Article Text

Abstract
Objective The lysosomal protease cathepsin B is upregulated in human pancreatic ductal adenocarcinoma (PDA) and represents a potential therapeutic target. Loss of cathepsin B delays tumour progression in mouse models of islet, mammary and intestinal carcinoma and decreases invasion and metastasis. This study examines the role of cathepsin B in the initiation, progression and metastasis of PDA.
Methods Cathepsin B germline knockout mice were crossed with animals expressing an endogenous KrasG12D allele in the pancreas, and mice were aged to evaluate the role of cathepsin B in pancreatic intraepithelial neoplasia (PanIN). A survival study was also performed with mice carrying an additional heterozygous conditional Trp53R172H allele. Cell lines derived from tumours were used to investigate the role of cathepsin B in vitro, and subcutaneous allografts investigated the cell autonomous and non-cell autonomous roles of cathepsin B in pancreatic cancer.
Results Constitutive cathepsin B loss resulted in delayed progression of both PanIN and PDA and a significant survival advantage in mice. Cathepsin B-deficient PDA cells and PanIN showed decreased proliferation and mitogen-activated protein (MAP) kinase signalling. The reconstitution of deficient cells with cathepsin B reversed these findings, which correlated with decreased levels of the active forms of the related protease cathepsin L. Conversely, acute ablation of cathepsin L activated the MAP kinase cascade in PDA cells.
Conclusions These results confirm that cathepsin B plays an important cell autonomous role in the progression of PDA and suggest that the regulation of cathepsin L by cathepsin B may be a means of stimulating cell proliferation in neoplasia.
- Cathepsin B
- cathepsin L
- pancreatic ductal adenocarcinoma
- metastasis
- Erk, cancer
- cadherins
- acini
- epithelial cell adhesion
- acute pancreatitis
- adhesion molecules
- pancreatic enzymes
- pancreatitis
- acute pancreatitis
- pancreatic disorders
- chronic pancreatitis
Statistics from Altmetric.com
- Cathepsin B
- cathepsin L
- pancreatic ductal adenocarcinoma
- metastasis
- Erk, cancer
- cadherins
- acini
- epithelial cell adhesion
- acute pancreatitis
- adhesion molecules
- pancreatic enzymes
- pancreatitis
- acute pancreatitis
- pancreatic disorders
- chronic pancreatitis
Significance of this study
What is already known about this subject?
Cathepsin B is of interest as a potential biomarker of early recurrence in human pancreatic cancer.
Cathepsin B loss in mouse models of islet, mammary and intestinal carcinoma results in decreased tumour initiation, proliferation, apoptosis, angiogenesis and metastasis. Cathepsin B produced by stromal cells is implicated in metastasis.
Cathepsin L is involved in termination of growth factor signalling in keratinocytes.
What are the new findings?
Cathepsin B loss decreases pancreatic intraepithelial neoplasia burden and proliferation in preneoplasms and tumours. The stromal content and vascularity of tumours is not affected by loss of cathepsin B.
Cathepsin B has a cell autonomous role in pancreatic ductal adenocarcinoma.
Cathepsin B loss impinges on mitogen-activated protein kinase signalling in vitro and in vivo. The decrease in phospho-Erk in the absence of cathepsin B correlates with an increase in levels of active cathepsin L.
Cathepsin B loss increases survival and decreases liver metastasis in mice with pancreatic ductal adenocarcinoma. This is the first report of improved survival in this model, and supports the development of selective cathepsin B inhibitors.
How might it impact on clinical practice in the foreseeable future?
The development of better cathepsin inhibitors might prove efficacious in a clinical setting and is worth exploring.
Introduction
Pancreatic ductal adenocarcinoma (PDA) is a lethal disease with a 5-year survival rate of <5%. It is the fourth leading cause of cancer-related deaths in the USA with an estimated 42 000 new cases and 35 000 deaths in 2009.1 Contributing to the dismal prognosis is the difficulty in detecting early tumours, with the majority of patients presenting with advanced and metastatic disease.2 3
The genetic and histological progression of pancreatic cancer has been analysed from resected tumour specimens, giving insight into the mechanism of disease initiation and progression. Activating mutations in the Kras oncogene have been identified as the initiating event, present in >90% of tumours. Other common genetic events include the inactivation of the Ink4a/Arf locus, point mutations in Trp53 and mutations or deletions of Smad4.3 Histologically, the model for disease progression describes increasing grades of preinvasive pancreatic intraepithelial neoplasia (PanIN), with the highest grade (PanIN 3) described as ductal carcinoma in situ, followed by frank carcinoma.4 5 A mouse model of PDA that recapitulates the progression of the human disease has been established with KrasG12D as the initiating mutation, a process that is accelerated by the concomitant expression of a point mutant Trp53R172H allele.6 7
Cathepsin B is a member of the cysteine cathepsin family of lysosomal proteases which includes 10 other members.8 It is ubiquitously expressed and is upregulated in several malignancies through different mechanisms including locus amplification,9 10 transcriptional regulation,11–13 alternative splicing14 15 and post-transcriptional regulation.16 In addition, its localisation is often altered in cancer cells, where it can be secreted or associated with the plasma membrane.17–19 Mouse models of islet,20 21 mammary22 and intestinal23 carcinomas have elucidated the importance of cathepsin B in various tumour types. In these models, cathepsin B loss impinges upon various facets of tumour development including initiation, proliferation, apoptosis, angiogenesis and metastasis. The contribution of cathepsin B to tumour development and metastasis is not solely cell autonomous. Indeed, tumour-associated stromal cells such as fibroblasts, macrophages and neutrophils express cathepsin B,24 and a role for microenvironmental cathepsin B in metastasis has been previously demonstrated in vivo in the transgenic MMTV-PyMT model of mammary carcinoma.19
To examine the role of cathepsin B in the initiation and progression of PDA, we used the oncogenic Kras-driven mouse models described above, concomitant with genetic ablation of cathepsin B. We found that cathepsin B plays an important role in tumorigenesis and suggested an interaction of cathepsin B with the related cathepsin L in this regard. Our results support the further investigation of cathepsin B as a therapeutic target in PDA.
Materials and methods
Mouse strains
The LSL-KrasG12D, LSL-Trp53R172H, Pdx-1-Cre and cathepsin B null strains of mice have been previously described.6 7 25 These strains were interbred to ensure a mixed C57BL6/129Sv background for survival studies. Mice were housed and maintained and experiments were conducted in compliance with UK Home Office regulations.
Cloning
The mouse cathepsin B cDNA26 was isolated from the pSG5 backbone using BamHI and EcoRI and was cloned into the pBabe-Hygro expression vector.
Cell culture
Cell lines derived from tumours were maintained in Dulbecco's Modified Eagle Medium (DMEM) +10% fetal bovine serum (FBS). Cells were transfected with pBabe-Hygro or pBabe-Cathepsin B using lipofectamine and selected with 200 μg/ml hygromycin for 7 days to generate stable clones. For harvesting protein, cells were plated at a density of 2×106 cells on a 10 cm dish, grown overnight and fed with DMEM +1% FBS for 24 h before use. Protein lysates were obtained using 1% SDS lysis buffer. Culture and immunofluorescence of cell lines in matrigel was carried out as previously described.27
For soft agar assays, 2×104 cells were suspended in 0.4% low melting point agar and layered over 0.8% agar in 6 cm dishes. Cells were fed every 3–4 days for 4 weeks.
Western blotting
Proteins were boiled in SDS loading buffer, separated on 4–12% Nupage gels (Invitrogen, Paisley, United Kingdom), and transferred onto PVDF membranes (Millipore). Membranes were blocked with 5% milk in TBS-Tween and were incubated with relevant primary and secondary antibodies. Densitometry was conducted using Adobe Photoshop.
Subcutaneous allografts
Cell lines were grown to confluence, trypsinised, washed in phosphate buffer solution (PBS) and resuspended at 1×107 cells/ml in sterile PBS. The mice were shaved and cells were injected subcutaneously into the flanks of C57Bl6 and cathepsin B null mice, with cathepsin B null cells injected into the left flank and cathepsin B-expressing cells into the right flank. Tumour growth was followed using calipers.
Statistical analysis
Kaplan–Meier statistics for survival curves were conducted using the Gehan–Breslow–Wilcoxon test. Proliferation, cathepsin activity, phospho-Erk, necrosis and mean vessel density data were analysed by the Mann–Whitney U test and metastasis incidence data were evaluated by the Fisher exact test. Graphpad Prism software was used to conduct statistical analysis.
Reagents, histology and immunohistochemistry, in vivo measurement of cathepsin activity and quantification of proliferation are described in the online supplement.
Results
Cathepsin activity is increased during disease progression
To evaluate the importance of cathepsins during the development of PDA, cathepsin activity was first measured in vivo using the cysteine protease-activatable probe prosense 680. In vivo cathepsin activity was increased in PanIN (p=0.0040) and PDA (p=0.0159) tissue compared with normal pancreas (figure 1A).
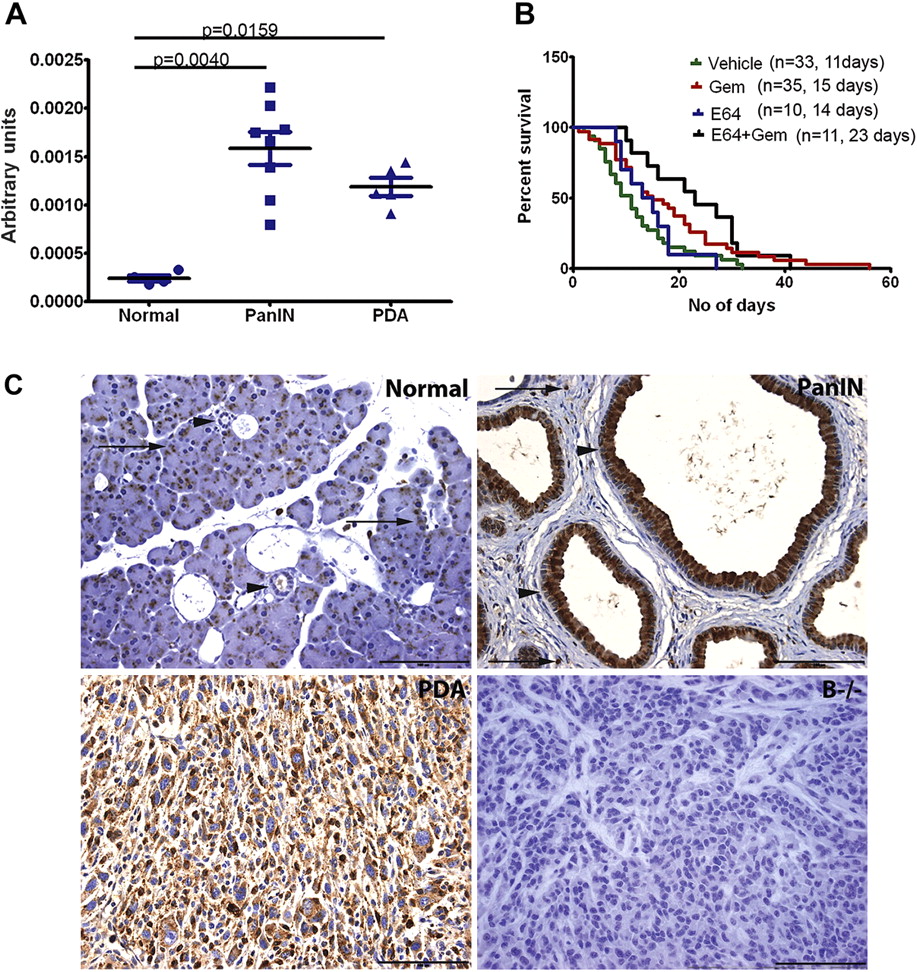
Increased cathepsin B expression during pancreatic cancer progression. (A) Cathepsin acitivity is increased in vivo in pancreatic intraepithelial neoplasia (PanIN) and tumour tissue compared with normal pancreas. (B) Inhibition of cathepsin activity with E64 in combination with gemcitabine (Gem) resulted in a median survival of 23 days, a significant increase over vehicle (11 days) and E64 as a single agent (14 days), and a 53% increase in survival compared with mice given gemcitabine alone (15 days). (C) Immunohistochemistry for cathepsin B shows that normal pancreatic tissue exhibits punctate staining in the acini (arrows) and low level staining in ducts (arrowheads). Cathepsin B expression is greatly increased in PanIN (arrowheads). Positive staining is also seen in stromal cells (arrows). Tumour cells are positive for cathepsin B. Cathepsin B null pancreatic ductal adenocarcinoma (PDA) tissue was used as a negative control. (Scale bar=100 μm).
Inhibition of cathepsin B increases survival in tumour-bearing mice in combination with gemcitabine
Since cysteine cathepsin activity was found to be increased in pancreatic tumours, the broad-spectrum cysteine protease inhibitor E64 was used as a therapeutic agent in mice bearing pancreatic tumours. E64 (50 mg/kg) was given daily intraperitoneally to tumour-bearing mice, either as a single agent or in combination with biweekly gemcitabine (100 mg/kg), until terminal disease progression. The median survival of mice receiving E64 alone was 14 days compared with 23 days for the combination arm (figure 1B). As has been reported previously in this system,28 the median survival for mice treated with gemcitabine monotherapy is 15 days (figure 1B). Although E64 alone did not result in an increase in survival, the combination of E64 and gemcitabine provided a significant increase in survival compared with vehicle (p=0.0037) or E64 alone (0.0271). The combination treatment also extended median survival on treatment by 53% compared with gemcitabine monotherapy, although this was not statistically significant (p=0.1078).
Cathepsin B expression is increased during disease progression
As has been previously shown, cathepsin expression was upregulated during disease progression in the pancreas.29 In our model, cathepsin B in the normal pancreas was confined to punctate areas in the acini consistent with acinar secretory granules (figure 1C, arrows) and low-level expression in ducts (figure 1C, arrowheads). In contrast, cathepsin B expression was increased in PanIN, with the apical cytoplasm of some PanIN staining strongly positive (figure 1C, arrowheads). It should be noted that staining in some PanIN was restricted to the perinuclear area of the cell (data not shown). In addition, stromal cells also expressed cathepsin B (figure 1C, arrows). Similar to PanIN, cathepsin B expression in PDA cells was strongly positive and distributed throughout the cytoplasm (figure 1C). Tumour tissue from a cathepsin B null mouse did not show immunohistochemical reactivity, confirming the specificity of the antibody detection (figure 1C). Cathepsin B is therefore expressed in the normal murine pancreas, with increased levels detected in both preneoplastic and neoplastic ductal cells.
Cathepsin B loss delays the onset of PanIN and decreases proliferation
To evaluate the role of cathepsin B in the initiation of PDA, cathepsin B null mice25 were crossed to mice harbouring the LSL-KrasG12D allele and transgenic Pdx-1-Cre recombinase.6 The pancreatic disease burden in mice with the relevant genetic combinations was compared at various time points. Cathepsin B ablation delayed but did not prevent PanIN formation, and the resultant preneoplasms were morphologically similar regardless of cathepsin B status (data not shown). At 2 months there was no significant difference in disease burden between pancreata from LSL-KrasG12D; Pdx-1-Cre; Cathepsin B+/+ (KC;B+/+) and LSL-KrasG12D; Pdx-1-Cre; Cathepsin B−/− (KC;B−/−) mice (figure 2A). At 4–6 months, pancreata from the KC;B−/− cohort had a higher fraction of normal lobules compared with the KC;B+/+ cohort (43.75±2.97% vs 19.89±3.65%; p=0.0002). In addition, there was a decrease in the number of PanIN 1-bearing lobules in the KC;B−/− cohort compared with the KC;B+/+ cohort (23.33±2.05% vs 41.77±6.52%; p=0.0178). There was no difference in reactive lobules or higher PanIN grades. At 8–10 months there was no significant difference between the two cohorts in any of the disease grades analysed, although the KC;B−/− cohort tended towards decreased PanIN 1 content compared with the KC;B+/+ cohort (figure 2A).

Loss of cathepsin B delays the onset of pancreatic intraepithelial neoplasia (PanIN) and decreases the burden and proliferation of PanIN. (A) Analysis of disease burden shows that, at 2 months, cathepsin B loss does not affect reactive acini or PanIN load. At 4–6 months, pancreata from cathepsin B null mice have more normal lobules and decreased PanIN1-containing lobules. At 8–10 months, there is no difference between cathepsin B null and cathepsin B-expressing pancreata. (B) Quantification of proliferation in PanIN by Ki67 immunohistochemistry reveals that loss of cathepsin B causes a significant decrease in proliferation in mice aged 4–6 months and mice aged 8–10 months. At 2 months there is a trend towards decreased proliferation, although this was not statistically significant. The Mann–Whitney test was used to analyse disease burden and proliferation data.
Cathepsin B loss led to decreased proliferation in PanIN. At 2 months, PanIN from KC;B−/− pancreata displayed a trend towards decreased proliferation (3.498±1.998 mean percentage proliferation), although this was not statistically significant compared with LSL-KC;B+/+ pancreata (5.267±2.125; figure 2B). At 4–6 months the difference in PanIN proliferation was statistically significant in KC;B−/− pancreata (6.095±3.064) compared with KC;B+/+ pancreata (11.040±3.756, p=0.0006; figure 2B). This effect on proliferation persisted at 8–10 months, with KC;B−/− mice (5.919±1.940) exhibiting a >50% reduction in proliferation compared with KC;B+/+ mice (12.750±3.201, p=0.0004; figure 2B). There was no difference in pancreatic apoptotic index between cohorts (data not shown).
The stromal compartment was assessed with Masson's trichrome stain to evaluate collagen deposition and probed with antibodies against alpha-smooth muscle actin to delineate activated myofibroblasts (see figure 1A in online supplement). No obvious stromal differences were evident between KC;B+/+ and KC;B−/− mice, suggesting that the differences in PanIN content and cellular proliferation may be cell autonomous.
Loss of cathepsin B decreases proliferation in an advanced model of pancreatic cancer
Following the initial observation of decreased proliferation in preinvasive lesions, the role of cathepsin B in pancreatic cancer progression was studied using a conditional point mutant Trp53R172H allele concomitant with oncogenic KrasG12D.7 Once again, cathepsin B null mice were bred into this combination of alleles along with Pdx-1-cre.
Cathepsin B loss did not prevent PDA formation, and the morphology of PDA in the LSL-KrasG12D; LSL-Trp53R172H; Pdx-1-Cre; Cathepsin B−/− (KPC;B−/−) cohort was comparable to the LSL-KrasG12D; LSL-Trp53R172H; Pdx-1-Cre; Cathepsin B+/+ (KPC;B+/+) cohort, including histological patterns ranging from ductal to sarcomatoid and solid (figure 3A). However, PDA cellular proliferation was measured by phospho-histone H3 content and was shown to be reduced in cathepsin B-deficient tumours (50.93±31.20 vs 66.15±22.61, p=0.0317; figure 3B).
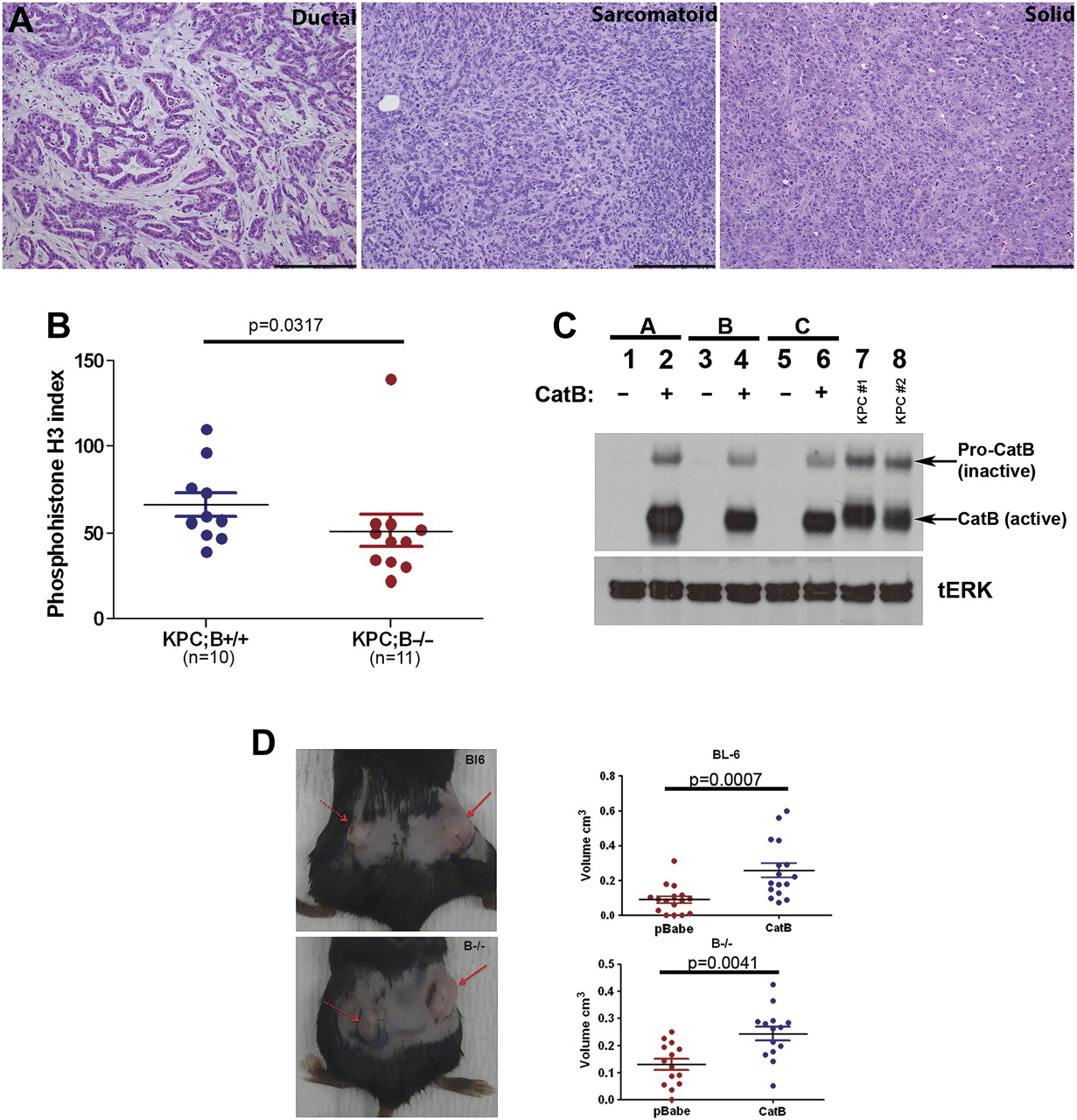
Cathepsin B loss inhibits proliferation in pancreatic tumours. (A) Tumours in cathepsin B null mice exhibit mixed histology with ductal, sarcomatoid and solid features, similar to KPC tumours (scale bar=200 μm). (B) Proliferation measured by phospho-histone H3 is significantly decreased in null tumours compared with wild-type mice for cathepsin B. (C) Reconstitution of cathepsin B in null tumour cell lines results in comparable expression to tumour cell lines from KPC;B+/+ mice (lanes 2, 4, 6 vs 7, 8). As expected, cathepsin B was absent in the null cell lines (lanes 1, 3, 5). (D) Pancreatic ductal adenocarcinoma (PDA) cells expressing recombinant cathepsin B (right flank, solid arrow) form larger subcutaneous tumours, with significantly increased volume, than isogenic cells lacking cathepsin B (left flank, dashed arrow) in both wild-type and cathepsin B null recipient mice.
In order to investigate the mechanism by which cathepsin B influences proliferation, a more detailed analysis of the tumours was carried out. There were no stromal differences between tumours from KPC;B+/+ and KPC;B−/− mice regarding Masson's trichrome and alpha-smooth muscle actin composition (see figure 1A in online supplement). In addition, the collagen content was quantified by picrosirius red staining, with no significant difference measured between KPC;B+/+ and KPC;B−/− tumours (see figure 1B in online supplement). There were also no significant differences in necrosis (figure 1C in online supplement) and mean vessel density measured with CD31 immunohistochemistry (figure 1D in online supplement).
Cathepsin B expression increases growth of subcutaneous allografts
To elucidate the mechanism of decreased proliferation in the cathepsin B null background, tumour cell lines were derived from KPC;B−/− mice and transfected with recombinant cathepsin B to allow comparison of isogenic cell lines. Evaluation of cathepsin B expression in these cell lines (figure 3C, lanes 2, 4, 6) revealed the presence of intracellular cathepsin B at levels comparable to KPC;B+/+ cell lines (figure 3C, lanes 7, 8). Cathepsin B was also expressed at the plasma membrane and secreted into the medium (data not shown).
Since cathepsin B did not appear to influence the stromal content of tumours, the cell lines were injected subcutaneously into wild-type (C57Bl6) and cathepsin B null mice to produce allografts to elucidate whether cathepsin B has cell autonomous or non-cell autonomous roles in tumour growth. Cathepsin B-transfected cells formed larger tumours in both wild-type (0.259±0.165 cm3, p=0.0007) and cathepsin B null recipient mice (0.243±0.094 cm3, p=0.0041) compared with their null counterparts (0.091±0.081 cm3 in wild-type mice and 0.130±0.078 cm3 in cathepsin B null mice; figure 3D). Cathepsin B therefore plays a prominent cell autonomous function in the development of pancreatic ductal tumour.
Loss of cathepsin B leads to decreased mitogen-activated protein kinase signalling
Cathepsin B reconstituted cells had increased signalling in the mitogen-activated protein (MAP) kinase cascade, as shown by increased levels of phospho-Erk when cells were grown on a monolayer (figure 4A). The increased phosphorylation of Erk in the presence of cathepsin B was more evident when the reconstituted cell lines were grown as spheroids in matrigel (figure 4B). As spheroids, cathepsin B expression also resulted in increased total protein tyrosine phosphorylation (figure 4B).

Cathepsin B loss leads to decreased mitogen-activated protein (MAP) kinase signalling and increased cathepsin L activation. (A) Cathepsin B reconstitution increases the levels of phospho-Erk when cells are grown in a monolayer (lanes 1 vs 2, 3 vs 4 and 5 vs 6). This trend is less evident when other members of the MAP kinase cascade are examined and total Erk is used as a loading control. Phospho-Erk values quantitated by densitometry have been normalized to levels of total-Erk. (B) Immunofluorescence analysis using isogenic cell lines grown in matrigel demonstrates that cathepsin B transfection increases the levels of phospho-Erk and phosphorylated protein tyrosine residues. (C) Lack of cathepsin B in vivo correlates with a decreased content of nuclear phospho-Erk (Scale bar = 50μm). (D) Cathepsin B deficiency correlates with higher levels of processed cathepsin L in tumors which correspond to the active single-chain (CatL s.c) and heavy chain (CatL h.c.) forms (KPC;B+/+ vs KPC;B−/−) and in cell lines 1 vs 2, 3 vs 4 and 5 vs 6 (arrows). (E) Acute cathepsin L knockdown in pancreatic ductal adenocarcinoma cell lines leads to increased phospho-Erk.
To further investigate this finding, phospho-Erk immunohistochemistry was performed on PanIN from KC;B+/+ and KC;B−/− mice. Although the overall intensity of cytoplasmic phospho-Erk staining did not vary between the cohorts, PanIN in KC;B+/+ pancreata exhibited a significantly higher percentage of phospho-Erk positive nuclei than KC;B−/− pancreata (93.48±1.79% vs 62.26±8.74%, p=0.0025; figure 4C).
Since cathepsin L was recently shown to attenuate mitogenic growth factor signalling and tumour progression,30 the expression of cathepsin L was examined in cathepsin B null tumours and cell lines. Cathepsin B null tumours (KPC;B−/−) were surprisingly found to contain higher levels of processed cathepsin L, which is consistent with the active single chain and heavy chain forms of cathepsin L than cathepsin B-expressing tumours (KPC;B+/+) (figure 4D, arrows). This finding was recapitulated in vitro, with decreased processed cathepsin L in cathepsin B-expressing cells (lanes 2, 4, 6) compared to their null counterparts (lanes 1, 3, 5) (figure 4D, arrows). To establish a direct link between cathepsin L and MAP kinase signalling in PDA, siRNA pools were used to acutely knock down cathepsin L in three PDA cell lines. In all three lines, cathepsin L knockdown was sufficient to increase the level of phospho-Erk (figure 4E). Cathepsin B expression therefore negatively modulates the levels of active cathepsin L in PDA cells, and this correlates with the activation of MAP kinase signalling.
Loss of cathepsin B prolongs survival and decreases liver metastasis in an advanced model of pancreatic cancer
Kaplan–Meier statistics showed that KPC;B−/− mice had a significantly increased median PDA-free survival of 199 days compared with 168 days in KPC;B+/+ mice (p=0.0113). Cathepsin B loss delayed the onset of PDA, as the earliest case of PDA-related morbidity in the KPC;B−/− cohort was 127 days compared with 90 days in the KPC;B+/+ cohort (figure 5A). KPC;B−/− mice developed the expected features of PDA including haemorrhagic ascites and liver and lung metastases.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cathepsin B loss extends survival and decreases liver metastases of mice with pancreatic ductal adenocarcinoma (PDA). (A) Cathepsin B null mice have a median PDA-free survival of 199 days, a significant extension over KPC mice with a median of 168 days. (B) Although cathepsin B is not required for metastasis to the liver, the number of metastases per liver is significantly decreased in null animals.
As cathepsin B has previously been implicated in metastasis,19 20 a comprehensive examination of liver tissue was conducted. Liver metastases developed and were histologically similar in KPC;B+/+ and KPC;B−/− cohorts (data not shown). The incidence of mice with liver metastases was decreased from 75% in the KPC;B+/+ cohort to 52.63% in the KPC;B−/− cohort, although this difference was not statistically significant (p=0.2928, Fisher exact test). Nonetheless, there was a significant reduction in the number of liver metastases in KPC;B−/− mice compared with KPC;B+/+ mice (1.421±2.090 vs 4.000±3.882, p=0.0359; figure 5B).
Discussion
The cysteine cathepsin family of lysosomal proteases is of great interest because of their overexpression and mislocalisation in a number of cancers.24 In pancreatic cancer in particular, cathepsin B has been reported to be overexpressed in primary tumours and associated metastases.31 In addition, cathepsin B is also of interest in PDA as a prognostic indicator of early recurrence after surgical resection.32 33
Following the observation that cathepsin activity is increased in preinvasive and invasive pancreatic cancer in mice, we tested the broad-spectrum cysteine protease inhibitor E64 in vivo. Previous studies conducted in vivo have shown promise in conjunction with chemotherapy,34 although bioavailability of the inhibitor may be problematic in specific cancer types.35 In our system, inhibition of extracellular cysteine proteases using E64 led to an increase in survival when combined with gemcitabine. Although the improved survival in combination treatment was not statistically significant compared with gemcitabine alone, the drug we used was not an optimal cathepsin inhibitor and the positive trend underlines the importance of understanding the specific contributions of different cathepsins in cancer.
This study examines the role of cathepsin B in the initiation and progression of pancreatic cancer. We have shown that cathepsin B expression increases during disease progression, and that cathepsin B loss delays the onset of PanIN and decreases the burden of PanIN. Cathepsin B loss also negatively impacts on proliferation in both PanIN and tumours.
The effect of cathepsin B loss on proliferation has been reported previously in other mouse models of cancer.21 22 Given that proliferation is consistently decreased at both early and late stages of disease in our model in the setting of cathepsin B loss, this is a likely mechanism of delaying tumour progression in these mice. Here we report that cathepsin B loss impinges on MAP kinase signalling both in vitro and in vivo, and may explain the defect in proliferation. In the setting of constitutive cathepsin B loss, we have shown that levels of active cathepsin L are surprisingly increased both in primary PDA tumours and cell lines. Recently, the role of cathepsin L in the termination of growth factor signalling has been demonstrated in keratinocytes.30 The increase of phospho-Erk in cathepsin B-expressing neoplasms and cell lines may therefore be a result of altered cathepsin L. In direct support of this model, cathepsin L knockdown in mouse PDA cell lines results in increased phospho-Erk. The exact mechanism by which cathepsin L is altered in response to cathepsin B expression remains to be elucidated. Nonetheless, our results show that the constitutive loss of cathepsin B leads to decreased phospho-Erk in pancreatic cancer, and we implicate the increased proteolytic processing of cathepsin L in this process.
Since cathepsin B is expressed in both tumour and stromal cells, it may have non-cell autonomous roles as a result of paracrine signals between the different cell types. This conclusion is consistent with the finding that intravenous injection of cathepsin B-expressing tumour cells into cathepsin B null mice results in lung metastases with decreased proliferation.22 Recent supporting cell culture evidence has been reported where the use of cathepsin B-specific inhibitors has led to decreased proliferation of endothelial cells.36 The allograft experiments presented here support a cell autonomous role of cathepsin B in PDA, as cell lines expressing the protease proliferate more rapidly than cathepsin B null cell lines irrespective of the background into which they are implanted. The relative contributions of intracellular and extracellular cathepsin B in this setting remain to be clarified. These experiments do not exclude a non-cell autonomous role for cathepsin B, as the stromal response in allografts is somewhat limited compared with that seen in in situ tumours.
Loss of cathepsin B confers a survival advantage to mice with PDA. This benefit is associated with decreased tumour proliferation and liver metastasis in tumour-bearing mice. This is the first report of extended survival due to a genetic deficiency in this mouse model of advanced pancreatic cancer. Our observation that early morbidity is absent in cathepsin B null mice indicates that optimal tumour development requires the activity of cathepsin B. A major characteristic of PDA is the profound deposition of extracellular matrix consisting of numerous components including collagens, laminin and fibronectin.37 38 The significance of cathepsin B in this setting is its ability to cleave collagen IV, laminin and fibronectin, as has been reported in normal and tumour tissue.39 In addition, mislocalisation of cathepsin B to the plasma membrane is reported to be induced by Ras oncogenes.40–42 This may reflect the importance of cathepsin B in the degradation of the extracellular matrix and the progression from ductal carcinoma in situ to frank cancer. These aspects of cathepsin B may also be involved in promoting metastasis. We therefore conclude that cathepsin B is a logical therapeutic target in pancreatic cancer and suggest that the development of specific inhibitors is warranted.
Acknowledgments
The authors thank Dr Terence Dermody for providing the mouse cathepsin B cDNA, Frances Connor, Paul Mackin, Lisa Young, Albrecht Neese and other members of the Tuveson laboratory for their input, and the histology core and biological resources unit staff at the CRI for their support.
References
Footnotes
See Commentary, p 790
Funding This work was supported by the Lustgarten Foundation, the University of Cambridge and Cancer Research UK, the Li Ka Shing Foundation and Hutchison Whampoa Limited, the National Institute for Health Research Cambridge Biomedical Research Centre, NIH (grants CA101973, CA111294, CA084291 and CA105490 to DAT) and the European Commission Seventh Framework Programme (FP7 Health 2010 2.4.1-6, contract number 256974). JM and MML were supported by Deutsche Krebshilfe/Dr Mildred-Scheel-Stiftung (109102), the Deutsche Forschungsgemeinschaft MA 4115/1-2/3 and the European Union (EU-FP-7: EPC-TM and EU-FP7-REGPOT-2010-1). TR was supported by European Commission FP7 grant 201279 (Microenvimet), the Deutsche Forschungsgemeinschaft SFB 850 project B7 and by the Excellence Initiative of the German Federal and State Governments (EXC 294).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.