Article Text

Abstract
Background Oncolytic viruses (OVs) have shown promise in containing cancer progression in both animal models and clinical trials. How to further improve the efficacy of OVs are intensively explored. Arming OVs with immunoregulatory molecules has emerged as an important means to enhance their oncolytic activities majorly based on the mechanism of reverting the immunosuppressive nature of tumor environment. In this study, we aimed to identify the optimal combination of different OVs and immunomodulatory molecules for solid tumor treatment as well as the underlying mechanism, and subsequently evaluated its potential synergy with other immunotherapies.
Methods Panels of oncolytic viruses and cells stably expressing immunoregulatory molecules were separately evaluated for treating solid tumors in mouse model. A tumor-targeted replicating vaccinia virus Tian Tan strain with deletion of TK gene (TTVΔTK) was armed rationally with IL-21 to create rTTVΔTK-IL21 through recombination. CAR-T cells and iNKT cells were generated from human peripheral blood mononuclear cells. The impact of rTTVΔTK-IL21 on tumor-infiltrating lymphocytes was assessed by flow cytometry, and its therapeutic efficacy as monotherapy or in combination with CAR-T and iNKT therapy was assessed in mouse tumor models.
Results IL-21 and TTV was respectively identified as most potent immunomodulatory molecule and oncolytic virus for solid tumor suppression in mouse models. A novel recombinant oncolytic virus that resulted from their combination, namely rTTVΔTK-mIL21, led to significant tumor regression in mice, even for noninjected distant tumor. Mechanistically, rTTV∆TK-mIL21 induced a selective enrichment of immune effector cells over Treg cells and engage a systemic response of therapeutic effect. Moreover, its human form showed a notable synergy with CAR-T or iNKT therapy for tumor treatment when coupled in humanized mice.
Conclusion With a strong potency of shaping tumor microenvironment toward favoring TIL activities, rTTVΔTK-IL21 represents a new opportunity worthy of further exploration in clinical settings for solid tumor control, particularly in combinatorial strategies with other immunotherapies.
One sentence summary IL21-armed recombinant oncolytic vaccinia virus has potent anti-tumor activities as monotherapy and in combination with other immunotherapies.
- oncolytic virotherapy
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information. Data sharing not applicable as no datasets generated and/or analyzed for this study. All data are published in this paper. Materials are available on reasonable request based on an appropriate MTA.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immunotherapy has recently emerged as a promising approach to treat tumor by using the power of the immune system.1 From the viewpoint of immunotherapy, tumors can be classified into two categories, the “hot” tumor where an inflamed microenvironment created by tumor cells and the stromal cells favors infiltration of lymphoid cells, and the “cold” tumor in which a hostile microenvironment rejects immune cells or renders them less functional or non-functional after recruitment.2 Thus, how to reverse the “cold” tumors into “hot” tumors becomes one of the most active fields in immunotherapy research and oncolytic virotherapy stands out as a major promising approach to serve this purpose by multiple mechanisms.3 The basic principle of this approach is taking advantage of defect of tumor cells in antiviral innate immunity, allowing virus preferentially replicate in and consequently kill the tumor cells. The tumor cells are not simply killed, but rather undergo immunogenic cell death, in which tumor-associated antigens and danger factors are released to engage both innate and adaptive arms of immune system for further destruction of tumor cells.4 The reshaping of the tumor microenvironment by oncolytic virus can be additionally facilitated by engineering the virus to express one or more immunomodulatory genes.5
To date, a wide range of oncolytic viruses (OVs) have been under active investigation, including both DNA and RNA viruses of different sizes and properties.6 Talimogene laherparepvec, also known as T-VEC, is the first oncolytic virus therapy approved by FDA for advanced metastatic melanoma,7 opening the door for the use of other OVs in cancer treatment. Generally, OVs are molecularly engineered in two ways to improve safety and efficacy. The first way is to ensure selective replication in tumor cells.8 One commonly used strategy involves deletion of non-essential viral genes, which function can be compensated by tumor cells but not normal cells. For instance, in the case of oncolytic vaccinia virus, deletion of virus-encoded thymidine kinase (TK), a protein critical for viral DNA synthesis, prevents effective replication in normal cells where TK levels are typically expressed at low level.9 The second way is to augment anti-tumor response by expressing therapeutic genes that either promote the cytotoxicity of tumor cells or enhance immune responses.10 The latter one has been extensively pursued with a main focus on immunomodulatory genes represented by cytokines to regulate the recruitment of T cells and their homeostasis. Indeed, T-VEC uses granulocyte-macrophage colony-stimulating factor (GM-CSF), a cytokine with known role in recruiting antigen-presenting cells like dendritic cells (DCs) as well as their maturation. Several other cytokines, including interlukin-2 (IL-2), IL-12, IL-7 and TNF, have been also employed for incorporation into viruses, which show improved therapeutic activity in mouse tumor models primarily owing to enhanced expansion of tumor-infiltrating lymphoid cells.5 In our systemic search for a potent cytokine that can arm oncolytic vaccinia virus to maximize their therapeutic efficacy, we had been considering IL-21 as an important candidate. As a member of the common gamma-chain (γc) family of cytokines, IL-21 is mainly secreted by activated CD4+ T cells and natural killer T (NKT) cells,11 and plays a pleiotropic role in regulating multiple types of immune cells, including B cells, T cells, natural killer (NK) cells and monocyte-derived DCs.12 Accordingly, IL-21 has been shown to possess tumor suppressor activity and, in a B16 melanoma model, its local administration conferred more potent anti-tumor efficacy than subcutaneous injection, leading to prolonged survival in treated mice.13 Moreover, a recent study reported the construction of an oncolytic adenovirus co-expressing IL-21 and chemokine CCL21 and its introduction into tumor cells is able to direct selective lysis by cytotoxic T cells (CTLs) in vitro.14 However, whether IL-21 alone is sufficient for CTL recruitment, and more importantly, whether IL-21 arming enhances the in vivo antitumor function of oncolytic virus, remains unknown.
A growing evidence supported that monotherapy is normally not sufficient to contain tumor progression and combinational strategies usually gain better performance.15 It has been envisioned that oncolytic virotherapy can be combined with other immunotherapy such as immune checkpoint blockade (ICB) and adoptive T-cell transfer therapy (ACT) to gain additive or even synergistic anti-tumor effect.16 Indeed, a number of recent studies reported a successful combination of oncolytic virotherapy and ICB in cancer treatment, with the former responsible for the initial recruitment of the immune cells while the later unleash the power of the recruited immune cells that otherwise would be suppressed by the original tumor microenvironment.17 18 In contrast, there are fewer studies coupling oncolytic virotherapy with ACT. Centered on T cells genetically modified to express a chimeric antigen receptor (CAR-T), ACT has shown promising results in the treatment of hematopoietic tumor.19 However, CAR-T approach has found much less success in treating solid tumors, largely attributed to the insufficient migration of CAR T-cells to the tumor site and the immune suppressive environment within tumor, which limits the persistence and function of tumor-infiltrating CAR T-cells.20 Theoretically, OVs and CAR-T approaches are complimentary to each other as OVs can facilitate the infiltration and anti-tumor functioning of CAR T-cells by priming the tumor microenvironment to attain a T cell–favorable state through the generation of inflammatory cytokine and chemokine, concomitant with the removal of immune suppression.21 Therefore, OVs–CAR-T combined strategies merit further investigation.
In this study, we initiated our request for a potent oncolytic virus by identifying the optimal virus type and the right immunomodulatory molecule to arm the virus for sculpturing the tumor microenvironment to promote immune response. This led to the construction of rTTVΔTK-IL-21 (rTTVΔTK-IL21) viruses, which were derived from vaccinia virus Tian Tan strain (TTV) by replacing its TK gene with either mice or human IL-21 gene. We first showed in three mouse xenograft tumor models that rTTVΔTK-IL21 was endowed with an enhanced capability to promote local enrichment of immune effector cells at tumor site, thereby being able to attain long-lasting tumor rejection and improved survival. Following this, we demonstrated an abscopal effect of rTTVΔTK-IL21, that is, suppressing tumor deposited distantly for the injection site likely through engagement of the systemic immune response. Finally, we explored a combination of virotherapy and immunotherapy in treating human tumors by coupling rTTVΔTK-IL21 with ACT approach in a sequential regimen. Using humanized mouse tumor model, we demonstrate a proof of principle that rTTVΔTK-hIL21 can synergize with CAR-T therapy or invariant nature killer cell (iNKT) therapy to deliver more effective cancer treatment. Together, our study identified IL21-armed TTV as a novel potent oncolytic virus and support its potential in partnering with ACT to surmount solid tumor challenge.
Materials and methods
Cell lines
The information of cell lines used in this study, including source and maintenance, is detailed in online supplemental information. The engineering of cell lines to express foreign genes, including mGM-CSF, m4-1BBL, mCXCL9, mCD86, mIL-21 or hCD19, was achieved by lentiviral vectors. The construction of lentiviral vectors, generation of lentiviral particles for infection, selection of stable clones and assay for cell growth were described in online supplemental information.
Supplemental material
Oncolytic viruses
The TTV752-1 was from our laboratory deposit as previously described.22 Generation and amplification of rTTV∆TK-mIL21, rTTV∆TK-hIL21 viruses, and following determination of their replication and cytotoxic effect in cultured tumor cells are described in online supplemental information. The information of other viruses used in this study is also provided in online supplemental information.
Generation of CAR-T cells
Human peripheral blood mononuclear cells (PBMCs), obtained from Shanghai Public Health Clinical Center, were sorted into CD4+ and CD8+ T cells by EasySep Human CD4+ T Cell Enrichment Kit (STEMCELL #19052) and EasySep Human CD8+ T Cell Enrichment Kit (STEMCELL #19053), respectively. After activation by anti-CD3/CD28 immunobeads in T-cell growth medium (TCM) for 24–36 hours, the purified T cells were transduced by lentiviruses in a novonectin (Novoprotein)-coated 48-well flat plate by spin infection. The next day, the cells were fed with fresh TCM. The immunobeads were removed 6–7 days after activation, and cells were expanded followed by 3-hour rest before being assayed. iNKT cell expansion iNKT cells were expanded from human PBMCs in X-VIVO-15 Medium (Lonza) containing 100 ng/mL KRN7000 (Enzo Life Science), 100 U/mL rhIL-2 (R & D Systems), 20 ng/mL rhIL-7 (R & D Systems) and 20 ng/mL rhIL-15 (R & D Systems) for 20 days as previously described.23 24
Xenograft animal models
All the mice were female mice 6–8 weeks old, with C57BL/6 mice, BALB/c mice and B-NDG mice purchased respectively from B&K Universal (Shanghai, China) and Biocytogen (Beijing, China). The mice were housed under specific pathogen-free conditions at the animal facilities of Shanghai Public Health Clinical Center, Fudan University (Shanghai, China) until virotherapy, and afterwards transferred to the Animal Biosafety Level 2 Laboratory. The volume of virus injected in was 100 µL. rTTV∆TK-IL21 monotherapy was conducted on C57BL/6 or BALB/c mice with tumors formed by subcutaneous injection of B16 (1×105), GL261 (1×106) or CT26 (1×106) cells into the right or left flank of experimental subjects. The combination strategy was evaluated in B-NDG mice bearing right flank located tumors formed by subcutaneous injection of NCI-H292 (2×106) cells. For oncolytic virotherapy, mice were intratumorally inoculated with PBS or rTTV viruses 10 days when the tumor volumes normally reached 50–100 mm3 after they were engrafted with tumor cells. Tumors were measured two to three times a week with calipers until the experimental endpoint (animal death or a tumor volume ≥2000 mm3, tumor volume=length×width2/2).
Immune cell depletion
One day prior to oncolytic virus treatment, 100 µg of rat anti-CD4 IgG (clone GK1.5), anti-CD8 IgG (clone TIB210), anti-NK IgG (clone PK136) or control IgG in 100 µL PBS was injected intraperitoneally. Antibody treatment was continued twice weekly throughout the experiment. Depletion was verified using flow cytometric assessment of splenocytes 24 hours after antibody administration.
Flow cytometry (FACS) analysis
The cell preparation and staining procedures, along with the list of used antibodies, are described in online supplemental information. All FACS analyses were performed by a BD LSRFortessa flow cytometer (BD Bioscience) and analyzed with FlowJo vX.0.7 (Tree Star).
Statistical analyses
All statistical analyses were performed using GraphPad Prism 6. Data are represented as the mean±SEM. Student’s t-test was used for non-paired comparisons of two groups. Difference between more than two groups were determined by one-way analysis of variance (ANOVA), and the Kaplan-Meier method with the log-rank test was used for survival analysis. A p value <0.05 was considered statistically significant.
Results
In vivo evaluation of therapeutic potential of virus vectors
To select the most powerful virus vector, we systematically assessed the oncolytic efficacy of a variety of oncolytic viruses in both cultured cells and a melanoma tumor model. The viruses we tested included wild-type replicative TTV serotype 752-1 (TTV752-1), influenza A virus PR8 strain (A/Puerto Rico/8/1934, H1N1), acute Armstrong lymphocytic choriomeningitis virus (LCMVARM), Zika virus strain MR766 (ZIKAMR) and adenovirus serotype 68 (AdC68). The amounts of viruses used for tumor injection were selected on basis of published reports as well as our own experience with virus challenge studies in mice and considered as sublethal doses.25–28 As a prelude to the in vivo study, we investigated the permissiveness of B16 cell culture to the tested viruses by infecting 1×106 cultured cells with one-tenth of viruses used for tumor injection, which was equivalent to 1×106 PFU, 2×104 PFU, 1×1010 VPs, 1×103 PFU and 5 TCID50 for TTV752-1, LCMVARM, AdC68, ZIKAMR and PR8, respectively. The post-infection medium of PR8-infected cells also contained 0.5 µg/mL TPCK-trypsin to facilitate virus replication. A substantial growth of TTV752-1, LCMVARM and AdC68 was observed between 12 and 36 hours post-infection, whereas ZIKAMR and PR8 only attained a moderate viral titer (figure 1A). The cytotoxicity analysis, on the other hand, showed that only TTV752-1 infection caused significant cytotoxicity (figure 1B). It has been reported previously that B16 cells were highly permissive to PR8 under higher multiplicity of infection. We consequently increased the infection dose of PR8 to 1×104 TCID50 and 1×105 TCID50, and found that, under these conditions, PR8 infection led to significant cytotoxicity along with effective viral growth (online supplemental figure S1A,B). Thus, although B16 cells are permissive to PR8, effective propagation of PR8 in these cells is influenced by the multiplicity of infection, which is likely attributed to the battle between virus and host interferon response.

Different oncolytic viruses exhibited different efficiency in virotherapy for B16 melanoma in mouse model. (A, B) Characterization of oncolytic virus candidates in cultured B16 cells. For infection, 1×106 cells were infected in 6-well format with TTV752-1 (1×106 PFU), LCMVARM (2×104 PFU), AdC68 (1×1010 VPs) or PR8 (5 TCID50). Supernatants or cells were collected at 12 hours, 24 hours and 36 hours after infection for viral titer determination (A); cytotoxicity was assessed at 72 hours after infection (B). Data are representative of two independent experiments. Error bars show the SEM of three biological replicates. (C) Treatment scheme. Mice were subcutaneously inoculated with 1×105 of B16 cells per mouse. When reaching about 50 mm3, tumors were directly injected with PBS (n=6), TTV752-1 (1×107 PFU, n=5), PR8 (50 TCID50, n=6), LCMVARM (2×105 PFU, n=5), ZIKAMR (1×104 PFU, n=6) or AdC68 (1×1011 VPs, n=6). (D) Kaplan-Meier survival curves of mice after virotherapy (*p<0.05, **p<0.01, log-rank test). (E) Responses of individual tumor to virotherapy in each treatment group.
For the assessment in vivo, mice were engrafted subcutaneously with B16 cells, followed by intratumoral injection of PBS (control) or different viruses (figure 1C). All mice in the control group exhibited rapid tumor progression and were dead or euthanized within 12 days after intratumoral injection. Among the virus-treated group, only LCMVARM and TTV752-1 group showed a growth rate of tumor notably lower than the control group. For the former, tumor was contained for approximately 12 days before regaining rapid growth. In contrast, for the majority of mice in the TTV752-1 group, tumor growth was controlled throughout the examination period, resulting in prolonged survival and reduced mortality (figure 1D–E). In addition, the TTV752-1 treatment showed a dose response in both time to progression and overall survival (online supplemental figure S2A-E). Compared with the highest virus dose (1×107 PFU), lower doses of TTV752-1 as of 1×105 or 1×106 PFU induced a reduced inhibition of tumor progression. Thus, we opted for TTV752-1 for developing new oncolytic virus in our study.
In vivo assessment of antitumor activities of immunoregulatory molecules
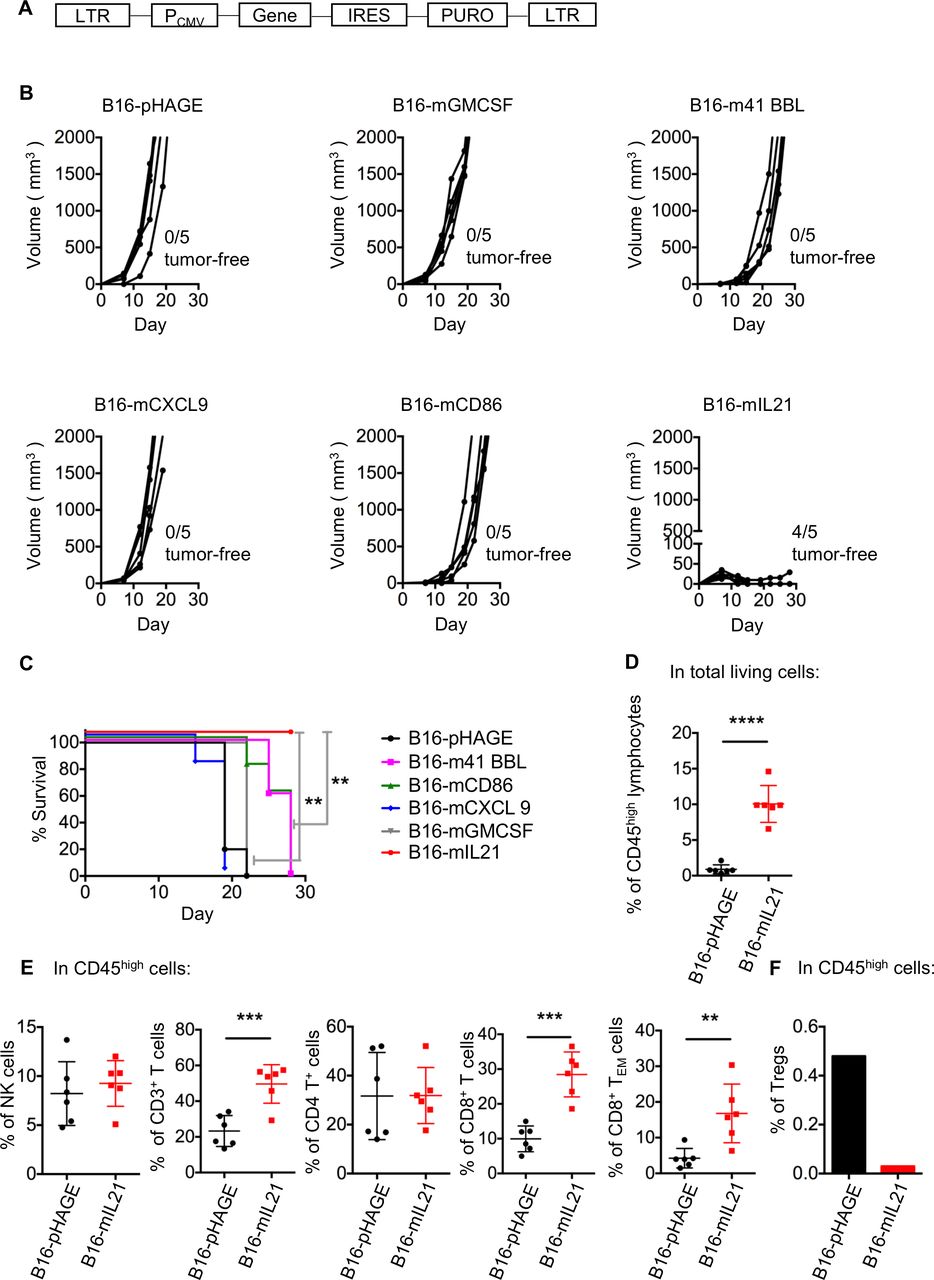
Potentiation of antitumor immune response would largely hinge on effective activation of antigen-specific T lymphocytes, a fine-tuned process best described by a three-signal model.29 To identify the most potent immunomodulatory molecule for integration into virotherapy, we tested five molecules with different roles in cellular immune response, including 4-1BB ligand (4-1BBL), CD86, C-X-C motif chemokine ligand 9 (CXCL9), IL-21 and GM-CSF, for their ability to limit tumor growth when expressed by tumor cells. Such ability should reflect their potency to engage the immune cells, especially T lymphocytes.30–34 To this end, we constructed a series of lentiviruses encoding individual immunomodulatory molecules, which were used for infection of B16 cell lines to establish stable cell lines (figure 2A). The identities of the resulting five cell lines were validated by the detection of surface protein expression by flow cytometry (online supplemental figure S3A). We also used the empty lentiviral vector to create a control cell line, namely, B16-pHAGE. The six stable clones were individually transplanted to syngeneic C57BL/6 mice, after which the tumor development was continuously monitored.

Local expression of IL-21 potently inhibited B16 melanoma growth in mice. (A) Schematic representation of the lentiviral vector used to create stable cell lines expressing individual immunomodulatory molecule. (B, C) Impacts of local expression of different immunomodulatory molecules on the B16 melanoma development in mice. 5×105 of the indicated cells were subcutaneously injected into the right flank of syngeneic mice, followed by measurement of tumor size (B) and survival curves (C) (n=5, **p<0.01, log-rank test). Data are representative of two independent experiments. (D–F) Effects of local expression of IL-21 on tumor-infiltrating lymphocytes (TILs). Mice were inoculated subcutaneously with 5×106 B16-pHAGE cells or B16-mIL21 cells. For the FACS analysis of TILs, B16-mIL21 tumors were assayed 21 days after inoculation while the B16-pHAGE tumors were assayed when end points were reached (animal death or tumor volume of ≥2000 mm3). (D) Frequency of CD45high lymphocytes. (E) Frequencies of CD3−NK1.1+ NK cells, CD3+ T lymphocytes, CD4+ T lymphocytes, CD8+ T lymphocytes, effector memory (CD44+CD62L−) T cells and (F) Tregs. Due to the scarcity of Tregs, for their measurements, the samples from mice receiving the same graft was pooled. n=6 per group; data are representative of two independent experiments; **p<0.01, ***p<0.001, Student’s t-test.
As compared with control B16-pHAGE group, cells expressing mIL-21 showed significantly compromised capability in driving tumor development with an 80% of rejection rate whereas there was only modest delay in tumor formation observed for both m4-1 BBL-expressing and mCD86-expressing cells (figure 2B). To our surprise, GM-CSF, which was widely applied in oncolytic virus therapy or tumor vaccines, was not observed to have an anti-tumor effect. There was also no discernible difference between CXCL9-bearing cells and control cells (figure 2B). In line with controlled tumor growth, the recipient mice of B16-mIL21 cells survived significantly longer than other treatment groups (figure 2C). In contrast, the in vitro growth rate of B16-mIL21 was similar to that of B16-pHAGE or the parental B16 cell line, demonstrating that mIL-21 expression did not, per se, influence the proliferation of B16 cells (online supplemental figure S3B). Collectively, these data revealed that, among the five signaling molecules tested, IL-21 stood out as the most promising candidate in the inhibition of in vivo tumor growth, potentially through enhancing anti-tumor immune response.
Next, we explore the impact of local IL-21 on tumor microenvironment. To this end, a total amount of 5×106 of B16-pHAGE or B16-mIL21 cells were subcutaneously injected into the right flank of mice. Due to the inhibitory effect of IL-21 on tumor growth, we allowed the mice receiving B16-mIL21 live for 21 days before tumor collection: the tumors were then normally large enough (50–100 mm3) to contain suitable amount of infiltrated immune cells (TILs) for FACS analysis. The B16-pHAGE tumors were collected at standard end points (animal death or tumor volume reaching 2000 mm3), thus earlier than 21 days. With gating strategy shown in online supplemental figure S3C, our FACS analysis revealed a significantly increase in the frequency of CD45high lymphocytes within B16-mIL21 tumors compared with B16-pHAGE tumors (figure 2D). Similar trends were observed for infiltrating CD3+, CD4+, CD8+ T cells as well as NK cells (CD3−NK1.1+ cells). Notably, the frequency of effector memory T cells (TEM) was also elevated in B16-mIL21 tumors relative to B16-pHAGE tumors (figure 2E). We also determined the population level of regulatory T cells (CD4+CD25+Foxp3+ Tregs). Due to the scarcity of Tregs, their measurements were taken on pooled sample from mice harboring the same tumor. Unlike active immune cells, Tregs were substantially downregulated in B16-mIL21–derived tumor compared with B16-pHAGE–derived tumor (figure 2F). Taken together, these results supported that IL-21–mediated inhibition of tumor growth may stem from its ability to reverse the suppressive nature of tumor microenvironment.
IL21-armed recombinant vaccinia virus induced enrichment of immune cells and led to long-lasting tumor regression in mouse xenograft models
We next generated a mouse IL-21 (mIL21)–armed rTTV using thymidine kinase–deleted version of TTV752-1, rTTV∆TK, as the backbone. Deleting TK gene is an effective strategy to enhance cancer targeting of vaccinia virus. We first examined the impact of IL-21 expression on viral replication and cytotoxicity in vitro. The result demonstrated that neither TK deletion nor the IL-21 insertion affected the virus-mediated cytotoxicity or the viral replication in cultured B16 cells (online supplemental figure S4A,B). Subsequently, we assessed the in vivo anti-tumor activity of rTTV∆TK-mIL21 in three different mouse tumor models. B16, GL261 or CT26 cells were subcutaneously injected into the right flank of mice respectively to establish orthotopic or heterotopic transplanted tumor models, followed by intratumoral injection of single dose of 107 PFU of rTTV∆TK or rTTV∆TK-mIL21. For the rTTV∆TK-mIL21 group, the tumor rejection rates for B16, GL261 and CT26 tumors were 8 out of 15, 4 out of 10 and 1 out of 7, respectively (figure 3A, online supplemental figure S5A,C). In contrast, none of the mice in rTTV∆TK and PBS groups showed complete tumor regression (figure 3A, online supplemental figure S5A,C). The superiority of rTTV∆TK-mIL21 in tumor suppression was also reflected by survival, as rTTV∆TK-mIL21 treatment rendered approximately 53% of B16-harboring mice and 40% of GL261-harboring mice survived 80 days after therapies. The CT26-harboring mice showed relatively poorer response to rTTV∆TK-mIL21 treatment, with 14% of treated animals surviving 40 days after treatment. In contrast, PBS-treated group all died within 30 days after treatment (figure 3B, online supplemental figure S5B,D). The rTTV∆TK group showed only moderate improvement in survival as compared with PBS-treated group (figure 3B, online supplemental figure S5B,D).

Introduction of IL-21 into rTTVΔTK significantly enhanced its tumor-suppressing activity. B16 cells (5×104) were subcutaneously implanted into the right flank of mice, tumors were allowed to reach approximately 50 mm3 and then subjected to intratumoral injection with PBS, TTVΔTK (1×107 PFU) or rTTVΔTK-mIL21 (1×107 PFU). (A) Tumor growth of individual injected tumors with ratios of tumor-free mice after treatment indicated (n=15 per group). (B) Cumulative survival curves (****p<0.0001, log-rank test). Data are representative of three independent experiments. (C–F) Tumor infiltration of immune effector cells. Tumors were established in mice by implantation of 1×105 B16 cells into the right flank, followed by intratumoral injection of PBS, TTVΔTK (1×107 PFU) or rTTVΔTK-mIL21 (1×107 PFU). Tumors were isolated 4 days later to analyze the levels of infiltrating immune cells by flow cytometry. The abundances of tumor-infiltrating immune cells and their subpopulations are shown in (C) CD45high, (D) NK, (E) CD3+, CD4+, CD8+ and (F) Tregs (n=6, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, NS: p>0.05, one-way ANOVA). Data are representative of three independent experiments. (G) Assessment of individual contribution of NK cells, CD4+ cells and CD8+ cells to the rTTVΔTK-mIL21–mediated control of tumor growth. Mice were treated as described above except that intraperitoneal administrations of cell-depleting antibody or control IgG antibodies were applied, beginning a day prior to the virus injection and continuing twice weekly throughout the experiment (n=6, **p<0.01 by one-way ANOVA; mean±SEM is shown).
To gain mechanistic insight into IL21-mediated enhanced anti-tumor activity of rTTV, we compared the presence of tumor-infiltrating immune cells among the three treatment groups at 4 days in B16 models after treatment (tumor treatment initiated when the tumor volumes normally reached 50–100 mm3) by flow cytometry. Consistent with that promoting immune cell enrichment in tumor microenvironment (TME) constitutes a major mechanism underlying the tumor-suppressing activity of oncolytic virus, the level of total TILs was significantly higher in rTTV∆TK-mIL21 group relative to PBS group. rTTV∆TK group also showed an increase in TILs as compared with PBS group; however, the difference was not statistically significant (figure 3C). The subsequent analysis of lymphocyte subsets showed a higher induction of NK cells, CD3+ T cells, CD4+ T cells and CD8+ T cells by rTTV∆TK-mIL21, indicative of a globally enhanced engagement of immune effector cells (figure 3D and E). On the contrary, the amount of Treg cells appeared to be reduced in rTTV∆TK-mIL21 group, compared with rTTV∆TK group (figure 3F).
We next sought to determine whether rTTV∆TK-IL21-mediated anti-tumor response involves a particular subset of immune cells. To this purpose, we developed an effective way to individually deplete NK cells, CD4+ T cells, and CD8+ T cells by injection of respective targeting antibodies (online supplemental figure S6). The result clearly indicated that the rTTVΔTK-mIL21–mediated tumor control was largely mediated by CD8+ T cells rather than by NK cells or CD4+ T cells (figure 3G).
Efficacy of rTTV∆TK-IL21 therapy with distant tumors
We next examined whether the local activation of anti-tumor immune response by rTTV∆TK-IL21 has a systemic impact, known as abscopal effect. To this end, we modeled metastatic disease using the bilateral flank B16 tumor model (figure 4A). Besides an expected remarkable inhibition of growth of injected tumors, rTTV∆TK-mIL21 treatment also significantly reduced tumor progression in distant tumors compared with control groups, suggesting rTTV∆TK-mIL21 to be effective in controlling distant metastatic disease (figure 4B). The fact that the expression of viral H3L protein was only detected in rTTV∆TK-mIL21–injected tumor (right flank) but not contralateral (left flank) tumor excluded the possibility that the inhibition of distant tumors was mediated by direct infection through virus spreading (figure 4C). We further analyzed the levels of infiltrated immune cells in distant tumors, and observed an effect of rTTV∆TK-mIL21 that resembled that observed in local immune response as shown earlier, featuring enhanced presence of CD45high, NK cells, CD3+ T cells, CD4+ T cells and CD8+ T cells, although all with much lower abundance. There was essentially no statistical difference between rTTV∆TK group and PBS group in the levels of these immune cell subsets (figure 4D–F). Interestingly, the abundance of intratumoral Treg cells in rTTV∆TK-mIL21 group was comparable with PBS group while significantly lower than rTTV∆TK group (figure 4G).

Intratumoral administration of rTTVΔTK-mIL21 changed immune status of noninjected distant tumors and suppressed tumor growth. (A) Scheme of treatment schedule. Tumors were established in mice by implantation of 1×105 B16 cells into the left and right flanks, followed by injection of PBS, TTVΔTK (1×107 PFU) or rTTVΔTK-mIL21 (1×107 PFU) into right flank tumor. (B) Growth of injected and distant tumors (n=5). *p<0.05, **p<0.01, ***p<0.001 by one-way ANOVA. Mean±SEM is shown. (C) Detection of TTV-expressing H3L protein in rTTVΔTK-mIL21 injected tumors and noninjected distant tumors by Western blotting. β-Tubulin protein was also detected as loading control. (D–G) Tumor infiltration of immune cells in noninjected distant tumors assessed by flow cytometry. The abundances of tumor-infiltrating immune cells and their subpopulations are shown in (D) CD45high, (E) NK, (F) CD3+, CD4+ and CD8+, and (G) Tregs (n=6, *p<0.05, **p<0.01, ***p<0.001, NS: p>0.05, one-way ANOVA). Data are representative of two independent experiments. (H, I) Populational levels of CD11c+ dendritic cells in injected tumors (H) and spleens (I) (n=6, *p<0.05, **p<0.01, ***p<0.001, one-way ANOVA). (J) Frequencies of IFNγ+CD8 T cells and TNFα+CD8 T cells (n=6, *p<0.05, **p<0.01, NS: p>0.05, one-way ANOVA).
Further, we examined the rTTV∆TK-mIL21–mediated engagement of dendritic cells and tumor-specific T-cell response. Our analysis revealed an upregulation of CD11c+ DCs in both injected tumors and spleens, consistent with enhanced antigen presentation to distant lymphoid organs (figure 4H-I, online supplemental figure S7A). For the assessment of tumor-specific T-cell response, the isolated TILs of injected tumors were restimulated with growth-arrested B16 cells and the percentage of IFNγ-producing cells and TNFα-producing cells were respectively detected by intracellular cytokine staining. The data clearly indicated that rTTV∆TK-mIL21 treatment triggered an enhanced tumor-specific T-cell response (figure 4J, online supplemental figure S7B). Taken together, the abscopal effect of rTTV∆TK-mIL21 was likely mediated by enhanced engagement of antigen-presenting cells both locally and systematically, in conjunction with recruitment and activation of tumor-specific immune effector cells.
Sequential combination of rTTV∆TK-IL21 and ACT therapy delivered more robust anti-tumor effect than monotherapy in vivo
The combination of virotherapy and immunotherapy emerged as a new promising avenue toward more effective cancer therapy. Thus, we examined whether there is synergy between rTTV∆TK-IL21 and ACT therapy in suppression of human solid tumors. For this purpose, we used B-NDG mice, which are immune deficient and suitable for engraft of human immune cells and tumor cells to study the in vivo effects of the human immune system on human tumors.35 We constructed the NCI-H292-CD19 cell line (online supplemental figure S8A), which stably expressed the CD19 antigen and served as the tumor target of anti-CD19 CAR-T cells (CD19 CAR-T) that were generated through lentivirus-mediated transduction of T cells isolated from healthy human PBMCs and amplified by in vitro culture (online supplemental figure S8B). NCI-H292-CD19 cells showed comparable in vitro growth with the parental NCI-H292 cells (online supplemental figure S8C). We also created an rTTV virus encoding human IL-21, namely, rTTV∆TK-hIL21, to match with the acting human immune cells. Mice were sequentially subjected to subcutaneous engraftment with the NCI-H292-CD19 cell line, intratumoral injection of either PBS or rTTV∆TK-hIL21, and finally intravenous infusion with either PBS or CD19 CAR T-cells (2×106 cells, 50% CAR+) (figure 5A). We observed little effect of CD19 CAR-T monotherapy on early tumor formation as the treated group, alike PBS-treated group, had rapid tumor progression within 10 days. The ensuing examination revealed a transient and moderate inhibition of CAR-T treatment on further tumor development (figure 5B and C). A significantly better result was obtained with rTTV∆TK-hIL21 group, in which the tumor progression was largely impeded during the first 20 days. However, a later tumor recurrence was found in some mice and the re-emerging tumor showed an aggressive phenotype (figure 5C). Remarkably, the combination of rTTV∆TK-hIL21 and CAR-T was substantially more effective in tumor control than rTTV∆TK-hIL21 and CAR-T individually applied, evidenced by consistent and long-lasting slower tumor growth along with the increased complete response rate (figure 5C). We analyzed the mouse blood 30 days after CAR-T cell infusion, and detected a small amount of CD19 CAR T cells, indicating the activation and persistence of CAR T-cells (online supplemental figure S8D).
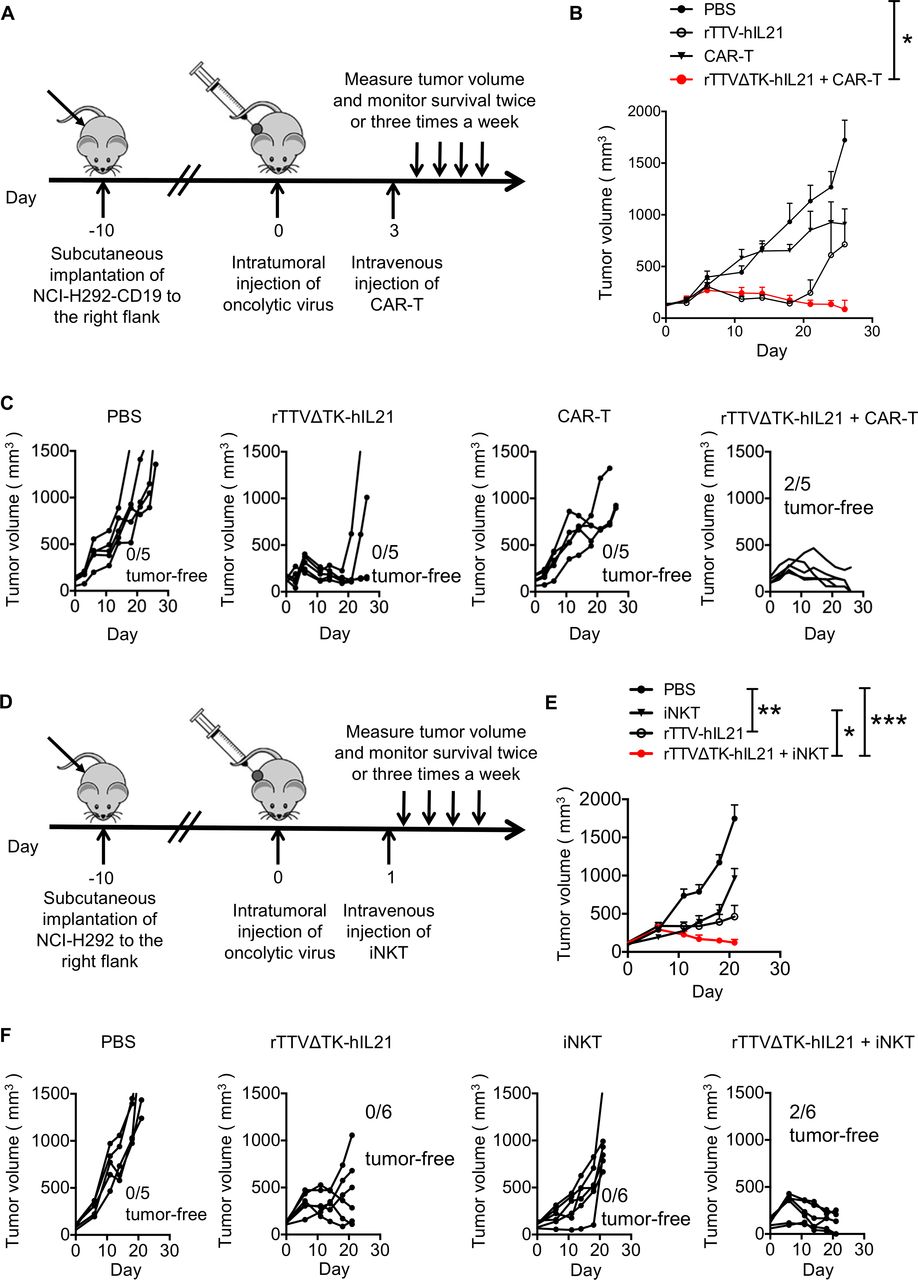
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
rTTVΔTK-IL21 worked synergistically with ACT in controlling tumor growth. (A–C) Strategy to combine rTTVΔTK-IL21 and CAR T-cells in cancer treatment and its anti-tumor efficacy in humanized mouse model. The combination was performed following a scheme shown in (A): NCI-H292-CD19 cells were subcutaneously implanted into right flank of mice 10 days before treatments. Tumors were directly injected with PBS or 1×106 PFU of rTTVΔTK-IL21 (on day 0) and 3 days later subjected to intravenous administration of PBS or 2×106 CAR T-cells. Tumor growth was subsequently measured, shown as cumulative data in (B) (n=5, mean±SEM is shown, *p<0.05, one-way ANOVA) and response of individual tumor in each treatment group (C). (D–F) Strategy to combine rTTVΔTK-IL21 and iNKT cells in cancer treatment and its anti-tumor efficacy in humanized mouse model. The treatment scheme (D) was the same as in (A), except that NCI-H292-CD19 cells were being replaced by NCI-H292 cells and CAR T-cells were being replaced by iNKT cells (5×106), which were intravenously administered 1 day after virotherapy. Shown in (E) is cumulative tumor growth data (n=6, mean±SEM is shown, *p<0.05, **p<0.01, ***p<0.001, one-way ANOVA) and in (F) are individual tumor responses in each treatment group. Data are representative of two independent experiments (B and C) or one experiment (E and F).
We also examined the use of rTTV∆TK-hIL21 to improve the therapeutic efficacy of iNKTs. To this end, we cultured CD3+Vα24+Vβ11+ iNKT cells in vitro (online supplemental figure S8E, 36% iNKT+), and the anti-tumor efficacy of its own and in sequential combination with rTTV∆TK-hIL21 was evaluated in NCI-H292-engrafted B-NDG mice following the same schedule for CAR-T assessment except for the interval between rTTV-hIL21 and iNKT administration being shortened to 1 day (figure 5D). The results showed a similar trend as seen with CAR-T assessment, with the rTTV∆TK-hIL21 and iNKT combination therapy showing a more effective control of tumor growth marked by eradication of some tumors than rTTV∆TK-hIL21 monotherapy, which itself demonstrated a significant tumor-suppressing activity compared with the largely ineffective iNKT monotherapy (figure 5E and F). Overall, these results indicated that rTTV∆TK-IL21 can work synergistically with ACT in controlling tumor growth, thus supporting the combination therapy consisting of sequential application of rTTV∆TK-IL21 and ACT as a new strategy for treatment of cancers.
Discussion
Cytokine-armed oncolytic viruses have been actively pursued as a new treatment for cancer. In this study, we first identified IL-21 in a screening of tumor-suppressing cytokine as the leading candidate that, when locally introduced into the tumor, potently inhibits tumor growth alongside reversal of the immunosuppressive TME. Consequently, we engineered an IL21-armed recombinant vaccinia virus, rTTV∆TK-IL21, and showed in mouse models that its intratumoral injection led to more effective inhibition of injected tumor and also noninjected distant tumors as compared with control rTTV∆TK virus. We further demonstrated a synergy between rTTV∆TK-IL21 and ACT in treating human cancers in humanized mouse model, supporting the potential of rTTV∆TK-IL21 as a new foundation of combination therapy strategy coupling virotherapy with immunotherapy.
Our characterization of different OV candidates provides strong support for the promise of TTV752-1 as an oncolytic virus. It showed robust replication in B16 cells. More importantly, among all the virus candidates under tested conditions, only TTV752-1 infection induced significant cell cytotoxicity, which is a prerequisite of an effective OV. We further found that TTV752-1 was capable of effectively killing two other tumor cell types, GL261 and NCI-H292 (data not shown), in line with the in vivo tumor treatment data later obtained with TTV752-1-derived IL-21-expressing OV. The broad tropism of TTV752-1 might stem from that its entry into cells does not require a specific receptor, but rather is facilitated by viral fusion with the plasma membrane. Its propagation in tumors might be also aided by its capability to replicate in hypoxic conditions.36 We would like to point out that our results did not necessarily deny the potential of other virus candidates as OVs as the oncolytic activities, which are influenced by multiple parameters including the amount of virus for infection, might be achieved in other settings. Nevertheless, by displaying robust oncolytic activities both in vitro and in vivo, TTV752-1 emerged in our virus screening as the optimal choice for the development of an effective OV.
Harnessing T-cell response represents a major theme of tumor immunotherapy design.37 This study stemmed from the speculation that overexpression of molecule related to T-cell activation in tumors might promote the activation of tumor-specific CD8+ T cells in TILs, thereby enhancing the anti-tumor immune response. The activation of T cells requires the TCR signal triggered by recognition of antigen presented on antigen-presenting cells and also is tuned by the second and third signals transmitted, respectively, by costimulatory receptor and cytokines. Therefore, our initial screening covered molecules representing each of the three signals for T-cell activation. GM-CSF primarily promotes the maturation of DCs and boosts the antigen presentation, whereas CD28/CD86 and 4-1BB/4-1BBL signaling pathways contribute to T-cell activation by transducing the co-stimulatory signals.30 38 Belonging to the third signal, IL-21 majors in promoting the maturation, differentiation and functionality of T cells.39 As a chemokine, CXCL9 is known to mediate recruitment of CD8+ T cells.40 Our analyses revealed a standout anti-tumor effect of IL-21 in comparison with other candidate molecules. It is a surprise that GM-CSF, which constitutes a T cell–stimulating module in the FDA-approved oncolytic virus T-Vec, has no detectable anti-tumor activity in our assay. A plausible explanation was provided by previous studies demonstrating that high dose of GM-CSF does not stimulate but rather suppress the immune response by inducing inhibitory Gr-1+CD11b+ myeloid-derived suppressor cells.41 42 The lack of observed tumor-suppressing activity for CXCL9 might be due to its complex action on both immune cells and tumor cells. Apart from immune cells, the receptor for CXCL9, CXCR3, is expressed on melanoma cells and consequently CXCL9 likely functions as a double-edged sword against melanoma by positively regulating both tumor and immune cells.43
By showing that local expression of IL-21, either by tumor cells themselves or by recombinant oncolytic virus as of rTTV∆TK-IL21, promoted the accumulation of effector T cells in tumor sites, we demonstrated the capability of IL-21 to reverse the inhibitory nature of TME. This conforms to the concept of certain types of therapies being more amenable to local versus systemic delivery, particularly those targeting tumor-resident cell populations.44 Our profiling of tumor-resident immune cells revealed that local IL-21 expression has a distinct advantage in reshaping the TME. It functions in both innate and adaptive immune compartments by increasing the abundance of NK cells along with CD8+ and CD4+ cells while lowering the level of inhibitory Treg cells. This is in sharp contrast to the action of other ILs, exemplified by IL-2, which promotes functions of both CD8+ T cells and Tregs and shows toxicity for the majority of treated patients.45 Our results corroborate a most recent study demonstrating a robust anti-tumor activity of intratumorally injected recombinant IL-21 protein.46
A potential concern of intratumor injection of rTTV∆TK-IL21 is that the therapeutic effect might be locally restricted, being unable to reach distant tumors. Our demonstration of induced inhibition of noninjected distant tumors by local rTTV∆TK-IL21 administration helped ameliorate this concern, supporting the potential of rTTV∆TK-IL21 in treating metastatic diseases. The observed enhanced presence of immune effector cells, along with the undetected viral protein expression, supported the notion that immune cells activated and expanded in rTTV∆TK-IL21–treated tumors are capable of entering the circulation and consequently being attracted to the distant tumor sites via chemokines secreted by tumor cells, where the activation of tumor antigen-specific T cells ensues. It should be noted that the induced levels of immune effector cells in distant tumors were substantially lower than in injected tumors, which might be responsible for the lack of complete regression despite significant control of tumor growth. How to further potentiate the abscopal effect of rTTV∆TK-IL21 treatment warrants future investigation, and a possible solution might be the combination with other therapeutics to further boost the initial pool of activated immune cells in the injection site.
IL-21’s capability to promote the activities of immune effector cells rationalized our design of a combination therapy of rTTV∆TK-IL21 and CAR T-cell. It was hoped that a priming of rTTV∆TK-IL21 can shift TME to a more hospitable environment for CAR T-cells to infiltrate, survive and proliferate in solid tumors. Using humanized B-NDG mice to establish human tumor models, we consequently demonstrated that rTTV∆TK-IL21 and CAR-T treatment had synergistic effects, with the combination treatment exhibiting a better anti-tumor performance than individual treatment. It is noteworthy, between the two monotherapies, that CAR-T was significantly less effective than rTTV∆TK-IL21, consistent with inefficacy of CAR-T in treating solid tumor as observed in clinical studies. A caveat of our humanized mouse model is the graft rejection after adoption of CAR T-cells, which would narrow the therapeutic window. In this regard, a truly humanized mice model or non-human primate model should be explored for the rTTV∆TK-IL21–CAR-T combination strategy before its clinical applications. It is also desirable to further optimize the administration scheme including the timing of rTTV∆TK-IL21 and CAR-T treatment. Nevertheless, our study provided proof of principle that rTTV∆TK-IL21 priming represents a novel strategy to boost the effectiveness of CAR T-cells in treating solid tumors. Our further demonstration of similar boosting effect with iNKT treatment implicates its generality in synergistic alliance with different types of ACT therapy.
In summary, we herein presented experimental evidence supporting strong antitumor activities of IL-21, which is underlined by its capability to promote immune effector cells while suppressing the Treg cells. In connection, we demonstrated IL21-carrying recombinant TTV as a novel virotherapy in cancer treatment and highlighted its potential in combination with ACT for an optimized dual-agent anti-tumor therapeutics. It is tempting to speculate that rTTV∆TK-IL21 might also work synergistically with other immunotherapies such as anti-PD-1 antibody for treating solid tumors. Further improvements on rTTV∆TK-IL21 might include deletion of additional non-essential viral genes to enhance biosafety and incorporation of additional immunomodulatory molecules to orchestrate a broader and stronger engagement of immune effector responses. Given the promise of rTTV∆TK-IL21 as an anti-tumor agent, it merits further exploration for cancer treatment in a clinical setting.
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as online supplemental information. Data sharing not applicable as no datasets generated and/or analyzed for this study. All data are published in this paper. Materials are available on reasonable request based on an appropriate MTA.
Ethics statements
Patient consent for publication
Ethics approval
Animal experiments were performed with the approval of the Institutional Animal Care and Use Committee of Shanghai Public Health Clinical Center (SPHCC). The study was reviewed and approved by the Ethics Committee of SPHCC.
Acknowledgments
We would like to thank Dr. Kun Fan (Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai, China) for kindly providing the murine glioma cell line GL261, and Dr. Yang Huang (Shanghai Public Health Clinical Center, Shanghai, China) for kindly providing the murine colon carcinoma cell line CT26 and the anti-H3L antibodies.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
TC and XD contributed equally.
Correction notice This article has been corrected since it first published. The provenance and peer review statement has been included.
Contributors JX conceived this study. JX, XZ, CZ, TC and XD designed the experiments. TC, XD, QL and NG performed the experiments. YC prepared the virus. TC and CZ prepared the manuscript. JX, XZ, CZ, TC and XD revised the manuscript.
Funding This work was supported by the National Key Research and Development Program (2016YFC1303402),the Chinese National Grand Program on Key Infectious Disease Control (2017ZX10202102-006) and Clinical Research Plan of SHDC(No. SHDC2020CR3002A)
Competing interests JX, XZ, XD, YC, TC and QL are inventors on patent application (Chinese Application No. 201910455932.8) submitted by Shanghai Public Health Clinical Center that covers the use of recombinant oncolytic poxvirus rTTV-IL21 against tumors.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.