
Abstract
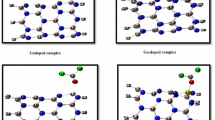
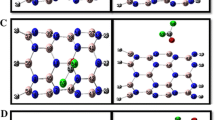
In this paper, we evaluated adsorption of phosgene gas molecule (COCl2), on the hexagonal-aluminum nitride (h-AlN) nanosheet by using first-principles van der Waals density functional theory calculations (vdW-DF) method. The nature of interaction between the phosgene molecule and h-AlN is discovered by geometries, adsorption energies, Mulliken, Hirshfeld as well as Voronoi charges analyses. The density of states (DOS) was calculated and the results show that HOMO/LUMO energy gap of h-AlN is significantly reduced upon the COCl2 adsorption. The projected density of states (PDOS) of the adsorption systems suggested that the enhancement of adsorption was owing to the hybridization between Al atom of h-AlN sheet and the O atom of phosgene molecule. Interestingly, the results reveal that the Eg of h-AlN is very sensitive to the presence of COCl2 molecule as its value reduces from 3.337 eV in pure h-AlN to 1.966 eV (41.08% change) after the COCl2 adsorption which would result in electrical conductance increment. Global reactivity descriptor values such as electronegativity (χ), global hardness (η), global softness (S), electronic chemical potential (μ), electrophilicity index (ω), and electro accepting power (ω+) were calculated. Additionally, the stability of the most stable phosgene/h-AlN complex was evaluated by means of DFT molecular dynamics (MD) simulation at room temperature under constant volume and temperature conditions with PBE method. Based on the DFT calculation results, the h-AlN nano sheet is expected to be potential novel sensor for detecting the presence of COCl2 gas.






Similar content being viewed by others

REFERENCES
S. C. Gad and H. L. Kaplan, Combustion Toxicology (CRC, Boca Raton, FL, 1990).
E. Hardy, Phosgene, Kirk-Othmer Encyclopaedia of Chemical Technology (Wiley, New York, 1971).
S. Virji, R. Kojima, J. Fowler, J. G. Villanueva, R. B. Kaner, and B. H. Weiller, Nano Res. 2, 135 (2009).
M. Burnworth, S. Rowan, and J. Weder, Chem. Eur. J. 13, 7828 (2007).
C. Tabtimsai, S. Keawwangchai, N. Nunthaboot, V. Ruangpornvisuti, and B. Wanno, J. Mol. Model. 18, 3941 (2012).
A. Kaushik, R. Kumar, S. Arya, M. Nair, B. Malhotra, and S. Bhansali, Chem. Rev. 115, 4751 (2015).
B. C. Wood, S. Y. Bhide, D. Dutta, V. S. Kandagal, A. D. Pathak, S. N. Punnathanam, K. G. Ayappa, and S. Narasimhan, J. Chem. Phys. 137, 054702 (2012).
X. Zhang, Z. Dai, Q. Chen, and Ju Tang, Phys. Scr. 89, 065803 (2014).
M. D. Ganji and M. Rezvani, J. Mol. Model. 19, 1259 (2013).
M. D. Ganji, N. Seyed-Aghaei, M. M. Taghavi, M. Rezvani, and F. Kazempour, Fullerenes Nanotubes Carbon Nanostruct. 19, 289 (2011).
P. Błoński and N. López, J. Phys. Chem. C 116, 15484 (2012).
M. Breedon, M. J. S. Spencer, and I. Yarovsky, J. Phys. Chem. C 114, 16603 (2010).
A. K. Geim and K. S. Novoselov, Nat. Mater. 6, 183 (2007).
Y. Zhang, J. W. Tan, H. L. Stormer, and P. Kim, Nature (London, U.K.) 438, 201 (2005).
F. Zhang, Q. Wu, X. Wang, N. Liu, J. Yang, Y. Hu, L. Yu, X. Wang, Z. Hu, and J. Zhu, J. Phys. Chem. C 113, 4053 (2009).
X. Zhang, Z. Liu, and S. Hark, Solid State Commun. 143, 317 (2007).
S. F. Rastegar, A. A. Peyghan, H. R. Ghenaatian, and N. L. Hadipour, Appl. Surf. Sci. 274, 217 (2013).
Y. Jiao, A. Du, Z. Zhu, V. Rudolph, and S. C. Smith, J. Phys. Chem. C 114, 7846 (2010).
P. Hohenberg and W. Kohn, Phys. Rev. B 136, 864 (1964).
W. Kohn and L. J. Sham, Phys. Rev. A 140, 1133 (1965).
P. Ordejon, E. Artacho, and J. Soler, Phys. Rev. B 53, 10441 (1996).
D. Sanchez-Portal, P. Ordejon, E. Artacho, and J. M. Soler, Int. J. Quantum Chem. 65, 453 (1997).
J. M. Soler, E. Artacho, J. D. Gale, A. Garcia, J. Junquera, P. Ordejon, and D. Sanchez-Portal, J. Phys.: Condens. Matter 14, 2745 (2002).
R. O. Jones and O. Gunnarsson, Rev. Mod. Phys. 61, 689 (1989).
J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996).
N. Troullier and J. L. Martins, Phys. Rev. B 43, 1993 (1991).
E. M. Fernández and L. C. Balbás, Phys. Chem. Chem. Phys. 13, 20863 (2011)
D. J. Carter and A. L. Rohl, J. Chem. Theory Comput. 8, 281 (2011)
M. Rezvani, M. D. Ganji, and M. Faghihnasiri, Phys. E 52, 27 (2013).
M. D. Ganji, M. Nashtahosseini, S. Yeganegi, and M. Rezvani, J. Mol. Model. 194, 1929 (2013).
M. Sabet and M. D. Ganji, J. Mol. Model. 199, 4013 (2013).
Z. Bagheri and A. Peyghan, Comput. Theor. Chem. 1008, 20 (2013).
Merck Index, 11th ed. 7310
M. D. Ganji, S. Jameh-Bozorgi, and M. Rezvani, Appl. Surf. Sci. 384, 175 (2016).
M. Rezvani, M. D. Ganji, and S. Jameh-Bozorgi, Appl. Surf. Sci. 360, 69 (2016).
F. L. Hirshfeld, Theor. Chim. Acta 44, 129 (1977).
C. F. Guerra, J. W. Handgraaf, E. J. Baerends, and F. M. Bickelhaupt, J. Comput. Chem. 25, 189 (2003).
G. J. M. Velders and D. Feil, Theor. Chim. Acta 84, 195 (1992).
G. J. M. Velders, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders, and T. Ziegler, J. Comput. Chem. 22, 931 (2001).
Z. M. Ao, J. Yang, S. Li, and Q. Jiang, Chem. Phys. Lett. 461, 276 (2008).
S. Li, Semiconductor Physical Electronic, 2nd ed. (Springer, USA, 2006).
Y. H. Zhang, Y. B. Chen, K. G. Zhou, C. H. Liu, J. Zeng, H. L. Zhang, and Y. Peng, Nanotechnology 20, 185504 (2009).
S. F. Rastegar, N. L. Hadipour, and H. Soleymanabadi, J. Mol. Model. 20, 2439 (2014).
R. G. Parr and Y. Weitao, Density Functional Theory of Atoms and Molecules (Oxford Univ. Press, New York, 1989).
T. Koopmans, Physica (Amsterdam, Neth.) 1, 104 (1934).
R. G. Parr and R. G. Pearson, J. Am. Chem. Soc. 105, 7512 (1983).
J. Martínez, Chem. Phys. Lett. 478, 310 (2009).
G. L. Gázquez, Struct Bond. 80, 27 (1993).
R. G. Parr, L. V. Szentpály, and S. Liu, J. Am. Chem. Soc. 121, 1922 (1999).
J. L. Gázquez, A. Cedillo, and A. Vela, J. Phys. Chem. A 111, 1966 (2007).
ACKNOWLEDGMENTS
The authors would like to acknowledge the financial support from the Office of the Vice-Chancellor in charge of research of Arak branch, Islamic Azad University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Banibairami, T., Jamehbozorgi, S., Ghiasi, R. et al. Sensing Behavior of Hexagonal-Aluminum Nitride to Phosgene Molecule Based on Van der Waals–Density Functional Theory and Molecular Dynamic Simulation. Russ. J. Phys. Chem. 94, 581–589 (2020). https://doi.org/10.1134/S0036024420030048
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024420030048