Article Text

Abstract
Objectives Non-alcoholic fatty liver disease (NAFLD) can persist in the stage of simple hepatic steatosis or progress to steatohepatitis (NASH) with an increased risk for cirrhosis and cancer. We examined the mechanisms controlling the progression to severe NASH in order to develop future treatment strategies for this disease.
Design NFATc1 activation and regulation was examined in livers from patients with NAFLD, cultured and primary hepatocytes and in transgenic mice with differential hepatocyte-specific expression of the transcription factor (Alb-cre, NFATc1c.a . and NFATc1Δ/Δ ). Animals were fed with high-fat western diet (WD) alone or in combination with tauroursodeoxycholic acid (TUDCA), a candidate drug for NAFLD treatment. NFATc1-dependent ER stress-responses, NLRP3 inflammasome activation and disease progression were assessed both in vitro and in vivo.
Results NFATc1 expression was weak in healthy livers but strongly induced in advanced NAFLD stages, where it correlates with liver enzyme values as well as hepatic inflammation and fibrosis. Moreover, high-fat WD increased NFATc1 expression, nuclear localisation and activation to promote NAFLD progression, whereas hepatocyte-specific depletion of the transcription factor can prevent mice from disease acceleration. Mechanistically, NFATc1 drives liver cell damage and inflammation through ER stress sensing and activation of the PERK-CHOP unfolded protein response (UPR). Finally, NFATc1-induced disease progression towards NASH can be blocked by TUDCA administration.
Conclusion NFATc1 stimulates NAFLD progression through chronic ER stress sensing and subsequent activation of terminal UPR signalling in hepatocytes. Interfering with ER stress-responses, for example, by TUDCA, protects fatty livers from progression towards manifest NASH.
- nonalcoholic steatohepatitis
- fatty liver
- hepatic fibrosis
- inflammation
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Non-alcoholic fatty liver disease (NAFLD) is a major cause of chronic liver disease.
Exposure to lipotoxic fatty acids can cause endoplasmic reticulum (ER) stress in hepatocytes.
Chronic unresolved ER stress drives NAFLD progression.
What are the new findings?
NFATc1 is highly activated in advanced NAFLD.
Lipotoxic fatty acids stimulate NFATc1 expression, nuclear localisation and activation in hepatocytes.
NFATc1 activation drives liver cell damage and inflammation through chronic ER stress sensing and activation of the terminal PERK-CHOP unfolded protein response (UPR) pathway.
Inhibition of chronic ER stress responses by tauroursodeoxycholic acid (TUDCA) blocks NFATc1 mediated terminal UPR signalling and prevents NAFLD progression.
How might it impact on clinical practice in the foreseeable future?
Our findings indicate a critical role of NFATc1 in chronic ER stress responses and NAFLD progression. Targeting unrestricted ER stress alleviates NFATc1-driven cell damage and therefore, our study provides the rational for current clinical trials aiming at the potential of TUDCA treatment in NAFLD.
Introduction
Non-alcoholic liver disease (NAFLD) is emerging as the leading cause of chronic liver disease with an estimated worldwide prevalence of more than 25%.1 2 NAFLD is often associated with metabolic disorders, for example, insulin resistance, type-2 diabetes or obesity, and usually presents as simple hepatic steatosis (NAFL). However, in about 20%, the disease can progress to non-alcoholic steatohepatitis (NASH), which carries an increased risk of developing cirrhosis, liver failure and hepatocellular carcinoma.3 4 In recent years, enormous efforts have been made to develop drug strategies for the treatment of NAFLD, but until now there are no approved drugs for the prevention or therapy of NASH in clinical use.
The lack of effective treatment is largely due to a still incomplete understanding of the molecular mechanisms responsible for the development and progression of the disease. Excessive accumulation of fatty acids and lipotoxic metabolites in hepatocytes is considered a key event in the initiation of NAFLD.5–7 It has been shown, for example, that sustained exposure to lipotoxic fatty acids can cause hepatocyte cell damage via induction of chronic endoplasmic reticulum (ER) stress and subsequent activation of the terminal unfolded protein response (UPR) pathway. UPR in turn activates the NLRP3 multiprotein inflammasome complex to foster hepatocyte cell death and inflammation in response to unresolved ER stress.7–10 Accordingly, high expression of NLRP3 inflammasome markers (NLRP3, Caspase-1, Caspase-11, interleukin (IL)-1β and IL-18) are found in liver biopsies from patients with NASH.11 12 However, the endogenous pathways responsible for chronic ER stress sensing and NLRP3 activation in fatty liver remain largely unknown.
Nuclear factor of activated T cells (NFAT) proteins comprises a family of calcium responsive transcription factors (NFATc1-NFATc4) involved in the regulation of adaptive cellular functions within and outside the immune system.12 13 Activation of NFAT proteins result in response to extracellular stimuli and in adaptation to multiple cellular stress signals. Moreover, ectopic activation of individual family members has been described in metabolic (eg, diabetes mellitus), inflammatory (eg, psoriasis) and malignant diseases (eg, pancreatic cancer and melanoma).12 14–21 When inactive, NFAT proteins reside in the cytosol in a hyperphosphorylated state. Following accumulation of cytosolic calcium, NFAT proteins become dephosphorylated by the phosphatase calcineurin and shuttle into the nucleus, where they regulate target gene signatures in concert with other site-specific transcription factors and chromatin remodelling proteins.12 Some recent investigations have reported higher NFAT expression and activation levels in chronic hepatitis and in liver cancer as well. It has been shown, for instance, that NFATc4 promotes PPARα expression in hepatocytes and stimulates liver fibrosis through activation of hepatic stellate cells.22–24
Here, we describe for the first time a causal role of NFATc1 in NAFLD progression. We show that NFATc1 fosters chronic ER stress sensing and cell damage responses via the NLRP3 inflammasome pathway. Furthermore, we provide experimental evidence that pharmacological interference with terminal ER stress responses may oppose NFATc1-driven progression of NAFLD.
Materials and methods
Human samples
Formalin-fixed, paraffin-embedded human tissues identified from patients with NASH were retrieved from the biobank of the Institute of Pathology, University Medical Center, Goettingen. Normal liver sections from patients with early steatosis were used as controls. All samples were used for immunohistochemical analysis of NFATc1 and analysed using imageJ software. Briefly, percentage of nuclear NFATc1 positive hepatocytes in 10 images per sample (Scale bar=50 µm) was calculated manually. NAFLD activity score (NAS) and degree of fibrosis were determined as described before.25 NFATc1 expression in patients with NASH was correlated with the NAS, the degree of fibrosis and serum levels of liver enzymes (ALT and AST) by simple linear regression.
Animal model
All animal experiments were approved and carried out according to the regulations of Federation of European Laboratory Animal Science Associations (Laves approval No. 33.9-42502-04-16/2189 and 33.9-42502-04-14/1633). Alb-Cre, NFATc1 and NFATc1Δ/Δ mice have been described previously.19 26–28 The c.a.NFATc1 knock-in strain was generated by cloning an N-terminal hemagglutinin (HA)-tagged constitutively active version of NFATc1 containing serine to alanine substitutions in the conserved serine-rich domain and all three serine-proline repeats into the ROSA26 promoter locus (Artemis Pharmaceuticals). The strains were interbred to generate Alb-Cre;NFATc1Δ/Δ and Alb-Cre;NFATc1c.a . (hereafter referred to as NFATc1Δ/Δ and NFATc1c.a ., respectively) mice. All mice were genotyped by PCR as explained before.19
RNA-Seq
Total RNA was isolated from AML12 cells transfected with either HA-tagged c.n.NFATc1 or control plasmid. Purity and integrity of RNA samples were validated by gel electrophoresis. TruSeq RNA Library Prep Kits (RS-122-2001 and RS-122-2002) from Illumina were used to prepare libraries following instructions from manufacturer and sequencing was performed by NGS Integrative Genomics Core Unit, University Medical Center Goettingen, Germany.
Obtained FastQ files were analysed by using usegalaxy.eu,29 FastQ files were analysed for quality control followed by mapping to murine transcriptome (mm9) using TopHat tool (V.2.1.1),30 with very sensitive bowtie2 settings, followed by HTSeq (V.0.9.1; -f bam -r pos -s reverse -a 10 -t exon -m union).31 Differential gene expression analysis was performed with DESeq2 (V.2.11.40.6),32 as well as principal component analysis (PCA). Heat maps were generated in GraphPad prims V.7.0. NFATc1-dependent differentially expressed genes (log2fold values≥0.5/≤−0.5; p≤0.05; base mean>10) were analysed in reactome (reactome.org) pathway database.
Detailed description of animal treatments and further materials and methods is included in the online supplemental information.
Supplemental material
Results
Nuclear NFATc1 activation in progressive human and murine NAFLD
To assess NFATc1 expression and activation in NAFLD progression, we performed immunohistochemical analyses in tissue samples from patients with healthy liver (n=8) and progressive NAFLD (n=46). These studies revealed absent or weak NFATc1 expression in healthy livers and a robust induction of nuclear NFATc1 expression in liver biopsies of patients with progressive disease, characterised by macrovesicular steatosis and pronounced lobular inflammation (figure 1A,B). Consistent with a role of NFATc1 in disease acceleration, we observed a significant correlation of intrahepatic NFATc1 expression with NAFLD progression, determined by the NAS, the degree of fibrosis and the levels of liver enzymes in blood samples from patients with NASH (figure 1C–F). Prompted by these findings, we expanded our analysis on NFATc1 activation in NAFLD progression and studied NFATc1 induction and nuclear localisation in a mouse model of progressive NAFLD. For this purpose, we fed mice with a high-fat western diet (WD) for 20 weeks (figure 2A). This model is an excellent tool for studying the incremental steps of NAFLD progression.33 In line with previous reports, WD treated mice developed hepatic steatosis with ballooned hepatocytes and a mild-to-moderate form of lobular inflammation with significant recruitment of CD45-positive immune cells and marked fibrosis (figure 2B–D). Moreover, and consistent with our findings in progressive human NAFLD, immunohistochemistry and western blot analysis confirmed a strong induction and nuclear accumulation of NFATc1 in hepatocytes of NASH livers (figure 2E,F). Taken together, these studies performed in progressive human and murine NAFLD provided first experimental evidence for a role of NFATc1 activation in disease acceleration.
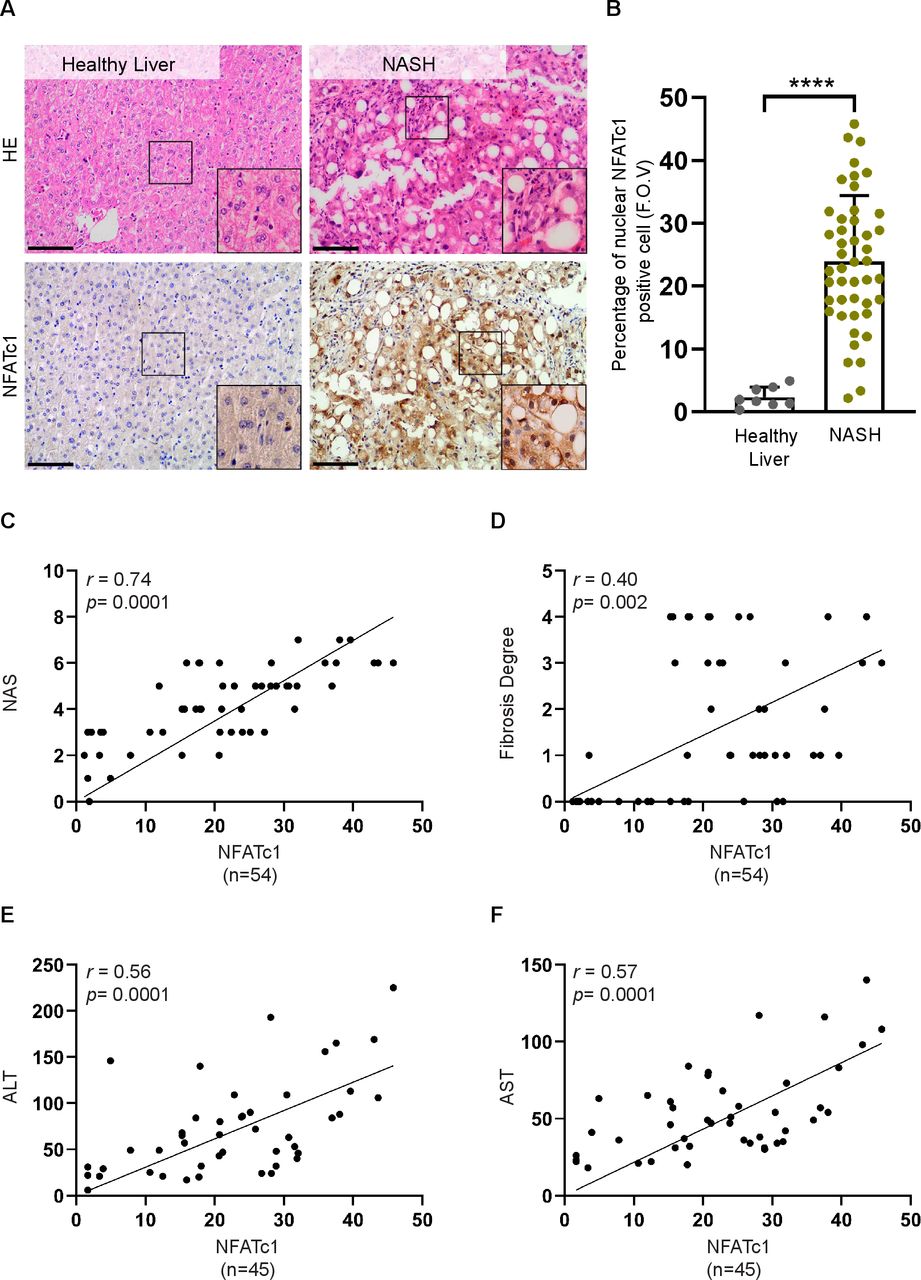
Hepatocyte-specific NFATc1 activation in progressive non-alcoholic liver disease (NAFLD). (A) Sections of healthy human liver (n=8) and non-alcoholic steatohepatitis (NASH) (n=46) were analysed by H&E staining and immunohistochemistry for NFATc1. Representative images are shown, scale bar=50 µm. (B) Percentage of nuclear NFATc1-positive hepatocytes per field of view in samples from healthy liver and NASH. Statistical analysis was performed by unpaired t-test. Data are shown as mean±SD, ****p≤0.0001. Simple linear regression analysis revealed a significant correlation of hepatic NFATc1 expression levels with (A) NAS (NAFLD activity score), (B) the degree of fibrosis, (C) ALT levels and (D) AST levels.
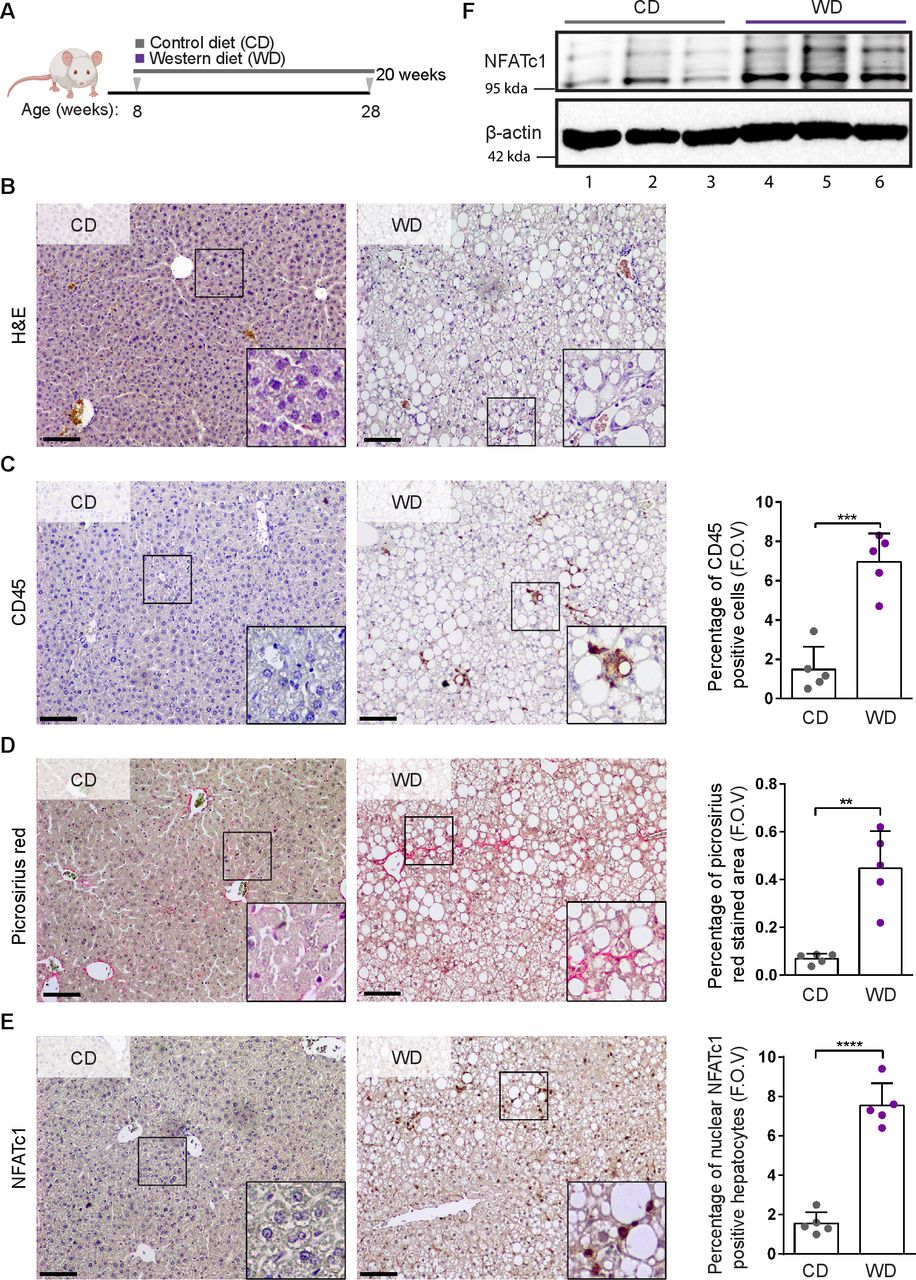
NFATc1 induction and non-alcoholic fatty liver disease (NAFLD) progression in western diet (WD) fed mice. (A) Schematic representation of the diet feeding protocol. Eight weeks old C57BL/6 wild-type mice were treated with either control diet (CD) or WD for 20 weeks. (B) Mice were sacrificed, and liver sections were analysed by H&E staining and immunohistochemical analysis for (C) CD45, (D) picrosirius red and (E) NFATc1 expression. Representative results are shown. Scale bars=100 µm. Quantification analyses were performed, and results were illustrated as percentage of CD45-positive cells, percentage of picrosirius red stained area and the percentage of nuclear NFATc1-positive hepatocytes in livers sections obtained from CD-treated and WD-treated mice (n=5). Statistical analysis was performed by unpaired t-test. Data are shown in mean±SD, **p<0.005, ***p<0.0005, ****p<0.0001. (F) Representative western blot of NFATc1 expression in liver tissue lysates of 20 weeks old mice treated with either CD (n=3, lane 1–3) or WD (n=3, lane 4–6). Each lane represents liver lysates from individual mice. WD-treated mice express high levels of active NFATc1, indicated by strong increase of the lower band.
NFATc1 activation drives fatty acids-induced steatohepatitis (NASH) and fibrosis
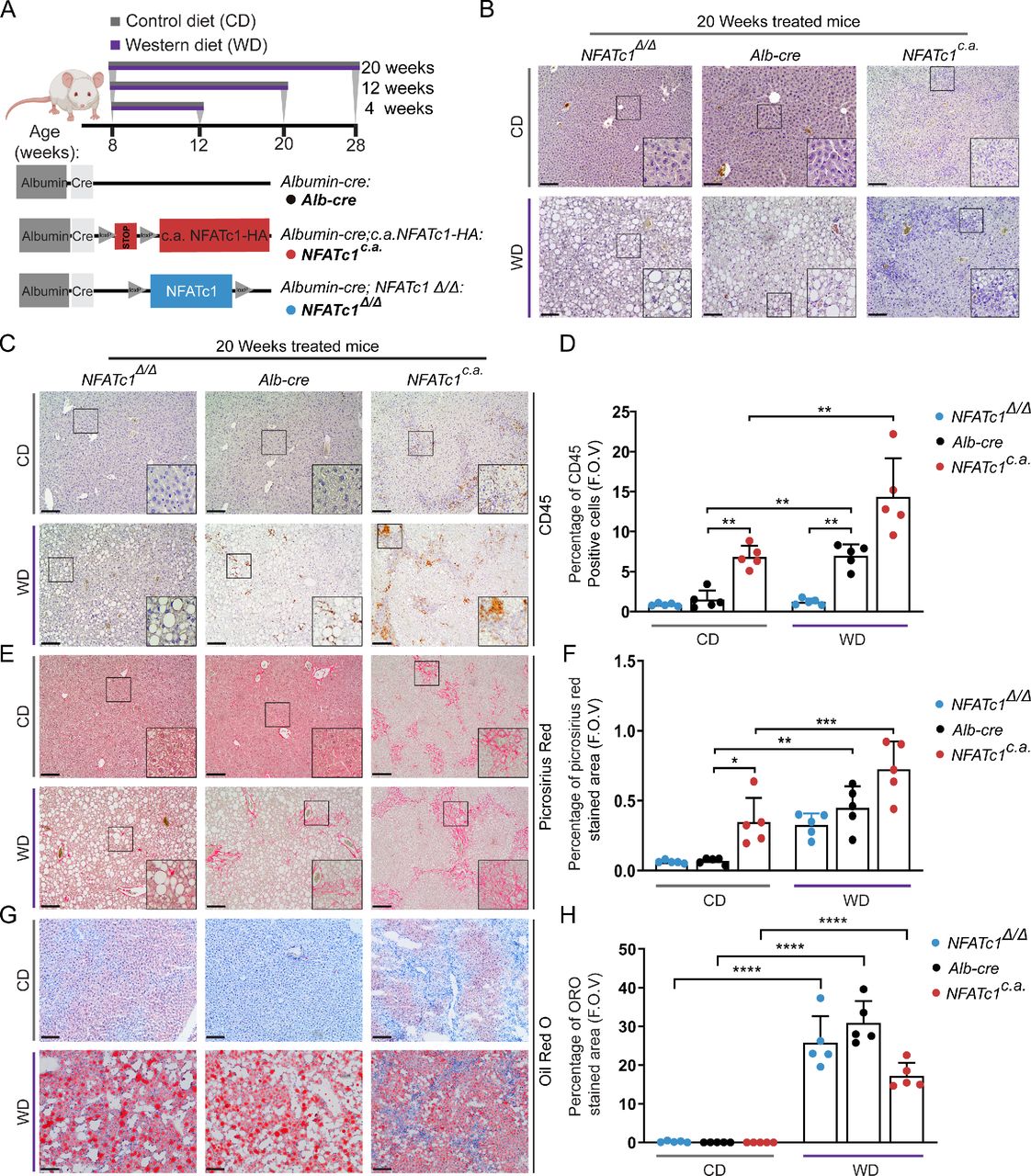
To study whether nuclear NFATc1 activation is indeed causally involved in the initiation and progression of NAFLD, we generated genetically modified mice with either hepatocyte-specific nuclear activation (Alb-Cre; NFATc1c.a . (NFATc1c.a .)) or depletion (Alb-Cre; NFATc1Δ/Δ (NFATc1Δ/Δ )) of NFATc1. Animals with endogenous NFATc1 expression (Alb-Cre) were used as controls. Eight-weeks old mice of all genotypes were fed with control diet (CD) or WD for defined treatment periods up to 20 weeks (figure 3A). As expected, treatment of Alb-Cre control mice with WD resulted in (1) increased liver and body weight (online supplemental figure 1A–C), (2) progressive steatosis with accumulation of intrahepatic triglycerides (figure 3B, (online supplemental figure 1D) and (3) an increasing inflammatory response with recruitment of CD45-positive immune cells and pronounced collagen deposition (figure 3B–H). Importantly, however, a comparable increase in inflammation, CD45-positive cell recruitment and collagen deposition was observed (figure 3B–F) in transgenic mice with hepatocyte-specific activation of nuclear NFATc1 (NFATc1c.a .), and this damage again increased on feeding with high-fat WD. Importantly and consistent with our observations in patients with NASH, we also measured elevated serum ALT levels in mice with NFATc1c.a. driven NAFLD progression (online supplemental figure 1E). In contrast, disease progression was greatly reduced in mice with hepatocyte-specific NFATc1 deficiency (NFATc1Δ/Δ ) and hence, NFATc1 depletion reduced the extent of liver fibrosis and inflammation and almost completely abolished the recruitment of CD45-positive cells (figure 3B–F). Of note, we could not observe NFATc1 addiction in the development of hepatic steatosis, such that neither ectopic expression nor genetic deletion of the transcription factor had a significant effect on the extent of hepatic fat accumulation after WD (figure 3G,H, online supplemental figure 1D,F). Collectively, these findings substantiate a causal role of NFATc1 in the progression of NAFLD and show that activation of the transcription factor in liver cells, for example, as a consequence of high-fat diet, results in pronounced inflammation and fibrosis.
Supplemental material

NFATc1 activation in hepatocytes drives liver inflammation and fibrosis. (A) Schematic depiction of genetically modified mice with hepatocyte-specific NFATc1 expression (Alb-Cre; NFATc1c.a . (NFATc1c.a .) or deletion Alb-Cre; NFATc1Δ/Δ (NFATc1Δ/Δ )) along with the feeding schedule for 4, 12 and 20 weeks, respectively. (B) H&E analysis of liver sections from CD (left) and WD (right) treated mice are shown (n=5). Scale bar=100 µm. Representative images of immunohistochemical analysis and quantification for (C–D) CD45, (E–F) picrosirius red staining and (G–H) oil-red-o staining in the livers of 20-week CD-treated and WD-treated NFATc1Δ/Δ , Alb-Cre and NFATc1c.a . mice (n=5). Statistical analysis was performed by two-way analysis of variance and data are shown as mean±SD where p values are *p<0.05, **p<0.005, ***p<0.0005.
Lipotoxic fatty acids induce NFATc1 signaling activation in hepatocytes
Free fatty acids (FFAs) are main contributors to the intrahepatic triglyceride pool and recent studies suggest that lipotoxic FFAs play a particularly important role in the development and progression of NAFLD. This applies in particular to the saturated FFA palmitate, which has a pronounced cell-damaging effect on liver cells and is found in particularly high concentrations in the blood of patients with NASH.34 To directly test whether exposure to palmitate induces NFATc1 expression in liver cells, we treated primary hepatocytes and AML12 cells with increasing concentrations of the highly lipotoxic fatty acid (online supplemental figure 2A and figure 4A–G). In fact, treatment with palmitate led to a remarkable and dose-dependent increase of NFATc1 expression in both cell models and on mRNA and protein levels as well (online supplemental figure 2A and figure 4A–D). By contrast, incubation with oleate, a non-lipotoxic FFA failed to induce NFATc1 expression in primary hepatocytes even at high doses and despite significant accumulation of liver cell steatosis, as evidenced by Oil-Red-O staining (online supplemental figure 2A,B). Palmitate and oleate are among the most abundant FFAs, accounting for more than half of total plasma FFAs, and it has been shown that the cell toxic effects of palmitate can be counteracted by coexposure to oleate. In line with this, coadministration of non-lipotoxic oleate in liver cells inhibited NFATc1 induction by palmitate, supporting the idea that induction of the Ca2+-regulated transcription factor in NAFLD progression is primarily a consequence of FFA-induced lipotoxicity—rather than fat accumulation per se. A close connection between lipotoxicity-induced cell stress and impaired Ca2+ signalling homeostasis has already been demonstrated in liver cells. Here, we extend these findings and show that exposure to palmitate initiates a dose-dependent shift of Ca2+ from ER stores to the cytosol of fatty liver cells (online supplemental figure 2C–E). A rise in cytosolic Ca2+ causes activation of various cell-type dependent stress response pathways,35 most notably the NFATc1 signalling and transcription factor. In fact, palmitate-induced accumulation of cytosolic Ca2+ is paralleled by robust NFATc1 activation in hepatocytes, evidenced by a particularly strong increase of the lower band that reflects the hypophosphorylated active status of the transcription factor (figure 4C,D, online supplemental figure 2A). Accordingly, reporter gene assays and immunofluorescence staining confirmed nuclear accumulation and increased transcriptional activity of NFATc1 in response to palmitate treatment in both cell lines (figure 4E–G; online supplemental figure 2F).
Supplemental material
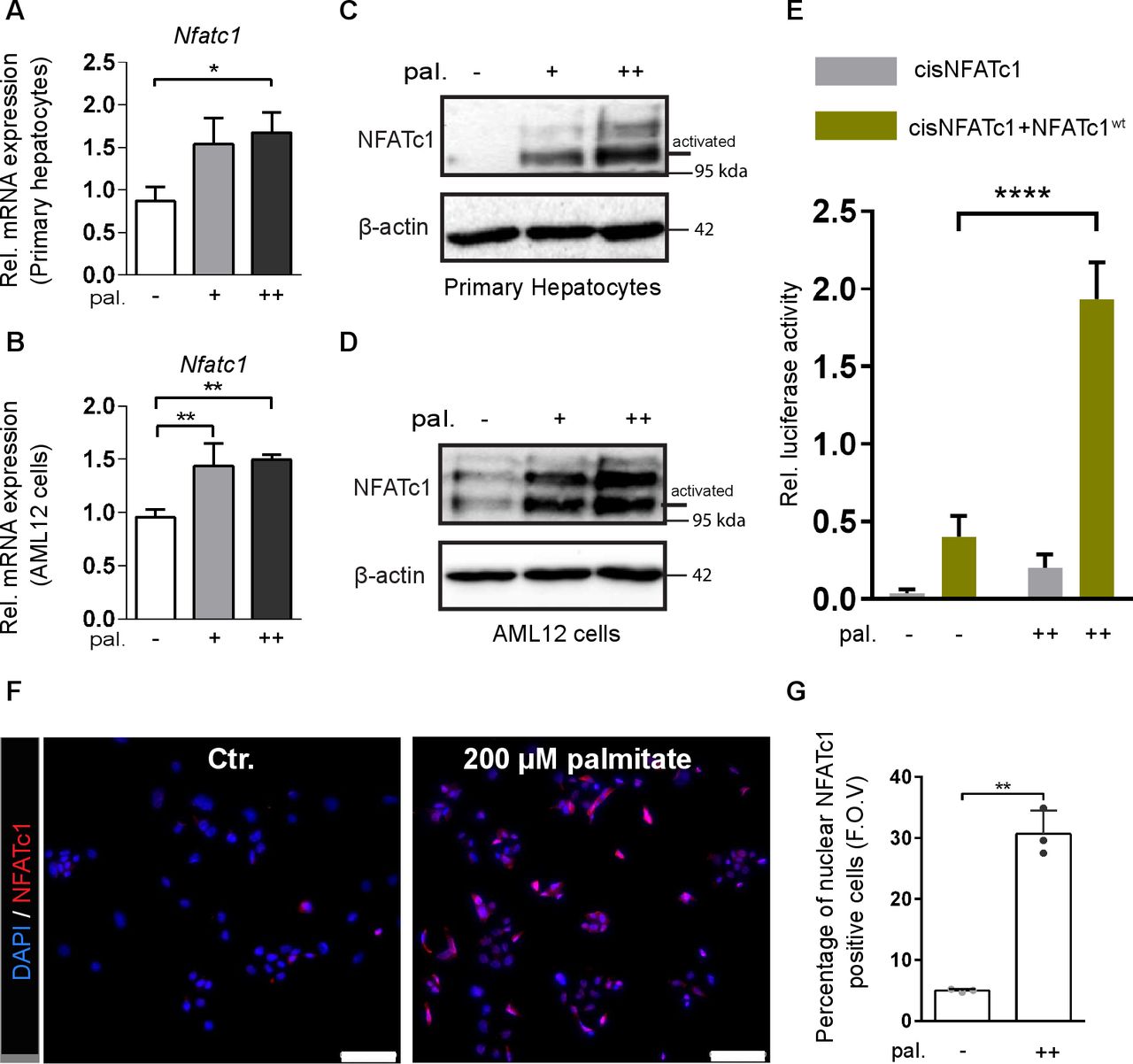
Lipotoxic fatty acids cause NFATc1 activation. (A–B) Expression of NFATc1 mRNA in (A) primary mouse hepatocytes and (B) AML12 cells following treatment with 100 µM (+) and 200 µM (++) palmitate (pal.) for 12 hours. NFATc1 gene expression was analysed by qRT-PCR and is shown as ‘relative mRNA levels’ compared with untreated control. (C–D) Induction of NFATc1 protein expression in (C) primary mouse hepatocytes and (D) AML12 cells treated with 100 µM (+) and 200 µM (++) palmitate (pal.) for 12 hours. The lower band represents the active state of NFATc1. (E) Dual luciferase reporter gene assay was performed in hepatocytes from Alb-cre mice to verify palmitate induced transcriptional activation of NFATc1. Cells were cotransfected with an NFAT responsive promoter luciferase reporter construct in combination with either an empty vector or NFATc1 wild-type (NFATc1wt) expression vector, and subsequently treated with 200 µM palmitate (pal.) for 24 hours. (F) NFATc1 immunofluorescence in AML12 cells demonstrating nuclear translocation of the transcription factor following treatment with 200 µM palmitate for 12 hours. Scale bar=100 µm. (G) Quantitative analysis of palmitate-induced nuclear NFATc1 localisation in AML12 cells. Statistical analysis was performed by one-way analysis of variance (ANOVA) (A,B), two-way ANOVA (E) and by unpaired t-test (G). Data are shown as mean±SD, *p≤0.05, **p≤0.005 and ****p≤0.0001.
NFATc1 promotes ER stress-induced UPR and immune signaling in liver
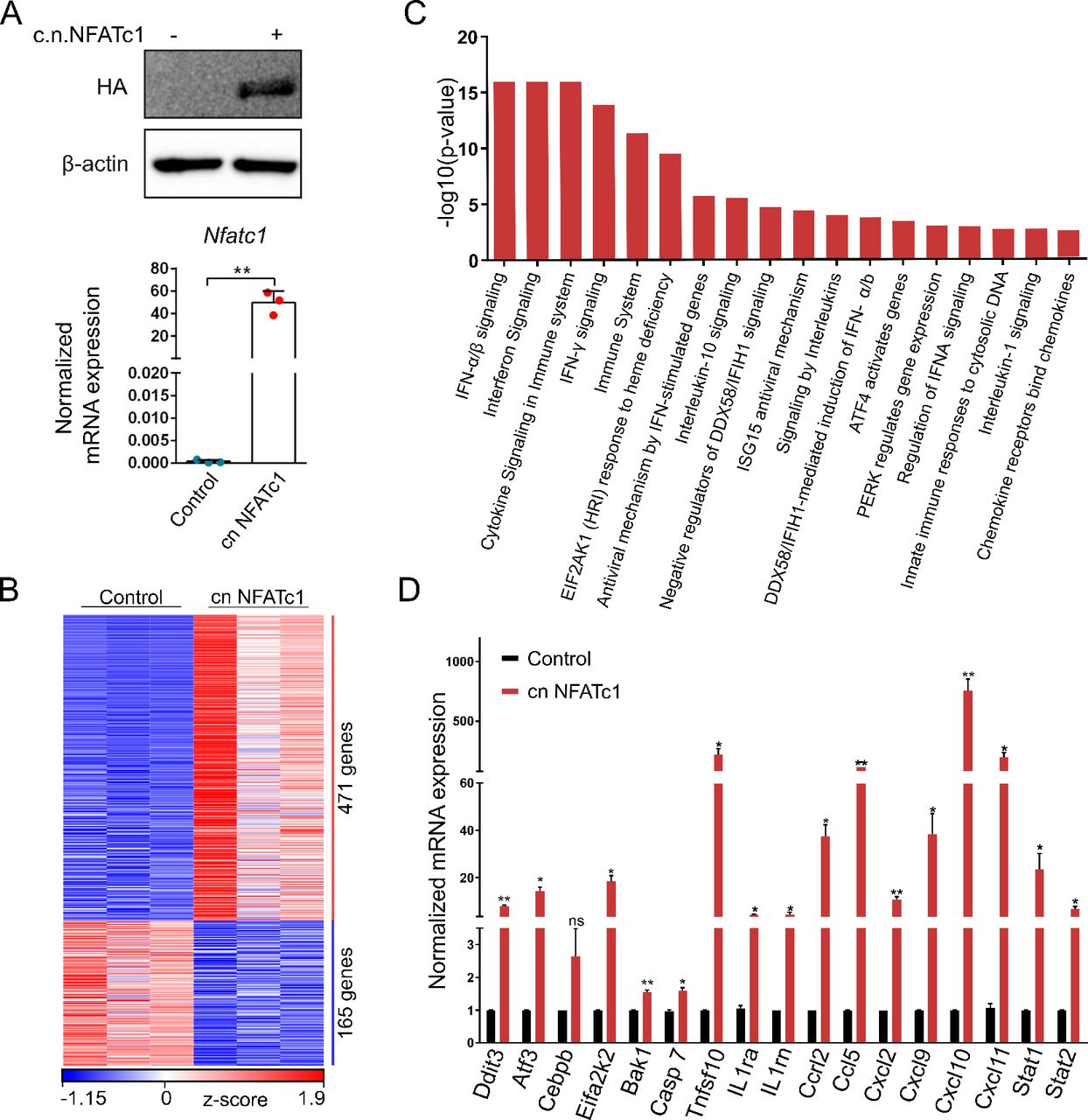
To explore the underlying mechanisms by which NFATc1 regulates NAFLD progression, we next studied NFATc1-dependent gene expression and performed transcriptome analysis in AML12 cells. For this purpose, we extracted mRNA from (HA-tagged) c.n.NFATc1 or control plasmid transfected AML12 cells and carried out RNA-Seq analysis. For transfection control, we performed western blot and qRT-PCR analysis (figure 5A). PCA demonstrated distinct clustering of NFATc1-induced and control profiles (online supplemental figure 3A). RNA-Seq analysis revealed 636 differentially expressed genes, of which 471 genes were upregulated following nuclear activation of NFATc1 (figure 5B). Moreover, reactome pathway analysis further demonstrated that the most significantly enriched pathways are involved in either interferon and cytokine/chemokine signalling, cell death regulation or cell stress responses (figure 5C). Specifically, NFATc1 activation led to a highly significant and reproducible induction of proinflammatory cytokines (eg, Ccrl2 and Ccl5) and chemokines (eg, Cxcl2, Cxcl9 Cxcl10, Cxcl11), inflammatory transcription factors (eg, Stat1 and Stat2) and cell death marker genes (eg, Bak1, Casp7 and Tnfsf10) (figure 5D). Noteworthy, we confirmed NFATc1-dependent regulation of the identified inflammatory cytokines and chemokines, for example, IL-1β, Ccl5, Cxcl9, Cxcl10 and Cxcl11 in the murine NAFLD progression model (online supplemental figure 3B,C).
Supplemental material

NFATc1 regulated gene signatures and signalling mechanisms. (A) Representative western blot and qRT-PCR showing successful NFATc1 transfection and expression in AML12 cells. (B) Heatmap depicting z-scores of significantly differentially expressed genes in RNA-Seq analysis on NFATc1 overexpression in AML12 cells. (C) Reactome pathway classification analysis demonstrating the most significantly regulated NFATc1 gene signatures in AML12 cells identified by RNA-Seq (log2fold values≥0.5/≤−0.5; p≤0.05; base mean>10). (D) qRT-PCR validation of differentially regulated candidate genes on NFATc1 activation in AML12 cells. Data are shown in mean±SD, p values are *p<0.05, **p<0.005. Statistical analysis was performed by unpaired t-test.
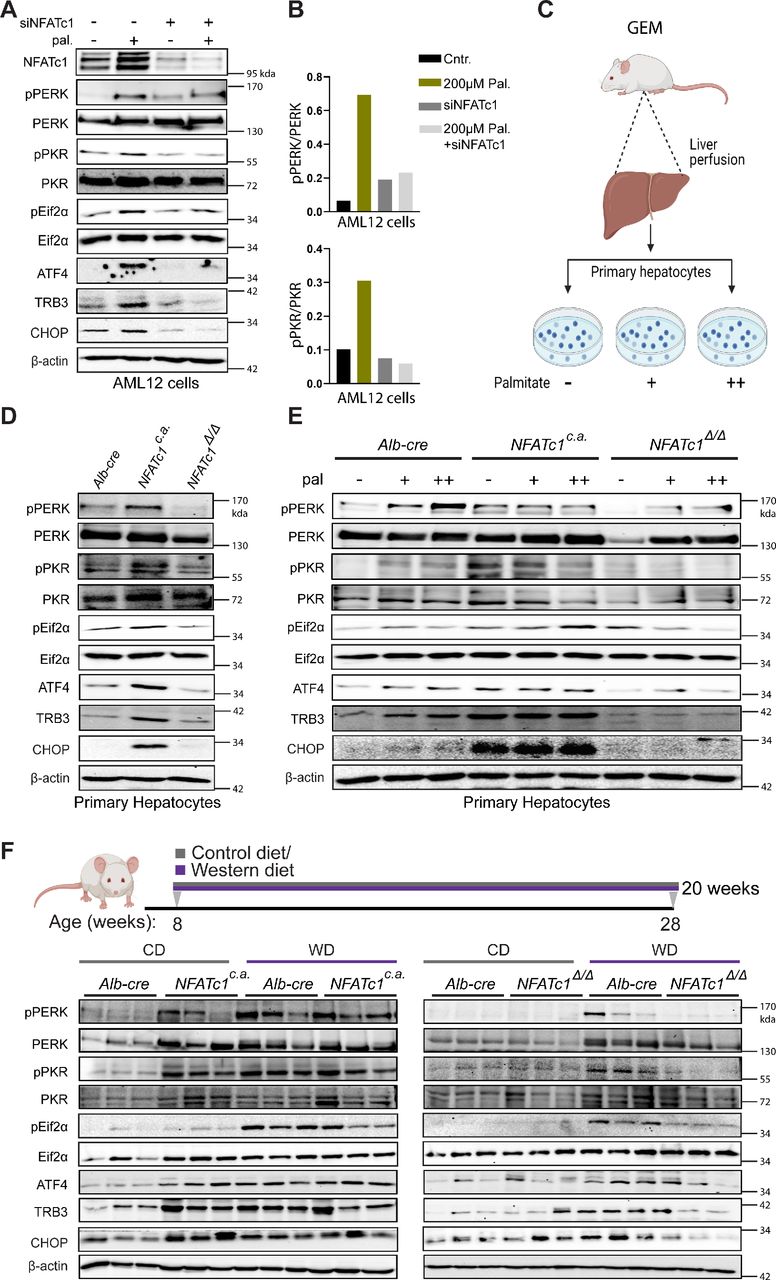
Most importantly, our transcriptome analysis also uncovered a strong and highly reproducible link between NFATc1 activation and induction of ER stress pathways, particularly the terminal PERK (protein-kinase RNA-like ER kinase) UPR. In fact, we found a significant (20-fold) induction of various signalling components, for example, Eif2ak2, Atf3 and Ddit3 (Chop) on NFATc1 activation (figure 5C,D). The UPR can be induced by activation of the three canonical ER-resident stress sensor proteins ATF6, IRE1 and PERK following perturbation of protein homeostasis in the ER lumen. On activation, UPR signalling controls multiple cell mechanisms to reduce protein synthesis and increase the protein folding capacity. However, while physiological UPR signalling allows cells to maintain cellular homeostasis, excessive UPR activation can lead to pathological changes, such as cell damage and death. It has been shown, for instance, that chronic ER stress sensing and UPR signalling can result from lipotoxic cell damage (eg, palmitate) and subsequently triggers cell death and inflammation in NAFLD progression.36–38 This is particularly true for PERK kinase-driven signalling through the Eif2α-ATF4-CHOP pathway.36 38 39 High levels of PERK and CHOP, for instance, are found in patients with NASH and specifically in those patients with a deleterious course of the disease.40 Here, we show that NFATc1 activation indeed induces PERK kinase signalling but also phosphorylation of PKR, another member of the eif2α kinase family, involved in cellular stress responses and UPR signalling (figure 6). In detail, NFATc1 activation—either genetically or following stimulation by palmitate—caused increased PERK (pPERK), PKR (pPKR) and Eif2α (pEif2α) phosphorylation and subsequent induction of the core downstream signalling components, for example, CHOP, both in cultured AML12 cells and primary hepatocytes (online supplemental figure 4A, figure 6A–E). Similar results were found in WD-treated mice (figure 6F).
Supplemental material

Nuclear NFATc1 promotes terminal unfolded protein response (UPR) signalling. (A) Immunoblot examination showing protein levels of NFATc1, pPERK, PERK, pPKR, PKR, ATF4, TRB3, p-Eif2α, Eif2α and CHOP in AML12 cells following 12 hours of palmitate (+=200 µM) exposure either alone or in combination with knock-down for NFATc1 (siNFATc1). (B) Densitometry graphs for pPERK/PERK and pPKR/PKR in AML12 cells treated with palmitate (200 µM) alone or in combination with siNFATc1. (C) Schematic illustration of primary hepatocytes isolation from transgenic mice with differential NFATc1 expression and subsequent palmitate treatment. (D) Representative western blot showing NFATc1-dependent protein levels of pPERK, PERK, pPKR, PKR, ATF4, TRB3, p-Eif2α, Eif2α and CHOP in primary hepatocytes. (E) Primary mouse hepatocytes were exposed to palmitate (+=100 µM and ++=200 µM) for 12 hours and alterations in pPERK, PERK, pPKR, PKR, ATF4, TRB3, p-Eif2α, Eif2α and CHOP levels were analysed by immunoblot. (F) Western blot analysis were conducted using liver tissue lysates from CD-treated and WD-treated genetically modified mice (GEM) models to determine fat-induced and NFATc1-dependent expression of pPERK, PERK, pPKR, PKR, ATF4, TRB3,p-Eif2α, Eif2α and CHOP.
Moreover, terminal UPR responses were strongly impaired following NFATc1 silencing (figure 6A–E, (online supplemental figure 4B–D) in vitro and in the murine NAFLD progression model, even on prolonged exposure to WD (figure 6F). Of note, NFATc1 activation appears not to be relevant for the activation of the other two ER stress sensors (IRE1/XBP1 and ATF6) in liver cells and accordingly we could not observe NFATc1-dependent expression differences (online supplemental figure 4E,F) on palmitate treatment. This observation is also in line with previous reports demonstrating that although IRE1/XBP1 and ATF6 pathways can also up-regulate CHOP, the PERK pathway predominates in NAFLD progression through selective upregulation of ATF4 translation, which subsequently induces transcription of CHOP to promote cell death.3 41
Together, these studies provide compelling evidence for a mechanistic link between lipotoxic fatty acids-induced NFATc1 activation in liver cells and the induction of the deleterious PERK/PKR-CHOP UPR pathway in progressive NAFLD.
NFATc1 depletion protects liver cells from ER-stress-induced inflammasome activation and apoptosis
Recent studies have unequivocally shown that chronic ER stress uses the PERK-CHOP signalling pathway to promote apoptosis and activation of the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome.37 NLRP3 is a multimeric protein complex that stimulates caspase-1 dependent cleavage of pro-interleukin-1β (pro-IL-1β) and gasdermin-D for induction of inflammation and a proinflammatory form of cell death, termed pyroptosis. Here, we examined whether terminal PERK-CHOP signalling activation promotes hepatocyte death and activation of NLRP3 driven pyroptosis in progressive NAFLD and if so, whether this is NFATc1 dependent. To this end, we analysed ER stress-induced NLRP3 inflammasome activation as well as caspase-1 mediated IL-1β and gasdermin cleavage in primary hepatocytes, AML-12 cells and transgenic mice livers with differential NFATc1 expression. Together, western blot, immunofluorescence and immunohistochemical analysis confirmed that lipotoxicity-induced UPR signalling promotes apoptosis (indicated by cleaved caspase-3) as well as NLRP3 inflammasome-induced cytokine release (indicated by cleaved IL-1β) and pyroptosis, as indicated by cleaved caspase-1 and increased gasdermin-D activation (C.GSDMD)) (figure 7A–F, online supplemental figure 4G,H). Importantly, UPR-driven inflammasome activation and cell death initiation require the presence of NFATc1 and therefore, genetic depletion of the transcription factor prevented palmitate-induced caspase-3 activation and blocked NLRP3-driven pyroptosis and IL-1β cleavage both in vitro and in the NAFLD progression model (figure 7A–F, online supplemental figure 4G,H). By contrast, and in line with our findings illustrated in online supplemental figure 2, non-lipotoxic oleate treatment neither induces NFATc1 activation nor terminal UPR signalling and subsequent NRLP3-inflammasome-induced cell death in primary hepatocytes (online supplemental figure 5).
Supplemental material
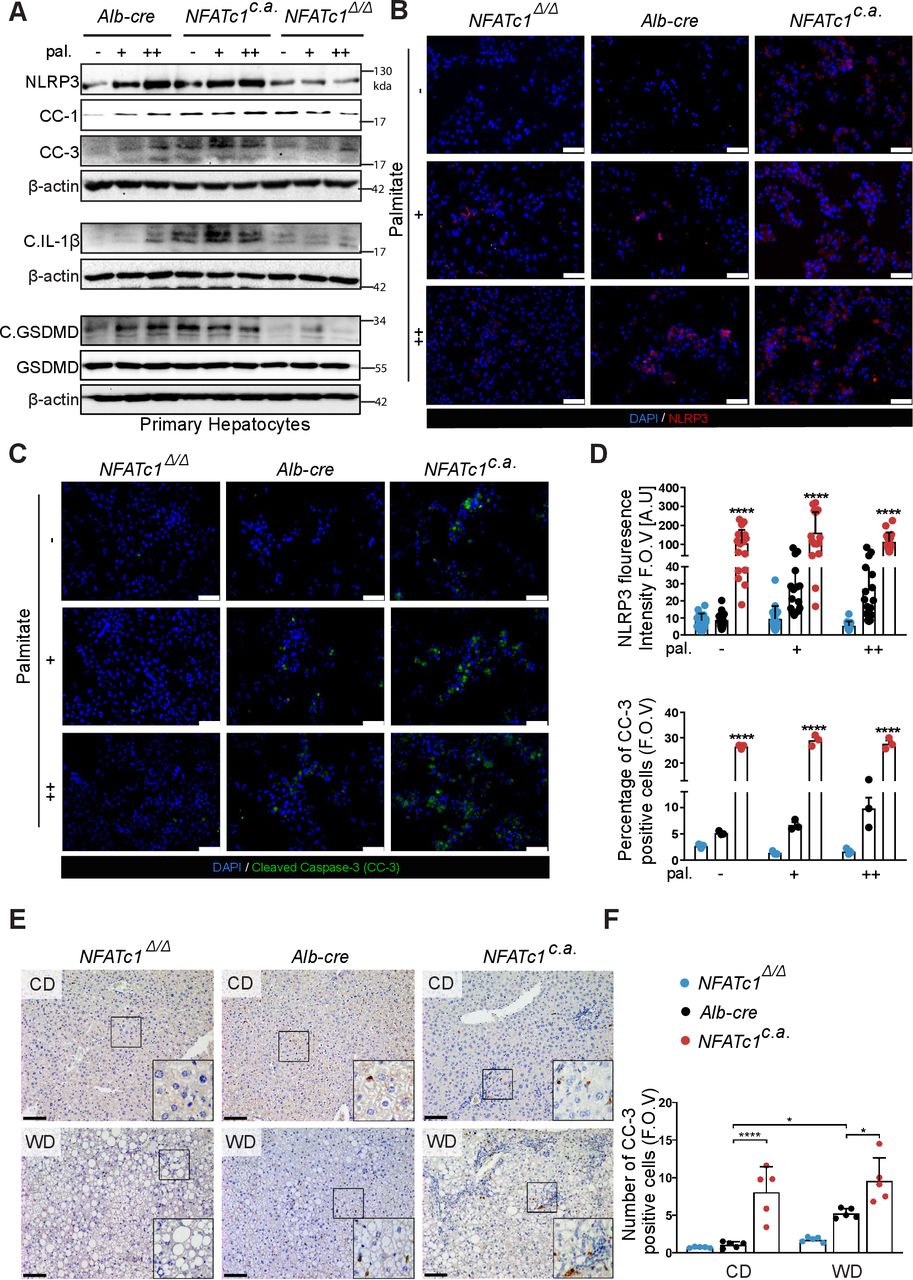
NFATc1-dependent cell death and inflammasome activation in vitro and in vivo. (A) Representative immunoblot displaying NFATc1-dependent changes in NLRP3, cleaved caspase-1 (CC-1), cleaved caspase 3 (CC-3), cleaved interleukin (IL)-1β, C.GSDMD and GSDMD (Gasdermin D) in primary hepatocytes following 12 hours of palmitate treatment (+=100 µM and ++=200 µM). (B) Immunofluorescence analysis of NLRP3 and (C) CC-3 in primary mouse hepatocytes after palmitate treatment. Scale bar=100 µm. (D) Graphs represent fluorescence intensity of NLRP3 and percentage of CC-3 positive hepatocytes (F.O.V). (E) Immunohistochemical analysis of CC-3 staining in liver sections of 20-week-treated mice. Scale bar=100 µm. (F) Quantitative analysis of CC-3 positive hepatocytes (F.O.V). Data are shown in mean±SD; p-values are *p<0.05, ****p<0.0001. Statistical analysis was performed using one-way analysis of variance (ANOVA) (D) and two-way ANOVA (F). CD, control diet; WD, western diet.
Collectively, these experiments performed in primary hepatocytes and transgenic mice emphasise a key role of nuclear NFATc1 in driving terminal ER stress responses to foster NAFLD progression.
Inhibition of chronic ER stress responses attenuates NFATc1-induced NAFLD progression
Based on these results, we tested whether pharmacological inhibition of ER stress can impede NFATc1-triggered disease progression in NAFLD. To this end, we analysed the impact of tauroursodeoxycholic acid (TUDCA), a well-established inhibitor of ER stress responses on NFATc1-dependent mechanisms and cell functions, both in primary hepatocytes and AML12 cells and in WD fed transgenic mice as well (figure 8, online supplemental figure 6A–C).
Supplemental material
Consistent with results presented in figures 6 and 7, ER stress induction by either palmitate treatment or following c.n.NFATc1 transfection resulted in terminal UPR signalling (eg, CHOP) activation and subsequent induction of the NRLP3 inflammasome effector pathway both in vitro and in vivo. Intriguingly, however, application of TUDCA sufficiently blocked ER stress-induced terminal UPR signalling responses and inflammasome activation. In detail, we observed a significant blockade of terminal UPR (indicated by loss of CHOP expression), inhibition of cell death induction (reflected by cleaved-caspase-3) and inactivation of NRLP3-inflammasome mediated cytokine release (eg, IL-1β), even in the presence of active NFATc1 (figure 8A,B, online supplemental figure 6A). Similar effects were confirmed in the NAFLD progression model, in which TUDCA-mediated inhibition of terminal UPR signalling and subsequent NLRP3 activation was associated with a tremendous reduction of inflammation, recruitment of CD45-positive immune cells and hepatic fibrosis despite expression of constitutive active NFATc1 (figure 8D–I, online supplemental figure 6D–E).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tauroursodeoxycholic acid (TUDCA) attenuates NFATc1-dependent unfolded protein response (UPR) signalling-induced inflammation and fibrosis in progressive non-alcoholic liver disease (NAFLD). (A) Immunoblot shows protein expression of CHOP, NLRP3 and CC-3 in Alb-cre primary hepatocytes treated with palmitate (++=200 µM) alone or in combination with increasing concentrations of TUDCA (100–500 µM) in comparison to control-treated cells. (B) Protein levels of CHOP, NLRP3 and CC-3 were assessed in AML12 cells with constitutive activation of NFATc1 and in the presence or absence of 500 µM TUDCA for 12 hours. Cells transfected with siNFATc1 were used as control. (C) Schematic representation of the preventive treatment scheme. (D) Immunoblot showing protein levels of CHOP and NLRP3 in liver tissue lysates of 20 weeks treated Alb-cre mice and NFATc1c.a . mice. (E) H&E staining and immunohistochemical analysis for (F) CD45 and (G) picrosirius red staining. Scale bar=100 µm. (H) Graph represents percentage of CD45 positive cells and (I) percentage of picrosirius red stained area (F.O.V). Data are shown in mean±SD, p values are *p<0.05, **p<0.005, ***p<0.0005, ****p<0.0001. Statistical analysis was performed by two-way analysis of variance.
Together, this study strongly supports an important role of NFATc1 in NAFLD progression and demonstrates that this function is based on the regulation of chronic ER stress responses and subsequent NLRP3 inflammasome activation. Moreover, we provide evidence that pharmacological inhibition of ER stress responses (eg, via TUDCA) can overcome NFATc1-driven progression of the disease.
Discussion
This study was designed to decipher key molecular mechanisms of NAFLD progression and hence to provide a rational basis for the development of new treatment strategies. We thereby focused on the calcium-responsive NFAT transcription factor family, which controls a plethora of cellular processes in inflammation-associated and metabolic disorders, for example, insulin resistance and diabetes, obesity and cancer.12 14 17 18 20 Here, we uncover an essential role of NFATc1 in the progression of NAFLD to NASH. Accordingly, NFATc1 expression and nuclear localisation are weak to absent in healthy livers but strongly induced in progressive NASH. Moreover, treatment with fatty acids, for example, palmitate or WD induces expression, nuclear translocation and transcriptional activity of NFATc1 both in primary and established hepatocytes as well as in mouse liver tissues. Furthermore, hepatocyte-specific activation of nuclear NFATc1 in mice livers—either following WD feeding or through genetic induction of the transcription factor (NFATc1c.a)—fosters rapid acceleration of liver damage, as evidenced by increased tissue inflammation with recruitment of CD45-positive cells and progressive formation of hepatic fibrosis. These results, together with the observation that hepatocyte-specific genetic depletion of NFATc1 prevents fatty acids-induced NAFLD progression beyond the stage of hepatic steatosis, provided strong experimental evidence for a critical role of the Ca2+ responsive transcription factor in progressive NAFLD.
Mechanistically, NFATc1 promotes NAFLD acceleration through unrestricted ER stress signalling responses in liver cells. The ER is responsible for proper protein folding,3 42 and it has been shown that defects in the calcium homeostasis or the ER protein folding machinery can cause ER stress and subsequent activation of the UPR pathway. The UPR pathway orchestrates a multitude of key regulatory mechanisms to repair ER stress including inhibition of protein synthesis and acceleration of ER protein degradation. In general, UPR activation is highly sufficient to repair transient and mild forms of ER stress.3 37 43–46 However, if ER stress is severe and unresolved, it can cause persistent activation of the UPR signalling pathway and eventually leads to cell death, inflammasome activation and accelerated organ damage. Recent studies demonstrated that fat accumulation can trigger chronic ER stress and subsequent activation of the terminal UPR signalling cascade in hepatocytes.3 7 11 37 47 48 Here, we show for the first time a fundamental role of the Ca2+ responsive transcription factor NFATc1 in ER stress-induced NAFLD progression. In detail, prolonged exposure to fatty acids induces expression, nuclear localisation and activity of NFATc1 in hepatocytes both in vitro and in mice with progressive NAFLD. Mechanistically, NFATc1 promotes ER stress sensing through terminal PERK-CHOP signalling and subsequently induces activation of NLRP3,49–51 a macromolecular inflammasome complex involved in cell death initiation and inflammation.7 36 38 48 The prominent function of the terminal PERK-CHOP pathway in chronic ER stress-driven NLRP3 inflammasome activation and apoptosis has been demonstrated in various disorders and especially in diseases related to metabolic disturbances.47 48 52 53 In NAFLD, for instance, CHOP activation induces the NRLP3 inflammasome complex to promote hepatocyte death and inflammation in response to unresolved ER stress.3 7 36 Our results not only confirm these previous observations, but in addition, demonstrate that NFATc1 activation is mandatory for fat-induced ER stress signalling through the PERK-CHOP branch and subsequent activation of the NRLP3 inflammasome pathway. Accordingly, genetic depletion of NFATc1 prevents liver cells from terminal UPR signalling, NRLP3 activation and cell death initiation both in primary hepatocytes and in liver tissues, even on longtime stimulation with high-fat diet.
Lastly, we assessed whether inhibition of chronic ER stress signalling can protect fatty livers from NFATc1-induced disease progression. For this purpose, we combined prolonged high-fat stimulation with application of TUDCA, a naturally occurring hydrophilic bile acid and taurine conjugate of ursodeoxycholic acid (UDCA), which is approved by Food and Drug Administration for the treatment of primary biliary cholangitis (PBC). Recent multicentre randomised clinical trials have shown that TUDCA presents the same level of safety and tolerability as UDCA for the treatment of PBC and may be even better to relieve symptoms of the disease, suggesting higher effectiveness of taurine conjugate in treatment of PBC.54 The efficacy of TUDCA in cholestatic diseases was primarily attributed to its choleretic and cytoprotective effects on hepatocytes by increasing bile flow and biliary acid secretion.55 Importantly, numerous preclinical studies have also demonstrated a remarkable therapeutic potential of TUDCA in non-cholestatic liver diseases and particularly in NAFLD, where it exerts strong cytoprotective effects through its ability to alleviate ER stress and to block terminal PERK-CHOP signalling.7 36 56 Our results strongly support these efforts and provide a mechanistic rationale for the proposed efficacy of TUDCA in the prevention of NAFLD progression. We show that application of TUDCA effectively interferes with acceleration of the disease through inhibition of NFATc1-mediated ER stress sensing and terminal UPR signalling activation. Consistently, TUDCA treatment blocked NFATc1-induced CHOP-NRLP3 inflammasome activation and consequently reduced the degree of inflammation, apoptosis and fibrosis in the liver, even on long-term feeding with high-fat diet.
Taken together, this study identifies the calcium signalling responsive transcription factor NFATc1 as a key player in NAFLD progression. We show that NFATc1 drives fat-induced NASH through promotion of chronic ER stress responses and activation of the NRLP3 inflammasome. This study not only contributes to a better understanding of NFATc1 signalling in NAFLD but provides a mechanistic rationale for recent clinical trials aiming at pharmacological interference with chronic ER stress responses to prevent disease progression.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
All animal experiments were approved by local animal care and use committee (LAVES) and carried out according to the regulations of Federation of European Laboratory Animal Science Associations (FELASA).
Acknowledgments
We are very thankful to Higher education commission Pakistan, for providing doctorate scholarship to Muhammad Umair Latif. We are thankful to Dr. Nai-Ming Chen for his inputs in project development and Sarah L. Hanheide for her contributions in mouse breeding. We thank Gabriela Salinas from NGS Integrative Genomics Core Unit, Goettingen for sequencing of the samples. All the schematic illustrations were prepared using BioRender.com. Finally, we thank all the laboratory members for insightful discussions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @ShivKSingh6
Contributors MUL performed most of the experiments, analysed and interpreted results. GES performed library preparation, RNA-seq analysis and figures designing. RR helped in the bioinformatics analysis for RNA-seq data. SM handled the mouse breeding and treatments. CSG and IS-T performed calcium measurement experiments in AML12 cells and AR performed in primary hepatocytes. KR facilitated in developing experimental plans and performed experiments. EH wrote all the approval applications for in-vivo experiments. SKS provided intellectual inputs and was involved in designing figures for the paper. AM facilitated in optimising primary hepatocyte isolation procedures. UJB provided support in IHC analysis and manuscript proofreading. PS provided human tissues as well as helped in analysing the IHC staining. SCB and HB provided human NASH patient samples. AN provided scientific input in planning experiments and data interpretation. IB provided scientific inputs for this study and helped in planning experimental models. VE was the principal investigator of the study and was responsible for study concept and design. Together with MUL, he was also responsible for manuscript writing. VE is the guarantor and responsible for the overall content of the study.
Funding This project was primarily funded by the DFG (KFO-5002). Beside this it was supported by the German Cancer Aid to Shiv K. Singh (70112999; Max-Eder group), to Albrecht Neesse (70113213; Max-Eder group) and to Eisabeth Hessmann (70112108), the DFG grant to Shiv K. Singh and Elisabeth Hessmann (KFO-5002), the DFG grant to Ivan Bogeski (SFB1190 P17) and the Volkswagen-Stiftung/Ministry for Culture and Science in Lower Saxony (MWK) to Volker Ellenrieder (11-25 76251-12-3/16).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.