Article Text

Abstract
Background E7046 is a highly selective, small-molecule antagonist of the E-type prostanoid receptor 4 (EP4) for prostaglandin E2, an immunosuppressive mediator of the tumor immune microenvironment. This first-in-human phase 1 study assessed the safety, tolerability, pharmacokinetics, pharmacodynamics, maximum tolerated dose (MTD) and recommended phase 2 dose of E7046.
Methods This first-in-human study enrolled 30 patients with advanced tumors of cancer types associated with high levels of myeloid infiltrates. E7046 was administered orally once-daily in sequential escalating dose cohorts (125, 250, 500, and 750 mg) with ≥6 patients per cohort. Tumor assessments were performed every 6 weeks. Paired tumor biopsies and blood samples, before and on treatment, were collected for pharmacokinetic and pharmacodynamic characterization of the treatment.
Results No dose-limiting toxicities were observed, and the MTD was not reached. E7046 had an elimination half-life (t1/2) of 12 hours, and drug exposure increased dose-dependently from 125 to 500 mg. Target modulation by E7046 was supported by changes in genes downstream of EP4 with concurrent enhanced antitumoral immune responses. A best response of stable disease (per irRECIST) was reported in 23% of patients treated with E7046 (n=30) (125 mg: n=2; 250 mg: n=2; 750 mg: n=3). Over half (4/7) of the patients with stable disease had treatment duration of 18 weeks or more, and three patients (3/15; 20%) achieved metabolic responses.
Conclusions In this first-in-human study, E7046 administered orally once daily demonstrated manageable tolerability, immunomodulatory effects, and a best response of stable disease (≥18 weeks) in several heavily pretreated patients with advanced malignancies. The 250 and 500 mg doses are proposed for further development in the combination setting.
Trial registration number NCT02540291.
- oncology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
A permissive tumor microenvironment is critical for tumor progression and metastasis.1 Targeting the tumor microenvironment, with a focus on reprogramming its immunosuppressive properties, represents a promising approach for developing novel immune-targeting anticancer therapies. Prostaglandins play a key role in mediating inflammatory responses, and their effects on the differentiation of monocytic cells and suppression of T-cell activation have been exploited by tumors to maintain an immunosuppressive tumor microenvironment.2 3
Prostaglandin E2 (PGE2) is a small-molecule derivative of the arachidonic acid pathway that regulates a number of biological processes via interaction with 1 of 4 E-type prostanoid receptors (EP1–EP4).4 PGE2 plays several important roles in the modulation of the tumor microenvironment: it promotes neoangiogenesis5 and the development of both protumoral tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs).6 7 In addition, PGE2 inhibits CD8+ cytotoxic T lymphocyte activity in tumors, thereby promoting tumor escape from immune responses.7
The PGE2 receptor EP4 is expressed primarily on myeloid cells, T lymphocytes, and tumor cells,8 and has emerged as a major contributor to PGE2-mediated enhancement of tumor survival pathways and as a suppressor of innate and adaptive antitumor immune responses.9 E7046 is a highly selective, investigational small-molecule inhibitor of EP4.4 10 Through selective antagonism of EP4, E7046 inhibits the differentiation of monocytic myeloid lineage cells to TAMs and MDSCs,4 which are known contributors to the formation and maintenance of an immunosuppressive tumor microenvironment.1 6 7 Other myeloid-modifying agents currently under development include macrophage colony-stimulating factor 1 receptor (CSF-1R) antagonists, which act by reducing the number of TAMs in patients.11 However, unlike CSF-1R antagonists, E7046 does not affect the survival of TAMs and MDSCs but instead modulates their phenotype.4
In preclinical studies, E7046 inhibited the growth of multiple mouse syngeneic tumor models in a manner that was dependent on the presence of both myeloid cells and CD8+ T cells.4 E7046 interferes with tumor-induced monocyte differentiation into immunosuppressive type 2 macrophages and MDSCs and promotes the differentiation of monocytes into antigen-presenting cells instead. Thus, E7046 promotes antigen presentation to T cells and facilitates T-cell accumulation at the tumor site.4 Moreover, in a preclinical model, there was a complete abrogation of the E7046-mediated antitumor effect in the absence of CD8+ T cells, highlighting the importance of these cells in E7046-modulated tumor immunity.4 E7046 represents a first-in-class investigational compound for cancer immunotherapy with a mechanism of action that is distinct from T-cell targeting immune checkpoint inhibitors and myeloid cell-reducing molecules. Here, we report the results from an open-label, first-in-human, phase 1 study of E7046 in patients with tumor types that are known to harbor high levels of myeloid infiltrates.12
Methods
Study design
This was an open-label, multicenter, phase 1 study of E7046 in patients with selected advanced malignancies. Patients were enrolled from one site in France and two sites in the USA. E7046 was administered by mouth once daily, continuously, in 21 day cycles. Patients were treated in sequential escalating-dose cohorts of ≥6 patients per cohort at each of four dose levels (125, 250, 500, and 750 mg). The E7046 starting dose of 125 mg was selected based on preclinical pharmacological studies, because it is the human equivalent of the minimally effective dose and it is <1/6th of the minimally toxic dose detected in preclinical toxicology studies. Thus, a dose of 125 mg was predicted to be safe and potentially efficacious in patients with advanced cancers. Patients continued study treatment until disease progression, development of unacceptable toxicity, or withdrawal of consent.
The primary objectives of this study were to assess the safety and tolerability of E7046, and to determine the maximum tolerated dose (MTD) and/or the recommended phase 2 dose (RP2D). The secondary objectives prespecified in the protocol were to evaluate the pharmacokinetic profile of E7046, as well as several efficacy end points according to immune-related Response Evaluation Criteria in Solid Tumors (irRECIST) including the objective response rate, time to response, duration of response, progression-free survival, disease control rate (ie, partial response+complete response+stable disease) and clinical benefit rate (ie, partial response+complete response+durable stable disease (≥24 weeks)). Patients who did not have a tumor response assessment for any reason were considered to be non-responders and were included in the denominator when the response rate was calculated. To evaluate response, tumor scans were performed every 6 weeks and assessed using irRECIST13 until disease progression, starting a new anticancer treatment, withdrawal of consent, or death.
Exploratory end points for this study included examination of the pharmacodynamic effects of E7046 on select biomarkers and immune cells in tumor infiltrate and in peripheral blood; measurement of metabolic response by 18fluorodeoxyglucose-positron emission tomography (18FDG-PET); and assessment of overall survival.
Patients
Patients had tumor types that typically harbor high levels of myeloid infiltrates, based on a bioinformatic analysis of data sets obtained from the Cancer Genome Atlas Data Portal.12 Patients were required to be ≥18 years old with advanced, non-resectable, or recurrent tumors for which no alternative standard therapy exists, including pancreatic adenocarcinoma, renal clear cell carcinoma, squamous cell carcinoma of the head and neck, non-small cell lung cancer, colorectal cancer, hepatocellular carcinoma, ovarian serous epithelial cancer, bladder transitional cancer, cervical cancer, and triple-negative breast cancer. Additionally, patients must have had ≥1 measurable lesion per irRECIST, and an Eastern Cooperative Oncology Group performance status of 0 or 1. Additional key inclusion criteria were adequate renal, bone marrow, and liver function, completion of all prior chemotherapy or immunotherapy (tumor vaccine, cytokine, or growth factor)≥4 weeks prior to study-drug administration; and completion of all prior definitive radiation therapy ≥6 weeks before study-drug administration. Key exclusion criteria included patients with another malignancy active within the previous 2 years, except for basal or squamous cell skin cancer, superficial bladder cancer, or carcinoma in situ of the cervix or breast that had completed curative therapy; any active autoimmune disease, including inflammatory bowel disease or a documented history of autoimmune disease, poorly controlled asthma, or history of syndrome that required systemic steroids or immunosuppressive medications; concurrent medical condition requiring the use of immunosuppressive medication, including immunosuppressive doses of systemic or absorbable corticosteroids (except inhaled or intranasal corticosteroids with minimal absorption); major surgery; or use of other investigational drugs within 4 weeks before study drug initiation; and prior exposure to drugs that are antagonists of CSF-1R.
Assessments
The MTD was defined as the dose level at which ≥2 of six patients experienced a dose-limiting toxicity (DLT). If ≤1 of six patients in all dose cohorts experienced a DLT, then the MTD was considered as not reached. DLTs were graded using the National Cancer Institute–Common Terminology Criteria for Adverse Events V.4.03, and were defined as drug-related toxicities (considered related, probably related, or possibly related to E7046) occurring during cycle 1, including non-hematologic toxicity ≥grade 3 (except diarrhea, nausea, and vomiting unless lasting >3 days despite optimal supportive care), confirmed (with a second measurement after 24 hours) non-hematologic, appropriately graded, laboratory findings of grade ≥3 that were grade ≤1 at baseline; hematologic toxicity, defined as grade 4 neutropenia for at least 5 days, grade 3 neutropenia with fever, grade 4 thrombocytopenia, or grade 3 thrombocytopenia with bleeding or lasting >7 days; any other toxicity, at the physician or study director’s discretion. Patients who missed ≥7 days of dosing in cycle 1 due to drug-related toxicity (but not qualifying for a DLT) were to be assessed as having experienced a DLT.
Safety
Safety assessments consisted of monitoring and recording all adverse events (AEs) as reported by the investigator, including all Common Terminology Criteria for AE V.4.03 grades and serious adverse events (SAEs). Regular laboratory evaluation of hematology, blood chemistry, and urine values; periodic measurement of vital signs; echocardiograms/multigated acquisition scans; electrocardiograms; and physical examinations were performed. Toxicities were managed by concomitant medication (as appropriate), treatment interruption, dose reduction, or treatment discontinuation.
AEs are presented by Medical Dictionary for Regulatory Activities preferred term, nested within primary system/organ/class. The incidences of treatment-emergent AEs (TEAEs) were categorized by treatment cohort and summarized by maximum severity and relationship to the study drug. TEAEs included any AEs that occurred during the entirety of treatment exposure.
Efficacy
To evaluate response, tumor scans were performed every 6 weeks and assessed using irRECIST until disease progression, starting a new anticancer treatment, withdrawal of consent, or death.13 Patients who did not have a tumor response assessment for any reason were considered to be non-responders and were included in the denominator when the response rate was calculated.
Pharmacokinetics
Blood samples were collected during cycle 1 on day 1 and day eight predose (0 hours), and at 0.5, 1, 2, 4, 6, 8, 10, and 24 hours postdose; and the plasma concentrations of E7046 and its acyl glucuronide metabolite (M1) were measured. The following pharmacokinetic parameters were calculated: maximum drug concentration (Cmax), time to reach maximum concentration following drug administration (Tmax), area under curve, and elimination half-life (t½), all using Phoenix software V.7.0.
Pharmacodynamic biomarkers
Paired tumor-core needle biopsies were collected before the first dose of E7046 and again during cycle 2 of E7046 treatment. Samples were assessed for immune-cell infiltration by both immunohistochemistry (using T-cell-specific (anti-CD3, anti-CD8) and macrophage-specific antibodies (anti-CD68, anti-CD163)) and gene-expression analysis (using a 92-gene TaqMan Low Density Array (TLDA) panel online supplementary file 1).
Supplemental material
Gene-expression profile analysis was performed on isolated mRNA from blood using a separate 92-gene TLDA panel (online supplementary file 1). Blood samples were collected at various time points (online supplementary file 1). Blood samples were also analyzed for the serum/plasma concentrations of 36 immune-related circulating factors (eg, cytokines and chemokines; online supplementary file 1) using global proteomics methods, ELISA, and a multiplex immunoassay platform (Meso Scale Discovery Platform).
Imaging biomarkers
The effect of E7046 on metabolic activity of the tumor was measured via 18FDG uptake using 18FDG-PET/CT scans performed at baseline, and again at the week 6 and week 12 tumor assessments for those patients with FDG-avid tumor lesions at screening. Metabolic responses were based on European Organization for Research and Treatment of Cancer recommendations.14 A partial metabolic response was defined as a reduction in maximum standardized uptake value (SUVmax-single voxel) of ≥25% after >1 cycle of drug. Images were assessed by a radiologist at an independent imaging core laboratory. Up to 10 target lesions per subject that were metabolically hyperactive (tumor-to-background ratio >2.0 and SUV >2.5 and were at least 1.5 cm in at least one dimension in the axial plane) were selected at baseline and then assessed at weeks 6 and 12. The SUVmax was reported for each lesion.
Statistical analyses
Paired samples were analyzed via the paired t-test using GraphPad Prism (V.7.0.2) software. Statistical analyses for survival statistics were performed using SAS software (V.9). The median survival time and the associated 95% CI were estimated using the Kaplan–Meier method. Biomarker results are presented as summary statistics and change from baseline.
Results
Patient characteristics
Overall, 30 patients were enrolled, with a median age of 58 years (range: 24–78 years) (table 1). Colorectal cancer was the most prevalent type of cancer in this study (43.3%), followed by pancreatic cancer (20.0%), and squamous cell carcinoma of the head and neck (13.3%). Four patients (13.3%) had received prior immunotherapy (pembrolizumab, n=3; nivolumab, n=1) (table 1).
Patient demographics and baseline characteristics
Safety
At least 1 TEAE was reported in 28 patients (93%). The most common TEAEs (all grades) were fatigue (37%), diarrhea (33%), nausea (30%), anemia (23%), and decreased appetite (23%; table 2). Grade ≥3 TEAEs occurred in 17 patients (57%) and are shown in table 3. Five patients (17%) discontinued treatment due to TEAEs, three of whom discontinued treatment due to TEAEs that were unrelated to study treatment (cancer pain and ascites (n=1 each; E7046 250 mg dose), large intestinal obstruction (n=1; 125 mg dose)). In the two patients who discontinued study treatment due to a treatment-related TEAE, one patient experienced hypersensitivity (n=1; 250 mg dose), and the other patient, who was in the 750 mg dose cohort, experienced two TEAEs (acute kidney injury and hyperuricemia) that led to discontinuation. Overall, treatment-related TEAEs occurred in 16 patients (53%) and grade ≥3 treatment-related TEAEs occurred in three patients (10%). Treatment-related SAEs were experienced by 10% (3/30) of patients (fever, rash, and hypersensitivity in one patient; hyperuricemia and acute kidney injury in one patient; and generalized rash in one patient). No patients experienced a fatal treatment-related TEAE.
Summary of adverse events
Grade ≥3 TEAEs
No DLTs were observed and the MTD was not reached. Slight increases in the incidence of the three most common TEAEs were observed in the 750 mg dose cohort compared with the combined incidence (fatigue: 42.9% vs 36.7%; diarrhea: 42.9% vs 33.3%; nausea: 42.9% vs 30.0%, respectively). However, the majority of TEAEs did not strongly correlate with increased dose of E7046. Furthermore, TEAE severity did not appear to correlate with increased dose of E7046, as there was no association between study-drug exposure and grade 3 or 4 TEAEs.
Pharmacokinetics
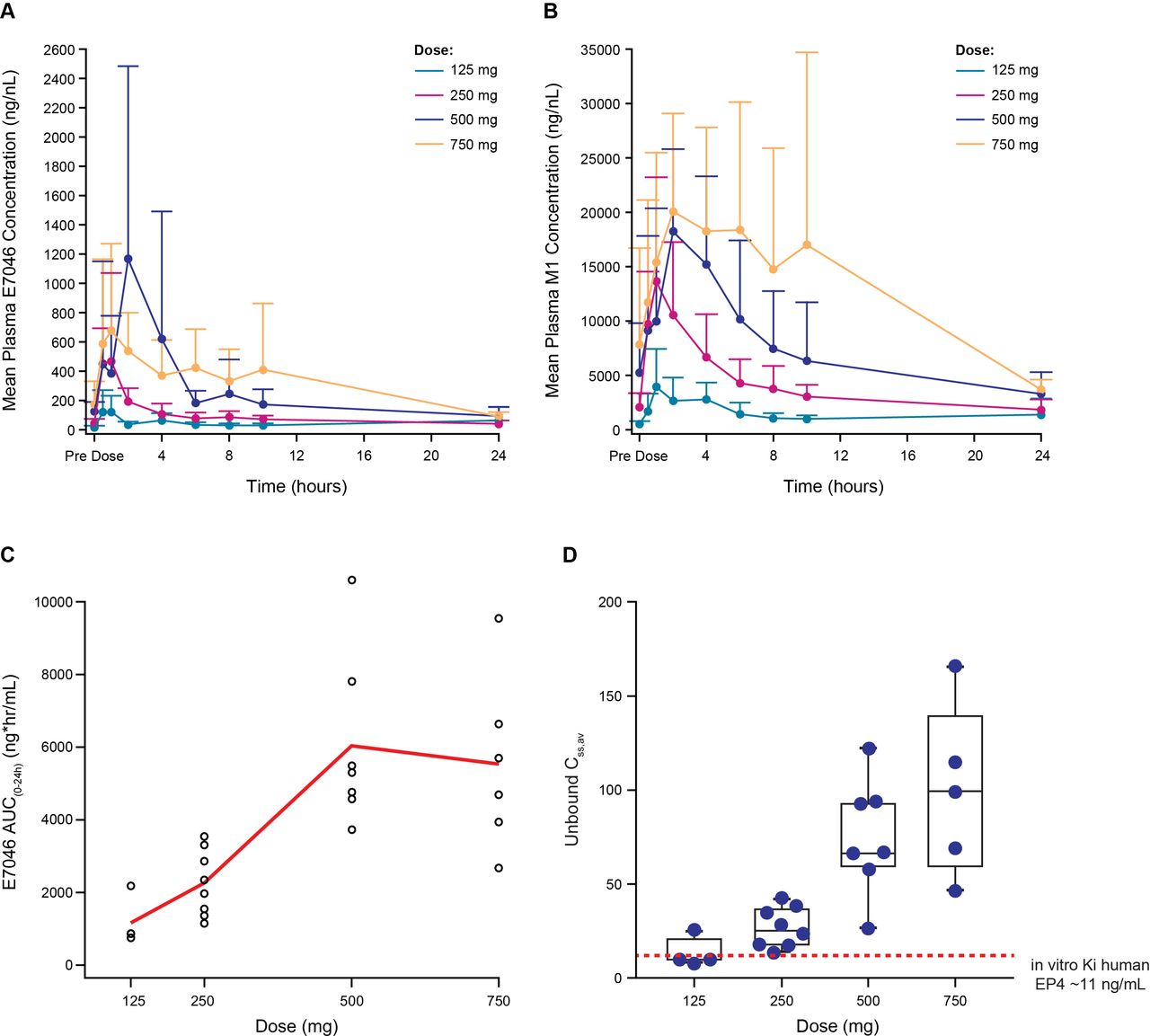
E7046 was rapidly absorbed across all doses (figure 1A; figure 1B); and time to maximum drug concentration was observed at ~2 hours postdose (online supplementary file 1). E7046 exposure was dose proportional up to 500 mg, with no incremental increase at 750 mg (figure 1C). Multiple dosing resulted in twofold to threefold accumulation, consistent with elimination (t1/2=12 hours). Free E7046 increased dose-dependently and reached or exceeded the preclinical Ki value4 of EP4 inhibition in all four doses (figure 1D). Overall, the pharmacokinetic profile of E7046 justifies daily dosing. E7046 is extensively metabolized to its M1 metabolite, which has negligible activity on EP4 (data not shown). Compared with E7046, exposure to the metabolite was >23 fold higher (figure 1B). The metabolite elimination parallels that of E7046 (t1/2 = 11 hours; twofold accumulation).
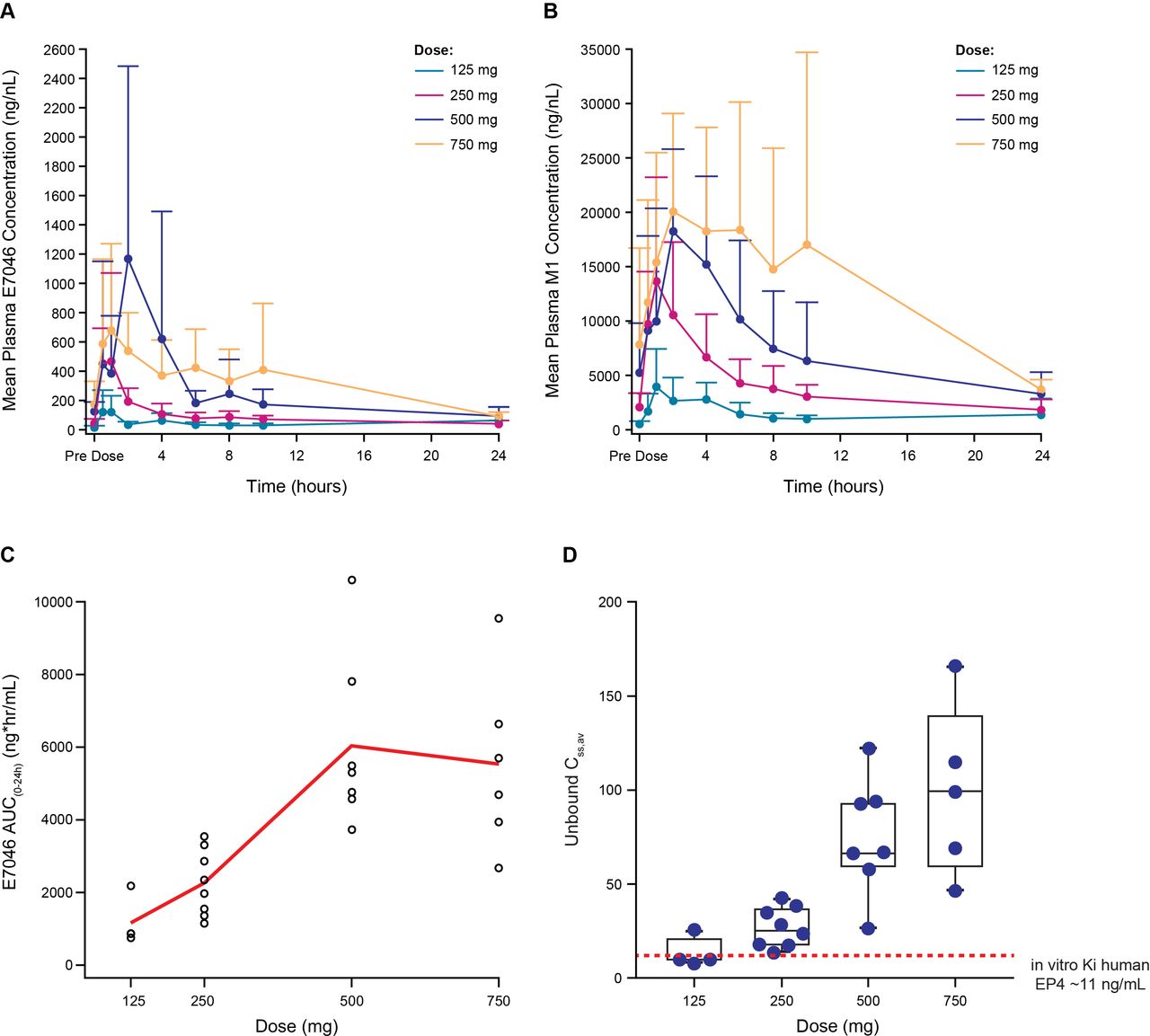
Pharmacokinetic profile of E7046 on cycle 1 day 8 following daily oral administration: (A) plasma concentration–time profiles for E7046; (B) plasma concentration–time profiles for M1 metabolite; (C) exposure–dose profile for E7046; (D) free E7046 concentration–dose profile for E7046a. aThe Ki value of human EP4 is indicated by a dashed line. AUC, area under the curve; EP4, PGE2-receptor E-type 4.
Efficacy
There were no objective responses by irRECIST. Stable disease was observed in seven of 30 (23%) patients, and the disease control rate was 23%. Among the seven patients with stable disease, five had treatment duration ≥18 weeks (figure 2A). The median duration of treatment was 5.9 weeks (range: 1.1–30). There were 10 patients (33%) who had a longer duration of treatment on E7046 compared with their most recent prior anticancer therapy (figure 2B). The efficacy outcomes for all dose groups are summarized inonline supplementary file 1. Metabolic responses as assessed by 18FDG-PET were evaluable in only 15 patients: three of these patients (20%) had a partial metabolic response—a representative case is shown in figure 2C.

Assessments of efficacy and treatment duration: (A) treatment duration by primary tumor location and dosage; (B) duration of treatment for prior therapy and E7046 by tumor type; (C) PET/CT scans showing metabolic responses in a patient with lung adenocarcinoma receiving E7046 treatmentc. aTwo patients in this group received prior immune checkpoint inhibitor therapy (but not as the most recent prior therapy). bPatient received at least one prior radiotherapy. cMetabolic response assessments were based on European Organization for Research and Treatment of Cancer recommendations.14 Partial metabolic response was defined as a reduction in SUVmax of ≥25% after more than one cycle of drug. α-PD-1, antibody to programmed death receptor-1; Nivo, nivolumab; PD, progressive disease; Pem, pembrolizumab; PET, positron emission tomography; SD, stable disease. SUVmax, maximum standardized uptake value.
Pharmacodynamics
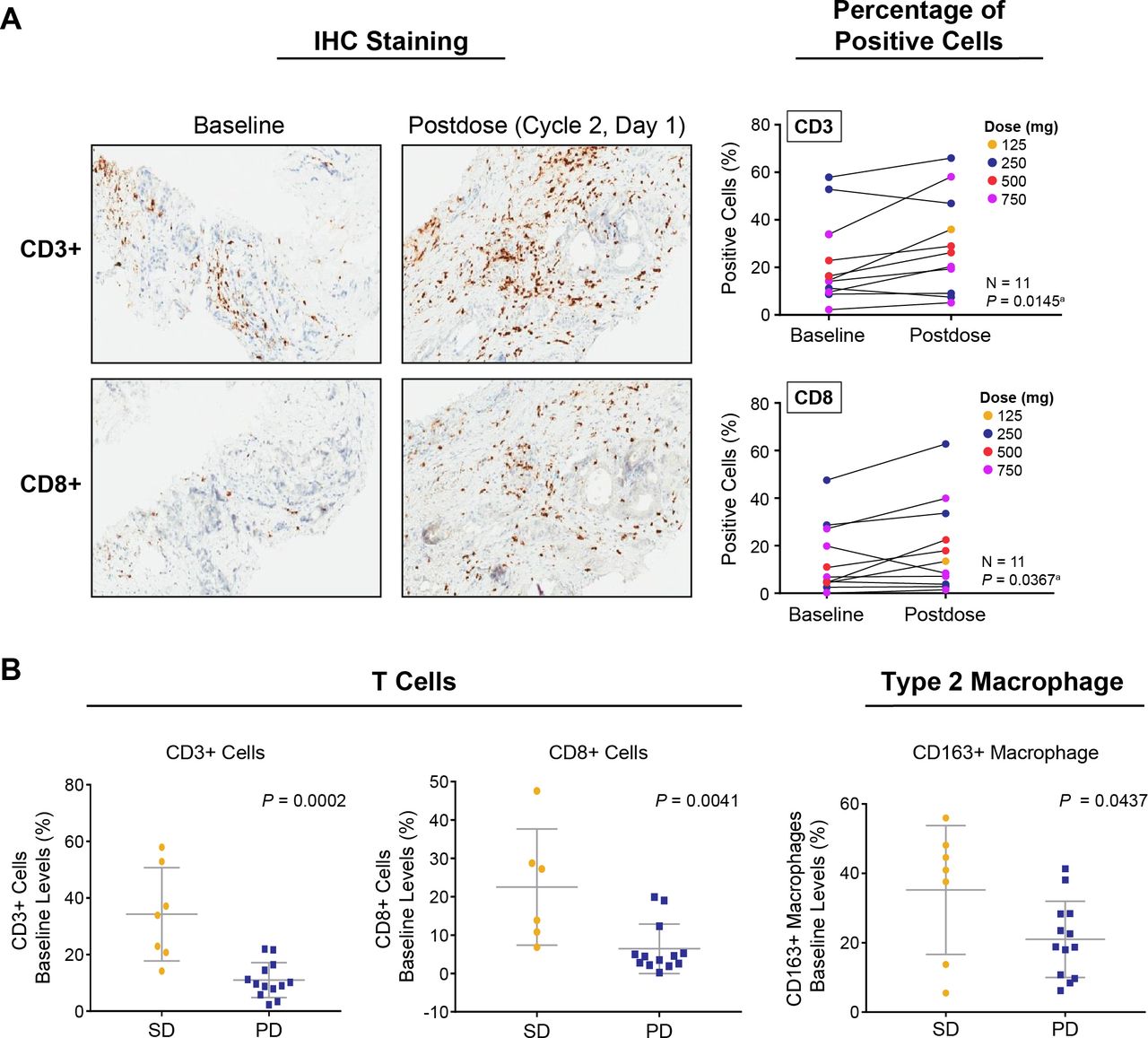
Paired tumor biopsies (before treatment and on cycle 2 day 1) were collected from 11 of 30 patients and analyzed by immunohistochemistry for immune cell infiltration. Following E7046 treatment, tumor tissues revealed a significant increase in CD3+ and CD8+ T cells (figure 3A). Immunohistochemistry specimens stained with the anti-CD68 antibody were not analyzed due to overlapping antigenicity in macrophages and tumor cells, which hindered the identification of the macrophage population. Of 11 paired tumor biopsies, 10 samples had sufficient tissue remaining for the gene-expression analysis of 92 EP4-regulated and immune-related genes. The analysis revealed changes in the levels of key EP4-regulated genes, including upregulation of tumor necrosis factor (TNF)-α and C-X-C motif chemokine ligand (CXCL)10, although without reaching statistical significance (online supplementary file 1). These results were consistent with preclinical findings where the EP4 agonist, PGE1-OH (100 nM), induced a dose-dependent down-regulation of TNF-α and CXCL10 in primary human peripheral blood mononuclear cells (PBMCs) (online supplementary file 1). Notably, patients with higher baseline tumor infiltrations of T cells (CD3+, CD8+) and of type 2 macrophages (CD163+) were significantly (p<0.05) more likely to achieve stable disease with E7046 treatment, rather than progressive disease (figure 3B).

Pharmacodynamic changes in pretreatment and post-treatment tumor tissues following E7046 treatment: (A) immunohistochemical staining of intratumoral T cells by CD3 and CD8 antigens from baseline to cycle 2 day 1; (B) the baseline levels of tumor T cells (CD3+, CD8+) and CD163+macrophages are associated with better clinical outcomes. aPaired one-tailed t-test. CD, cluster of differentiation; IHC, immunohistochemistry; PD, progressive disease; SD, stable disease.
Blood gene-expression analysis revealed that 16 genes out of a 92-immune-gene panel were modulated (upregulated or downregulated) on E7046 therapy at cycle 1 day 15 compared with baseline (figure 4A). Among these genes, five known EP4-regulated genes were modulated, as assessed by paired t-test: T-cell exhaustion marker, eomesodermin (EOMES); prostaglandin E2 receptor type 4 (PTGER4); indoleamine 2,3-dioxygenase 1 (IDO1); programmed death ligand (PD-L) 1 (CD274); and PD-L2 (PDCD1LG2) (figure 4B). Out of a panel of 36 serum cytokines and chemokines, 14 were modulated by E7046 treatment at cycle 1 day 15 compared with baseline (figure 4C): C-C motif chemokine ligand (CCL)5, CXCL1, CXCL2, CXCL10 (IP-10), renin, eotaxin (CCL11), MCP-1 (CCL2), MCP-4 (CCL13), interleukin (IL)−7, IL-8 (CXCL8), IL-10, IL-12p40, IL-13, and TNF-β. Notably, serum levels of the T-cell recruiting chemokines CXCL10 and CCL5 were significantly upregulated compared with baseline (p=0.0015 and p=0.0292, respectively; figure 4D). In addition, no significant changes were found for the presence of major immune cell subsets in the blood (T cells, B cells, or natural killer cells and MDSC populations) after E7046 treatment, using eight-color flow cytometry analysis (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pharmacodynamic changes in expression of blood biomarkers following E7046 treatmenta: (A) volcano plot of gene expression-levels from baseline to C1D15 as determined by a 92 gene TLDA panel (criteria for signal detection ≥1.2-fold change in expression and p value<0.1); (B) relative transcriptional changes of five genes from baseline to C1D15; (C) volcano plot of changes in serum levels of cytokine/chemokines using a 36-analyte MSD panel; (D) relative changes in serum levels of CXCL10 and CCL5 from baseline to C1D15 as determined by the MSD platform. aPaired t-test was used for statistical calculation. ARG1, arginase 1; CCL, C-C motif chemokine ligand; CD, cluster of differentiation; C1D15, cycle 1 day 15; CXCL, C-X-C motif chemokine ligand; CX3CL1, C-X3-C motif chemokine ligand 1; EOMES, eomesodermin; IDO1, indoleamine 2,3-dioxygenase 1; IFN, interferon; IL, interleukin; IP, interferon γ-induced protein; ITGAM, integrin subunit alpha M; L, ligand; MCP, monocyte chemoattractant protein; MSD, Meso Scale Discovery; PD-L, programmed death ligand; PDCD1LG2, programmed cell death 1 ligand 2; PTGER, prostaglandin E receptor; PTGES, prostaglandin E synthase; PTPRC, protein tyrosine phosphatase receptor type C; RCN, relative copy number; RORC, RAR-related orphan receptor; TLDA, TaqMan Low Density Array; TNF, tumor necrosis factor; VCAM, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor.
Recommended phase 2 dose
The RP2D of E7046 was determined to be ≤500 mg as the exposure to study drug increased dose proportionally up to 500 mg, with no additional increase in exposure observed at the 750 mg dose. Although no objective responses were observed, the best overall response of stable disease was observed across several dose levels examined (n=2, 125 mg; n=2, 250 mg; n=3, 750 mg). At the lowest two doses of E7046 (125 and 250 mg), 25% of patients (n=2/8, in both cohorts) achieved a best response of durable stable disease for ≥18 weeks. Thus, based on the exposure to study drug and clinical activity, the RP2D was determined to be ≤500 mg and preferably higher than 125 mg. In PK/PD correlation analyses, linear trends were observed between E7046 exposure with T-cell infiltration in tumor biopsies (CD3+ T cells), and with expression of EP4-regulated genes EOMES and CD274 (which encodes PD-L1) in blood (online supplementary file 1), suggesting that higher exposure may possibly translate to increased biological activity. Hence, two doses—250 mg and 500 mg—were chosen as the RP2D for future clinical investigation.
Discussion
E7046 treatment was associated with manageable toxicity in patients with advanced malignancies. No DLTs were reported, and no apparent correlation between TEAE incidence and exposure was observed, indicating that safety did not limit the RP2D selection (within the range of 125 to 750 mg examined in this study).
Pharmacodynamic biomarker analyses showed that treatment with E7046 resulted in significant changes in the circulating gene-expression levels of several EP4-regulated genes including decreased expression of EOMES, PTGER4 (gene encoding the EP4 receptor) and IDO1; and increased expression of CD274 (gene encoding PD-L1) and PDCD1LG2 (gene encoding PD-L2). Additionally, increased expression of the following EP4-regulated cytokines2 7 15–18 was observed: IL-10, IL-8, IL-12p40, IP-10 (CXCL10), CCL5, and CXCL2. These serum biomarker changes indicated that E7046 successfully antagonizes EP4 in the clinical setting and underscores the unique mechanism of action of E7046. Moreover, these results are consistent with preclinical studies, wherein E7046 promoted the differentiation of myeloid cells to antigen-presenting cells and the recruitment and activation of cytotoxic T cells. Finally, in this first-in-human study, patients treated with E7046 had increased serum levels of two T-cell recruiting chemokines (CXCL10 and CCL5) that were accompanied by enhanced accumulation of cytotoxic T cells in the tumor tissue. Taken together, these data support the hypothesis that E7046 reverses the immunosuppressive effects of PGE2 and ultimately enhances the host antitumoral immune response, although further research is needed.
Increased expression of PD-L1 and PD-L2 are among the signature downstream effects of the interferon (IFN) response.19 The upregulation of the genes encoding PD-L1 and PD-L2 in the blood of E7046-treated patients indicates an activation of the IFN response in these patients. This result is consistent with preclinical findings—that demonstrated stimulation of EP4 suppressed the IFN signaling pathway in human PBMCs (online supplementary file 1). On the other hand, the expression of IDO1, another IFN response gene, decreased with E7046 treatment, suggesting additional mechanism of EP4 signaling in IDO1 gene expression other than the IFN pathway. In this context, PGE2 was shown to be a direct driver of IDO1 expression in both human tumor cells and dendritic cells.20 21 Equally interesting, expression of EOMES and PTGES4 (target of E7046) were also downregulated by E7046. EOMES has been reported to be a PGE2–EP4-regulated gene22 and plays an important role in T-cell differentiation and exhaustion.23 The dose-dependent reduced expression of EOMES by E7046 provides clinical evidence that EP4 signaling might directly regulate T-cell exhaustion in cancer patients. This hypothesis is further supported by an earlier report that EP4 was one of the few molecules that were highly and specifically upregulated in exhausted T cells from melanoma patients.24 Although the precise mechanism of the aforementioned modulations requires further investigation, altogether, these results suggest a multifaceted role of EP4 signaling blockade by E7046 in regulating antitumoral immune responses.
A notable finding from this study is the observed concurrent increase in both the serum levels of a key effector T-cell recruiting chemokine, CXCL10, and the increased infiltration of T cells in tumors. CXCL10 and TNF-α are two important effector molecules of the EP4 signaling pathway, and their expression was dose-dependently suppressed by PGE1-OH, an EP4 agonist, in vitro (online supplementary file 1). Examination of their expression in paired tumor biopsies showed upregulated expression of CXCL10 and TNF-α on E7046 treatment, although without reaching statistical significance. When an individual case was examined (ie, the only patient demonstrating a partial metabolic response (31%) of FDG-PET signal reduction where all measured PD biomarker data were available), we found concurrent increases in the percentage (31%) of CD8+ T cells and the expression (~20%) of CXCL10 in the tumor, as well as an increase (53%) in circulating CXCL10 protein. Additionally, there appeared to be an increase in CD163 staining postbaseline, which suggests an increased accumulation of type 2 macrophages. However, this increase was accompanied by enhanced infiltration, and total macrophage staining could not be quantified. Therefore, further investigation is warranted to determine whether the increased CD163 staining was a direct effect or an indirect feedback response to enhanced T-cell infiltration following E7046 treatment.
Although the data set is small, these clinical biomarker results support the proposed mechanism of action that intratumoral blocking of EP4 signaling by E7046 promotes T-cell recruitment by releasing the PGE2-induced suppression of CXCL10. In addition, higher baseline infiltration of T cells and type 2 macrophages was found to be associated with stable disease versus disease progression in E7046-treated patients. Thus, baseline levels of these immune cell populations may have a role as predictive biomarkers, and their correlation with clinical response warrants further investigation.
Tumor metabolism was explored as a potential early response biomarker in the study. This was based on preclinical studies, in which treatment of 4T1 breast and Pan02 pancreatic syngeneic tumor models with E7046 decreased the FDG-PET signal compared with vehicle treatment. This reduction was associated with antitumor activity (data not shown). In this study, the three patients who achieved partial metabolic responses measured by FDG-PET had the longest duration of treatment (145–208 days) among all the patients, suggesting a potential relationship between metabolic response of E7046 and clinical benefit. Additionally, analysis of the duration of treatment between E7046 and most recent prior anticancer therapy revealed that 10 patients (33%) had a longer duration of treatment on E7046. Typically, as patients with cancer progress through various lines of therapy, the duration of therapy becomes shorter with subsequent treatments as the cancer generally advances. Therefore, a longer duration of therapy compared with previous treatments may potentially indicate that the current treatment is benefiting the patient. Of note, a similar type of analysis has been used previously.25
Several myeloid cell-targeting agents are currently under clinical investigation including CSF-1R antagonists.26–28 CSF-1R is expressed on tumor-associated cells, including TAM and MDSCs. The intratumoral presence of CSF-1R+ macrophages correlated with poor survival rates in various cancers.26 Unlike modulation of the differentiation of infiltrated myeloid cells by E7046, the CSF-1R kinase inhibitor, JNJ-28312141, substantially decreased macrophage infiltration in preclinical models by reducing their survival.28 In a phase 1/2 study, the CSF-1R inhibitor JNJ-40346527 demonstrated clinical activity in patients with relapsed or refractory classical Hodgkin’s lymphoma, with an objective response rate of 5% (1/21 patients had complete response), and 55% (11/21) of patients achieved stable disease.27 These data suggest that modulation of the tumorous myeloid cells through either inducing cell differentiation or reducing cell survival may translate to clinical anticancer activity. However, the full anticancer potential of these myeloid cell modifying agents may be better realized by use in combination with other cancer therapies, such as immune checkpoint inhibitors.
Combination therapies of E7046 with radiation29 or immune checkpoint inhibitors4 have been shown to have greater efficacy than single-agent E7046 in preclinical models. For example, E7046 and anti-CTLA-4 combination therapy significantly reduced tumor volumes compared with all other treatments examined and resulted in complete response for 12.5% (1/8) of mice treated that bear highly immunosuppressive anti-PD-1-nonresponsive 4T1 breast tumors.4 Treatment with E7046 increased levels of PD-L1, a known regulator of immune responses.30 Thus, these preclinical data suggest that combination therapy of E7046 with monoclonal antibodies to the PD-1/PD-L1 pathway may also represent a potential clinical approach to control cancer progression, particularly in tumors regulated by both PD-L1 and PGE2.
Altogether, available clinical and preclinical data suggest that E7046 has the potential for further clinical investigation in combination with other types of cancer therapies including checkpoint inhibitors such as PD-1/PD-L1, radiation therapy, and other anticancer agents. Two dose levels of E7046 (250 and 500 mg) are proposed to be evaluated further in the combination setting. Two of the three patients who achieved partial metabolic responses were being treated with a 250 mg dose. However, pharmacodynamic biomarkers (EOMES, CD3, PD-L1) displayed a linear response to increasing doses of E7046, suggesting that a higher dose may be beneficial. A dose of 500 mg was selected to be further evaluated since there was no additional increase in exposure observed with a 750 mg dose. The 250 mg dose may offer better tolerability in the combination setting but the 500 mg dose has the potential for higher biologic activity, based on greater T-cell accumulation in patients’ tumors at the higher dose. Additionally, should E7046 dose reduction be required, the 125 mg dose was biologically active as a single agent in this study, and may be efficacious in the combination setting.
Conclusions
In patients with advanced malignancies, treatment with E7046 was well tolerated with no DLTs observed at the four dose levels studied in this first-in-human study. Therefore, safety concerns did not appear to limit dose selection. The 250 and 500 mg doses were both observed to enhance T-cell accumulation in the tumors and achieve stable disease in some patients. Over half (4/7) of the patients with stable disease by irRECIST criteria had a treatment duration of 18 weeks or more and three patients (3/15; 20%) achieved metabolic responses. In addition, results from this study support the proposed mechanism of action of E0746 and demonstrated that higher baseline levels of T cells, along with type 2 macrophage tumor infiltration, were associated with stable disease and, therefore, warrant further investigation. A phase 1b study of E7046, at the RP2Ds of 250 and 500 mg, in combination with radiochemotherapy in patients with locally advanced rectal cancer is currently recruiting patients (NCT03152370).
Acknowledgments
Medical writing support was provided by Tarah M. Connolly, PhD, of Oxford PharmaGenesis Inc, Newtown, PA, with funding provided by Eisai Inc. Imaging analysis for FDG-PET was performed by VirtualScopics (now BioTel Research), Rochester, NY.
References
Footnotes
Twitter @AnaingMD
Contributors All authors read and approved the final manuscript. DSH, AP, GIS, AV, AN, AM, and FM-B gathered patient data; contributed to study design, data interpretation, critical review of article drafts, and approval of the final version. OA, LR, TAB, MR, ML, SD, AYS, PS, LX, VB-P, IT, CEO, XB contributed to study design, data interpretation, critical review of article drafts, and approval of the final version.
Funding This work was supported by Eisai Inc., Woodcliff Lake, NJ, USA; Eisai Inc., had a role in writing the manuscript, study design and collection, analysis, and interpretation of data; editorial assistance, provided by Tarah M. Connolly, PhD, Oxford PharmaGenesis Inc, Newtown, PA, USA, was funded by Eisai Inc.
Competing interests DSH has received research grant funding from AbbVie, Adaptimmune, Amgen, AstraZeneca, Bayer, BMS, Daiichi-Sankyo, Eisai, Fate Therapeutics, Genentech, Genmab, Ignyta, Infinity, Kite, Kyowa, Lilly, LOXO, Merck, MedImmune, Mirati, MiRNA, Molecular Templates, Mologen, NCI-CTEP, Novartis, Pfizer, Seattle Genetics, and Takeda; DSH received travel, accommodations, and expenses from LOXO and MiRNA; DSH held a consulting or advisory role for the following: Alpha Insights, Axiom, Adaptimmune, Baxter, Bayer (advisory boards and Speakers’ Bureaux), Genentech, GLG, Group H, Guidepoint Global, Infinity, Janssen, Merrimack, Medscape, Numab, Pfizer, Seattle Genetics, Takeda, and Trieza Therapeutics. DSH discloses the following other ownership interests: Molecular Match (Advisor), OncoResponse (founder), and Presagia (Advisor). AN has received research funding from NCI, EMD Serono, MedImmune, Healios Onc. Nutrition, Atterocor, Amplimmune, ARMO BioSciences, Eli Lilly Karyopharm Therapeutics, Incyte, Novartis, Regeneron, Merck, BMS, Pfizer, CytomX Therapeutics, Neon Therapeutics, Calithera Biosciences, TopAlliance Biosciences, Kymab, PsiOxus, and the Immune Deficiency Foundation (spouse); AN has served on an advisory board for CytomX Therapeutics and Novartis; AN has received travel and accommodation expenses paid for by ARMO BioSciences. GIS has received research funding from EL, Merck KGaA/EMD-Serono, Merck, and Sierra Oncology. GIS has served on advisory boards for Pfizer, EL, G1 Therapeutics, Roche, Merck KGaA/EMD-Serono, Sierra Oncology, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Astex, Almac, Ipsen, Bayer, Angiex, and Daiichi Sankyo. The Dana-Farber Cancer Institute has received funding from Pfizer and Array BioPharma for the conduct of investigator-initiated clinical trials of palbociclib led by GIS. GIS holds Patent 9872874, entitled, “Dosage regimen for sapacitabine and seliciclib,” and also has a pending patent related to his work on CDK4/6 inhibition entitled, ‘‘Compositions and Methods for Predicting Response and Resistance to CDK4/6 Inhibition.’’ FMB has no conflicts of interest to disclose. AP is a consultant/advisory board member for Puretech, Driver, Foundation Medicine, and Eisai; AP has institutional research funding from Array, Plexxikon, Guardant, BMS, MacroGenics, Genentech, Novartis, OncoMed, and Tolero; AP has received travel support from Eisai. AM has received honoraria and consulting fees from Eisai. XB is an employee of H3 Biomedicine, a subsidiary of Eisai. LR, TAB, MR, SD, PS, LX, IT, ViB-P, and CEO are employees of Eisai or Eisai Ltd. ML, ÖA, and AYS were employees of Eisai at the time the study was conducted.
Patient consent for publication Not required.
Ethics approval The study was approved by each Institutional Review Board or Independent Ethics Committee in accordance with International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice, and Federal Regulations. In addition, the study adhered to guidelines set forth in the Declaration of Helsinki. Informed consent was obtained from all patients in the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. The data will not be available for sharing at this time as the data are commercially confidential. However, Eisai will consider written requests to share the data on a case-by-case basis.