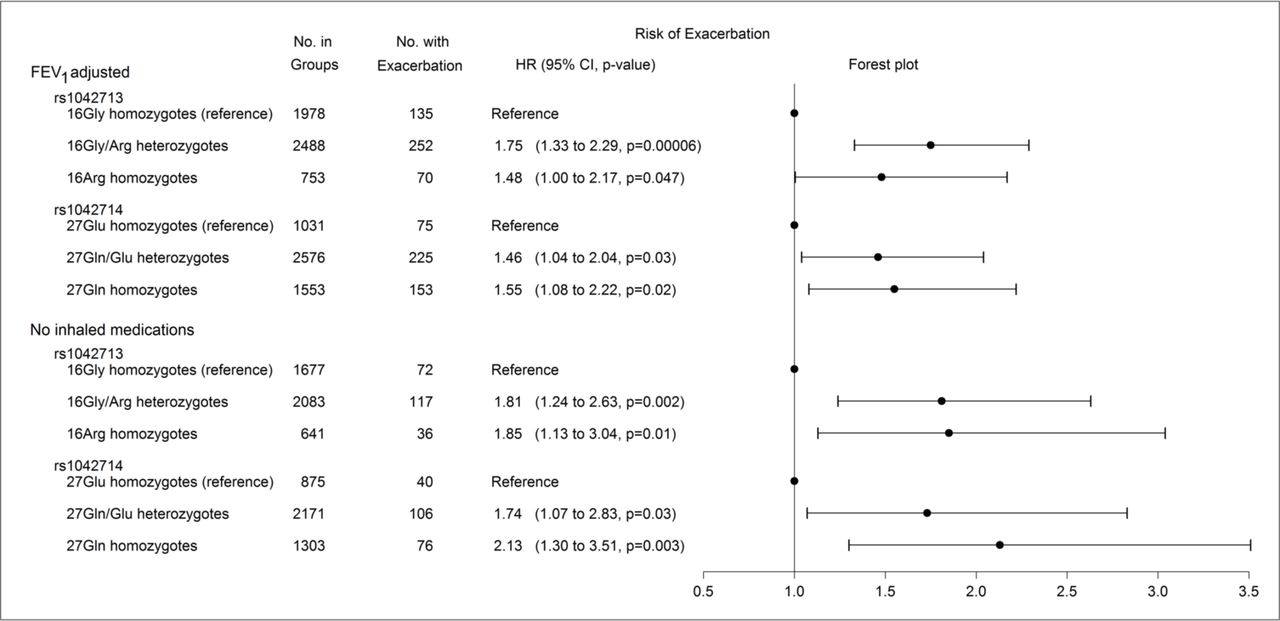
All estimates were robust and significant after adjustment for FEV1% predicted, as shown in the upper part of figure 2. No significant interactions with baseline use of long-acting β2-agonists were observed (p>0.40 for both single nucleotide polymorphisms). Furthermore, exclusion of all users of inhaled medications in 1 year prior to the baseline examination gave consistent and significant estimates, as shown in the lower part of figure 2. Adjustment for gender in the Gly16Arg model only changed results slightly, and the HRs were 1.76 (1.18 to 2.63, p=0.005) for 16Gly/Arg heterozygotes and 2.00 (1.22 to 3.30, p=0.006) for 16Arg homozygotes compared with 16Gly homozygotes.

Influence of Gly16Arg (rs1042713) or Gln27Glu (rs1042714) on risk of severe exacerbations in the Copenhagen General Population Study. Upper part shows a competing risks regression model adjusted for forced expiratory volume in one second in percentage of predicted value. Lower part shows an unadjusted competing risks regression model but excluding users of inhaled medicines in 1 year prior to the baseline examination. From left to right: number of individuals with corresponding genotype (‘No. in groups’), number of individuals with severe exacerbations during follow-up (‘No. with exacerbation’), HRs with 95% CIs and p values (‘HR (95% CI, p value)’), and a corresponding forest plot (‘forest plot’).
Subgroup analyses
Table 2 shows distribution of mutual Gly16Arg and Gln27Glu genotypes in the CGPS. Only four, one and zero individuals carried the genotypes Gly16Arg-Glu27Glu, Arg16Arg-Gln27Glu and Arg16Arg-Glu27Glu, respectively, reflecting linkage disequilibrium.21 Similarly, online supplementary table S6 shows genotype distribution in the full CGPS cohort.
Distribution of Gly16Arg (rs1042713) and Gln27Glu (rs1042714) genotypes among 5153 individuals with Chronic Obstructive Pulmonary Disease (COPD) in the Copenhagen General Population Study and successful genotyping of both single nucleotide polymorphisms
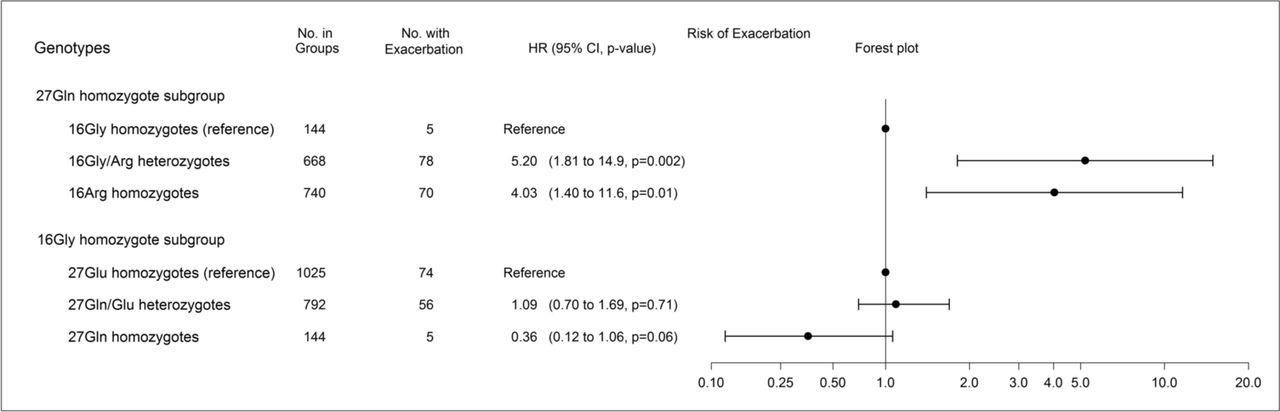
Among 27Gln homozygotes only, the HRs for severe exacerbations were 5.20 (1.81 to 14.9, p=0.002) for 668 16Gly/Arg heterozygotes and 4.03 (1.40 to 11.6, p=0.01) for 740 16Arg homozygotes, compared with 144 16Gly homozygotes as shown in the upper part of figure 3. Surprisingly, among 16Gly homozygotes only, the Gln27Glu genotype did not significantly influence risk of exacerbations and HRs were 1.09 (0.70 to 1.69, p=0.71) for 792 27Gln/Glu heterozygotes and 0.36 (0.12 to 1.06, p=0.06) for 144 27Gln homozygotes compared with 1025 27Glu homozygotes as shown in the lower part of figure 3.
{kind=link}
{kind=link}
{kind=link}
Upper part shows influence of Gly16Arg (rs1042713) among 27Gln (rs1042714) homozygotes only, and lower part shows influence of Gln27Glu among 16Gly homozygotes only. Estimates obtained from competing risks regression analyses adjusted for forced expiratory volume in one second in percentage of predicted value. From left to right: subgroups (‘genotypes’), number of individuals with corresponding genotype (‘No. in groups’), number of individuals with severe exacerbations during follow-up (‘No. with exacerbation’), HRs with 95% CIs and p values (‘HR (95% CI, p value)’), and a corresponding forest plot (‘forest plot’). The x-axis in the forest plot has been log10 transformed.
Discussion
Among subjects with COPD identified from a general population sample, we found that common β2-adrenergic receptor genotypes influence risk of severe exacerbations in COPD, potentially mainly by genetic influence of the 16Arg allele in rs1042713.
Previous studies have shown that Gly16Arg and Gln27Glu polymorphisms do not influence airflow limitation in itself.23 Our study shows that although these genotypes may not be involved in susceptibility to developing airflow limitation, they seem important to a prominent feature of the disease, namely exacerbations. The 16Arg allele in rs1042713 is common and is carried by more than 60% of individuals with COPD in our study. Therefore, our observations could be important to prognosis for many patients with COPD and could perhaps play a small part in future genotype based precision medicine, although most of the potential genetic contribution to risk of severe exacerbations remains to be defined.
Green et al studied Chinese hamster fibroblasts more than 20 years ago and showed that following a 24 hours agonist exposure with isoprotenerol, the 16Arg allele caused β2-adrenergic receptor down-regulation compared with 16Gly homozygote wildtype carriers.24 During acute COPD exacerbations patients are often treated with high doses of as-needed short-acting β2-agonists. This treatment could cause acute receptor down-regulation in individuals carrying the 16Arg allele, thereby increasing resistance to acute as-needed treatment and susceptibility to severe exacerbations requiring hospital admission. This mechanism could be part of the explanation for our findings suggesting 16Arg as a risk allele of COPD exacerbations requiring hospital admission in individuals with COPD who were not receiving treatment with maintenance medication.
In recent post-hoc analyses from a randomised controlled trial, it was shown that the Gly16Arg polymorphism caused a differential effect on treatment response to long-acting β2-agonists. In that study, the 16Arg allele was associated with reduced risk of exacerbations compared with 16Gly homozygotes in users of long-acting β2-agonists with or without inhaled corticosteroids.4 A possible explanation for this apparent discrepancy to our findings is that whereas all patients are intensely treated in an efficacy trial, individuals with COPD in the present cohorts are mostly undiagnosed at the baseline examination and undertreated with maintenance medicines both before and after the examination.25 Furthermore, the above mentioned randomised controlled trial was not designed to study single nucleotide or genotype effects per se. Importantly, in our study the observed influence of β2-adrenergic receptor genotypes per se on risk of exacerbations was robust towards exclusion of all users of inhaled medicines, and no interactions with baseline use of long-acting β2-agonists were observed.
The large size of the CGPS is an important strength in this genetic study, providing us with sufficient power to perform subgroup genotype analyses accounting for linkage disequilibrium.26 Each individual carries a non-random β2-adrenergic receptor genotype combination influencing individual susceptibility to exacerbations. To assess this, we did a subgroup analysis among 27Gln homozygotes only and observed that 16Gly/Arg heterozygotes and 16Arg homozygotes had a similar very high risk of exacerbations compared with 16Gly homozygotes. We speculate that this observation could indicate that the genetic influence on severe exacerbations is mainly due to a dominant genetic effect of the 16Arg allele. Importantly, though, interpretations of these subgroup genotype analyses should be considered in the light of the small number of events recorded in these analyses. For the 27Gln homozygote subgroup analyses we only recorded five exacerbations in the 16Gly homozygote reference group which increase the likelihood of both false positive and false negative findings. On the other hand, this reference group was rather large comprising 144 individuals, and the observed effects size of the 16Arg allele was very large and highly significant and could be clinically important. It could be argued that a possible limitation to our study is that haplotype analyses were not performed instead of subgroup genotype analyses. However, if the true causal connexion between the two β2-receptor polymorphisms studied is truly driven by just 16Arg then the haplotype-based approach may actually perform worse than the approach reported in the present study.27
In single-polymorphism analyses, a significant increased risk of exacerbations for the 27Gln allele in rs1042714 was observed in the CGPS, and this influence was also significant in replication analysis in the CCHS. To our knowledge, this is the first candidate gene study showing replicated influence of a single nucleotide polymorphism in the β2-adrenergic receptor locus on future risk of COPD exacerbations. However, because of the large size of the CGPS we were able to perform novel subgroup genotype analysis in 16Gly homozygotes only. Surprisingly, this analysis did not support our initial observation but provided interesting new information. In presence of linkage disequilibrium, the single nucleotide polymorphism analysis did not consider that among 27Gln homozygotes 91% were carriers of the 16Arg allele. Similarly, among 27Gln/Glu heterozygotes 69% were carriers of 16Arg (table 2). These individuals were compared with 27Glu homozygotes as reference group where only one-half per cent were carriers of 16Arg. Thus, the presence of 16Arg in most 27Gln homozygotes and 27Gln/Glu heterozygotes could cause the impression of 27Gln as a risk allele. However, subgroup genotype analyses were suggestive of 27Gln/Gln as a protective genotype of severe exacerbations, although the estimate did not reach statistical significance.
Trends for the Gly16Arg polymorphism were similar in both COPD cohorts, although failing to reach statistical significance in the smaller CCHS. We speculate that the complex distribution of the 27Gln allele in 16Gly homozygotes and 16Gly/Arg heterozygotes could potentially lower observations of true effect sizes and dilute observed statistical influence in a smaller COPD cohort. In genetic studies, the outcome definition is particularly important. Although a much larger replication cohort would have been preferred, we consider inclusion of a replication cohort with exacerbation outcome measured identically an important strength to our study. A possible limitation to our study, though, was that the smoking prevalence was different in the two cohorts with more current smokers in the CCHS. This could potentially explain some of the observed difference in effect size for 16Gly/Arg between the two cohorts. Although baseline demographics did not indicate an association between single-nucleotide polymorphisms and smoking, we did two post-hoc sensitivity analyses in both cohorts testing for a possible interaction with smoking on exacerbation risk and we adjusted our main analyses for smoking. There was no evidence of interactions and our main results remained similar after smoking adjustment (online supplementary table S5).
Further strengths to our study include the complete national discharge diagnoses registry data on severe exacerbations known for a high validity with a positive predictive value of approximately 92%.14 Nevertheless, results from a previous study in the CGPS indicate that most exacerbations are treated with oral corticosteroids with or without antibiotics without admission to hospital, indicating that the negative predictive value of our severe exacerbations is likely rather low.28 Strengths to our study also include important clinical baseline characteristics of COPD, supporting the notion of largely unconfounded genotypes in our COPD cohorts, including the two most important predictors of severe COPD exacerbations, FEV1 and previous exacerbations. A possible limitation, though, was that we observed a difference in FEV1% predicted across Gly16Arg in the total CGPS cohort. However, the clinical difference was very small and all estimates for the 16Arg allele remained similar and significant after adjustment for FEV1% predicted. β2-adrenergic receptors are well represented in the airway smooth muscle,26 and we therefore speculate that the 16Arg allele could cause a slight difference in smooth muscle tone which would increase susceptibility to exacerbations in COPD, thereby explaining our findings. Similarly, a possible limitation was the small difference in gender distribution across genotype in the COPD population of The CGPS, but results were not weakened when adjusting our analyses for gender.
Furthermore, in our genetic studies potential confounders typically will not influence the results as genotypes are distributed at random during meiosis irrespective of the lifestyle adapted by the person later in life. Nevertheless, although the p values for genetic influence on potential confounders in our study suggest there are no significant differences in other variables such as age, infections and inhalants between genotype groups, slight differences could still influence risk of exacerbations, which we naturally cannot totally exclude, particularly in the case of a small sample size, and residual confounding could still be present.
Further strengths include the Hardy-Weinberg equilibrium assessment, and because of the almost exclusive Danish ethnicity in our cohorts, population stratification bias is likely not present. However, inclusion of individuals with only Danish descent could be a limitation to generalisability of our results.
Another possible limitation to our study is exclusion of asthma by self-report. Although this method is well acknowledged in epidemiological studies, we cannot rule out that some individuals with asthma could have been included. A study has shown that in 1182 steroid treated young asthma patients who were frequent users of β2-agonists (albuterol or salmeterol), the 16Arg allele increased risk of asthma exacerbations.29 In that study, this influence of 16Arg was not observed in those individuals who were not exposed to β2-agonists, and a recent meta-analysis in children with asthma supports the hypothesis that the effect of the 16Arg allele on risk of asthma exacerbations could be dependent on medication use.30 However, possible inclusion of individuals with asthma seems less likely to explain our findings of a genetic influence of the 16Arg allele per se on risk of severe exacerbations in COPD.
Further limitations include analyses of only two adrenoreceptor polymorphisms. A previous study found a significant influence of 164Thr/IIe (rs1800888) on pulmonary function measures.23 Therefore, we studied this rare variant in the large CGPS but did not see any effect on prospective risk of severe COPD exacerbations (online supplementary table S5). Furthermore, although our candidate gene approach has biological plausibility since these missense mutations have functional supportive evidence, non-coding variation could be important, and future comprehensive assessment of variation in the ADRB2 locus and multi-allele risk scores are necessary.
The CGPS is an ongoing study, currently collecting data in the second examination initiated in 2014. These data could be used in a future study using even longer follow-up periods and further increasing statistical power using repeated measurement analyses. Importantly, though, findings such as ours based on epidemiological cohort studies should be replicated in other, independent and preferably even larger study samples. Furthermore, conclusions should also be based on data from randomised controlled studies including patients randomised to receive inhaled COPD maintenance treatment. However, we suggest that such studies should be based on an a priori hypothesis including stratification by genotype subgroup, that is, 27Gln homozygotes and 16Gly homozygotes, and not post-hoc analyses. Such randomised controlled trials could assess whether the observed increased risk of severe exacerbations associated with 16Arg and the suggested protective effect of 27Gln are similar in patients receiving regular COPD treatment.
In conclusion, among subjects with COPD identified from a general population sample, we found that common β2-adrenergic receptor genotypes influence risk of severe exacerbations in COPD, potentially mainly due to genetic influence of the 16Arg allele in rs1042713. These observations call for future studies with inclusion of β2-adrenergic receptor genotypes as potential biomarkers of prognosis and studies of pharmacogenetics in COPD.
References
Footnotes
Contributors Study concept and design: TSI, JV, BGN, LR; acquisition of data: PL., BGN; analysis and interpretation of data: TSI, JLM, JV, BGN; critical revision of the manuscript: all authors; statistical analysis: TSI, JLM; obtained funding: PL, JV, BGN; study supervision: TSI, JV, BGN. All authors had full access to all the data in the study. TSI, BGN, and JLM take responsibility for the integrity of the data and the accuracy of the data analysis, and for the submission.
Funding Supported by the Capital Region of Copenhagen, the Danish Heart Foundation, the Danish Lung Foundation, the Velux Foundation, and Herlev and Gentofte Hospital. JV is supported by the NIHR Manchester Biomedical Research Centre.
Competing interests TSI has received a fee for speaking from AstraZeneca. JV has received honoraria from Chiesi, GlaxoSmithKline, Almirall, AstraZeneca, Boehringer-Ingelheim, and Novartis for consulting and for presenting at meetings and symposia, none of it related to the topic of this study. LR and JLM: none to declare. PL has received honoraria from AstraZeneca, Boehringer Ingelheim, Chiesi, Novartis, Teva, and GlaxoSmithKline for presenting at meetings and symposia, none of it related to the topic of this study. BGN: none to declare.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement No data are available.