
Abstract
Over the past few years, the attention of the whole world has been riveted to the emergence of new dangerous strains of viruses, among which a special place is occupied by coronaviruses that have overcome the interspecies barrier in the past 20 years: SARS viruses (SARS), Middle East respiratory syndrome (MERS), as well as a new coronavirus infection (SARS-CoV-2), which caused the largest pandemic since the Spanish flu in 1918. Coronaviruses are members of a class of enveloped viruses that have a lipoprotein envelope. This class also includes such serious pathogens as human immunodeficiency virus (HIV), hepatitis, Ebola virus, influenza, etc. Despite significant differences in the clinical picture of the course of disease caused by enveloped viruses, they themselves have a number of characteristic features, which determine their commonality. Regardless of the way of penetration into the cell—by endocytosis or direct fusion with the cell membrane—enveloped viruses are characterized by the following stages of interaction with the target cell: binding to receptors on the cell surface, interaction of the surface glycoproteins of the virus with the membrane structures of the infected cell, fusion of the lipid envelope of the virion with plasma or endosomal membrane, destruction of the protein capsid and its dissociation from the viral nucleoprotein. Subsequently, within the infected cell, the newly synthesized viral proteins must self-assemble on various membrane structures to form a progeny virion. Thus, both the initial stages of viral infection and the assembly and release of new viral particles are associated with the activity of viral proteins in relation to the cell membrane and its organelles. This review is devoted to the analysis of physicochemical mechanisms of functioning of the main structural proteins of a number of enveloped viruses in order to identify possible strategies for the membrane activity of such proteins at various stages of viral infection of the cell.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Most processes in cell membranes are mediated by specific proteins. Protein–lipid interactions in the cell regulate membrane topological rearrangements, such as fusion and division of cells and their organelles, endo- and exocytosis, which accompanied by the formation, budding and transport of membrane vesicles. These interactions require the coordinated work of a large complex of proteins that determines the complexity of their study and experimental modeling [1‒4]. Although the membrane fusion requires cooperative protein–lipid interactions, the mechanical properties of the lipid matrix surrounding membrane proteins determine the energy of the formation of intermediate structures through which membrane fusion is realized [5]. Experiments conducted by L.V. Chernomordik and G.B. Melikyan on flat lipid bilayers in the Laboratory of Bioelectrochemistry of the Institute of Electrochemistry of the Academy of Sciences of the USSR, headed by Y.A. Chizmadzhev, have demonstrated that the fusion process consists of the following sequential stages: (1) establishment of the tight contact between membranes, (2) formation of the stalk (or bridge) between adjacent monolayers, (3) expansion of the stalk, leading to the formation of a trilamellar structure (or hemifusion diaphragm) and, finally, (4) its rupture, indicating the end of the fusion [6]. It is shown that the rate of these stages essentially depends on the spontaneous curvature of the lipid monolayers. Thus, the negative spontaneous curvature of the contacting monolayers (a typical example is dioleoylphosphatidylethanolamine) promotes stalk formation, while the rupture of the hemifusion diaphragm is accelerated in the presence of positive spontaneous curvature (i.e., lysolipids) in the distal monolayers. The proof of this fusion scheme has been obtained by V.S. Markin and M.M. Kozlov, members of the Laboratory of Bioelectrochemistry [7]. The theory quantitatively describes the entire set of experiments conducted at that time. A detailed description and relevant literature can be found in the review [8].
Further studies of the membrane fusion required the consideration of the membrane protein component. The first works in this direction have been aimed at studying synaptic fusion and the action of SNARE complex proteins [9, 10]. However, the large number of proteins included in this complex makes it difficult to clarify the physical-chemical mechanisms of their function and to establish the energy of the intermediate stages. For this reason, the study of virus-induced fusion, which in a number of systems is mediated by a single protein of known structure (e.g., hemagglutinin (HA) in the case of influenza virus) has come to the fore [11]. Several HA molecules are thought to form a fusion rosette and then lipid protrusions (dimples). On their tops, after merging, monolayer junctions (stalks) are formed, which lead to the monolayer (and subsequently, to the complete) fusion. In various model systems based on the hemagglutinin-expressing cells [12], it was shown that the effect of the lipid composition of the target membrane on fusion is well described within the stalk theory. In the same works, it was shown that the role of HAs is the formation of a complex, a “rosette” of fusion proteins, within which cell membranes merge to a distance sufficient for the subsequent spontaneous membrane fusion. Thus, the fusion theory assumes that the action of hemagglutinin molecules is necessary for the local merging of lipid bilayers of cell membranes, which fuse spontaneously similar to two planar bilayer lipid membranes (BLM). However, experimental observations obtained in cellular systems, such as the absence of lipid exchange through small fusion pores, are poorly described within the proposed model. Moreover, recent electron tomography data on influenza virus fusion with lipid vesicles show the formation of structures with only the target membrane bending, while the viral membrane remains nearly undeformed [13]. These facts lead to the conclusion that the classical stalk theory, which well describes fusion in the case of compositionally symmetric and purely lipid membranes, may not be applicable to the protein-mediated fusion.
In enveloped viruses, a shell of capsid or matrix proteins is located under the outer lipid membrane. These proteins constitute the majority of viral proteins. Interacting with the viral genome and surface glycoproteins, they play an important role in the assembly of viral particles and the production of progeny virions in the infected cell [14–16]. Matrix proteins are multifunctional: on the one hand, they maintain the integrity of the virus, on the other, they disassemble to release genetic material into the cytoplasm during infection and assemble to form new virions. In all these processes, matrix proteins interact with lipid membranes—the virus outer envelope or the cell plasma membrane. This interaction determines the life activity of the virus, although its molecular mechanisms remain an open question. In particular, the influenza A virus penetrates the cell by endocytosis, entering the cellular endosome as a result. The main trigger for the fusion of the viral particle and the endosomal membrane is the change of the pH of the endosomal milieu to 4–5, leading to conformational transitions in the HA and to the destruction of the capsid formed by the matrix protein M1. The drop of pH inside the virus occurs due to the action of M2 proton channels located in the viral envelope. In experiments conducted under the guidance of Yu.A. Chizmadzhev it was shown that blocking M2 channels with amantadine leads to “freezing” of viral fusion at the stage of a small fusion pore with a dimeter of about 1 nm, so that the genetic material of the virus is not released into the cell cytoplasm [17]. Thus, on the one hand, the disintegration of the envelope of matrix proteins is a critical step in the viral infection. On the other hand, matrix proteins must assemble on plasma membrane of the infected cell into the structure of a new virion. The process of assembly and budding of the progeny virion implies local deformation of cell membrane, in which matrix or capsid proteins should also play an important role. Nevertheless, the specific physico-chemical mechanisms defining the self-assembly of the viral capsid and changes in the cell membrane topology often remain unclear.
VIRUS-INDUCED MEMBRANE FUSION
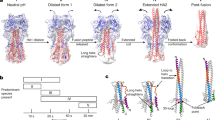
The envelope virus genome and capsid (nucleocapsid) proteins are surrounded by an additional envelope, a bilayer lipid membrane that is acquired from the host cell during virion release from the infected cell [18] (Fig. 1). Enveloped viruses use membrane fusion to enter the cell [19]. For this process, viruses use the function of fusion proteins. Conformational rearrangements of these proteins initiated by a certain trigger (e.g., binding to a receptor on the cell surface or a change of pH inside the cell endosome) initiate the fusion between the virus lipid membrane and cell membrane [20]. These proteins do not require energy-supplying molecules for activity [21].

Structure of enveloped viruses. (a) Coronavirus SARS-CoV-2; (b) influenza A virus; (c) Newcastle disease virus; (d) Human immunodeficiency virus. Abbreviations: HA, hemagglutinin; NA, neuraminidase; RNP, ribonucleoprotein; M1, M1 protein; M2, M2 proton channel.
The entry of any virus into the cell begins with its binding to certain receptor molecules on the cell surface [22] (Fig. 2). Membrane fusion is the most important stage in the life cycle of enveloped viruses. Initially, the genetic material of the virus is separated from the cell cytoplasm by two membranes: the viral and the cell. As a result of the fusion of these membranes, the cell cytoplasm and the internal space of the virion containing the viral genetic material are combined. Some enveloped viruses fuse directly with the plasma membrane, while others enter the cell through endocytosis.

Examples of life cycles of enveloped viruses. (a) Influenza A virus; (b) Newcastle disease virus; (c) Human immunodeficiency virus.
Membrane fusion is an important part of the cell life activity, mediating many vital processes. The most representative example of such a process is the synaptic transmission, during which synaptic vesicles containing neurotransmitters fuse with the presynaptic plasma membrane [23]. In addition, organelles also merge during cell fusion, for example, in the processes of fertilization, carcinogenesis, etc. [24–27]. Certain fusion proteins are involved in these processes [28, 29]. For example, during hormone and neurotransmitter secretion, the SNARE protein complex catalyzes membrane fusion [9, 10]. Intercellular fusion is also provided by specific proteins, which, however, are detected only in some cases, but for most remain unknown [30]. Fusion proteins are responsible for the deformation of interacting membranes, followed by their fusion and mixing of initially separated aqueous volumes. The function of these proteins in the fusion process in each case determines their structure, number, presence of auxiliary proteins, etc. As usual, viral membrane fusion is performed by one or two proteins; in particular, the influenza A virus has only one fusion protein—hemagglutinin [31]. Membrane fusion, as any other membrane topological rearrangements, requires energy for the mechanical deformations of the lipid matrix. Thus, protein–lipid interactions determine the energy of viral-induced membrane fusion and, therefore, regulate this stage of the infection process. Theoretical estimates show that the characteristic height of the fusion energy barriers is several tens to hundreds of kBT, and such barriers cannot be overcome only by thermal fluctuations [32]. The mechanical energy reserved in the fusion proteins covers the energy costs.
Many experimental data show that membrane fusion can occur spontaneously if substances connecting the two interacting membranes (e.g., Ca2+) or dehydrating their contact (e.g., polyethylene glycol) have been added between the fusing membranes [33, 34]. These data suggest that bringing two membranes into a close contact and overcoming hydration repulsion are the most energy-consuming phases of fusion, which cannot occur spontaneously due to the energy of thermal fluctuations of lipids. As further fusion in such model systems occurs without the presence of any proteins, it can be concluded that the energy barriers on the rest of the fusion trajectory should not be measurably affected. Thus, the function of fusion proteins is largely similar to enzymes: they can reduce the basic energy activation barrier for fusion and possibly accelerate the rate constant of the reaction.
Type I viral fusion proteins (e.g., influenza A virus HA and HIV Env gp120/gp41 protein) are the most well studied [21]. At the beginning of the infection viral fusion proteins bind specific receptor molecules on the cell surface. Immediately thereafter (or when the pH inside the endosome changes), the fusion protein attacks the cell membrane with a fusion peptide, which is a specific N-terminus consisting of about 20 amino acids [35]. The fusion peptide releases from the hydrophobic pocket of the ectodomain of HA and then incorporates into the lipid bilayer of the target membrane as an effective anchor. In the process of ongoing conformational rearrangements, the fusion protein is refolded, attaching the target cell membrane with the embedded fusion peptide to the viral membrane [36]. These membranes come into close contact, merge, and form a fusion pore, through which the viral genome can be released into the cytoplasm. At this stage, the fusion protein folds into a “post-fusion” conformation when its transmembrane domain contacts the fusion peptide [31]. If the fusion peptide does not attain the target membrane, it can integrate into the viral membrane next to the transmembrane domain of the fusion protein, also bringing it into the post-fusion conformation. The height of the fusion protein in this state is about 10 nm [37].
Fusion peptides of viral proteins are generally amphipathic [38, 39]. They can partially integrate into the cell membrane, providing both an anchor and a lever for the forces and moments, as well as deform the target membrane locally. The depth of the fusion peptide integration regulates its fusion activity [39]. However, replacement of the transmembrane domain of the fusion protein with a lipid anchor prevents the extension of the fusion pore [40, 41]. Hence, both the fusion peptide and the transmembrane domain of the respective protein are necessary for the efficient fusion. In fact, fusion proteins form protrusions on the viral and the cell contact membranes. The intense hydration repulsion acting on the tops of these protrusions leads to the lateral shift of the polar lipid heads from the region of a tight contact [42–44]. With a certain probability, these local protrusions of the opposite membranes can touch, forming a stalk [44].
A number of theoretical models have shown that the main energy barrier for the interaction of membrane protrusions induced by fusion proteins strongly depends on the distance between these membranes [32, 45]. This distance is defined by the thickness of the layer of fusion proteins in the post-fusion conformation, whose fusion peptides have not reached the target cell membrane. For hemagglutinin, this thickness is about 10 nm, as shown by electron microscopy [37]. Thus, fusion proteins must locally overcome this distance for bringing the fusion membranes closer over a small contact area, providing a relatively low energy barrier for hemifusion. According to various estimates, the energy required for such membrane rearrangement is on the order of several tens of kBT [32, 45, 46]. For comparison, the energy stored in a single hemagglutinin trimer is about 60 kBT, i.e., 20 kBT per hemagglutinin monomer [47]. Roughly, the same energy estimation is also obtained for the HIV gp41 trimer [48]. This indicates that effective membrane fusion requires the cooperative action of several fusion proteins that form the fusion rosette. In the case of influenza A virus, the required number of HA trimers in the fusion rosette is estimated to be between 3 and 9 [49, 50]. However, in the case of HIV, it is known that one trimer of the gp41 protein may be sufficient for the fusion process [50].
A specific feature of type I fusion proteins is the intensive deformation of the target membrane. The viral membrane is deformed less due to the presence of a capsid protein envelope in contact with it [37]. The deformation of the target membrane is caused by the integration of fusion peptides into it. Such interactions have been considered in numerous theoretical works [51–53]. In particular, it is shown that the study of the cooperative fusion rosette formation should consider the dependence of the elastic energy on the mutual orientation of fusion peptides rather than on the distance between them, as positions of viral fusion proteins are rather strictly fixed because of their transmembrane domains that interact with the viral capsid. The fusion peptides released by viral fusion proteins are also anchors, through which fusion proteins can exert mechanical forces and moments on the target membrane [11, 54]. Thus, lipid matrix protrusions required for membrane fusion can be formed either by elastic deformations of embedded fusion peptides or by direct application of forces of fusion proteins, which tighten the two fusing membranes [55, 56].
The shallow integration of amphipathic fusion peptides into the target membrane is similar to the formation of positive spontaneous curvature, which is induced, for example, by lysolipids, and leads to pore formation [57], but not to stalk. Thus, the physico-chemical mechanism of amphipathic peptide functioning for fusion proteins cannot be based only on the modification of the target membrane’s contact monolayer. Apparently, it necessarily requires the application of constricting forces to the two membranes. To decrease the energy barrier of stalk formation, fusion peptides must effectively induce negative spontaneous curvature [57]. For this purpose, their insertion depth into the contact monolayer of the target membrane should be increased. Calculations made in [58, 59], which explicitly take into account the depth of fusion peptide insertion, fully support this view. Moreover, there is an experimental evidence for the HIV fusion protein peptide that its depth of integration into the contact monolayer of the target membrane increases the probability of fusion [39].
In the case of HIV, fusion can be catalyzed by several Env proteins down to a single protein trimer [50]. The key difference is that in addition to fusion, the Env proteins are also responsible for the virus reception, i.e., its binding to the cellular plasma membrane [60]. The gp120 protein recognizes the CD4 receptor and the coreceptor CXCR4 or CCR5, which are large transmembrane proteins. Binding to the receptor triggers a conformational rearrangement of the Env glycoprotein. This type of viral fusion trigger additionally limits the number of simultaneously activated gp120/gp41 trimers due to the relatively low surface density of CD4 molecules in the plasma membrane of T-lymphocytes. This differs from the case of influenza A virus, whose hemagglutinin activation is caused by a decrease of the pH in the late endosome, i.e., all virion hemagglutinin trimers are activated almost simultaneously.
Two possible mechanisms of action of fusion proteins have been commonly proposed. The first one is based on the assumption that integrated fusion peptides modify the elastic properties of the target membrane, especially its spontaneous curvature [61]. It is assumed that the modified spontaneous curvature in the ring zone of the fusion rosette may be the trigger for the formation of a protrusion in the target membrane. The membranes come into close contact at the top of the protrusion, which significantly facilitates the fusion. According to the second mechanism, rather than modifying the target membrane, fusion proteins induce bending moments, resulting in the formation of protrusions. Proteins also generate forces by directly and mechanically bringing two tightly attached membranes into close contact [31]. The analysis in [62] showed that it is the second mechanism that provides the formation of a highly symmetric fusion rosette by organizing the cooperativity of the mechanical efforts of several fusion proteins. In contrast, the first mechanism uses rosette symmetry to explain protrusions on the fusion membranes but cannot explain the nature of such symmetry. Therefore, the directed mechanical activity of fusion proteins controls the entire fusion process. This activity may be the result of the cooperative effect of various fusion protein subunits, including those sites involved in the interaction with the receptor. This may explain the observed difference in the cooperative action of influenza A virus and HIV fusion proteins. Therefore, the development of elastic models of the membrane fusion has provided answers to many questions about the mechanisms of viral-induced membrane fusion, in particular, about possible triggers of the cooperative effects of viral fusion proteins.
CAPSID PROTEINS OF ENVELOPED VIRUSES AND FORMATION OF PROGENY VIRIONS
Under the outer lipid membrane of enveloped viruses, there is a capsid of matrix proteins. Interacting with the viral genome and surface glycoproteins, they play an important role in the assembly of viral particles and the production of progeny virions in the infected cell [14–16]. Matrix proteins are multifunctional at different stages of the virus life cycle. On the one hand, they maintain virion integrity and architecture; on the other hand, they perform a controlled disintegration of the capsid to release viral genetic material into the cytoplasm; on the third—organize the assembly of progeny virions. In all these processes, matrix proteins interact with the lipid membranes of the virus or cell. This interaction is a determinant in the virus life activity, although the molecular mechanisms are still unknown. The nature of such protein–lipid interactions, as well as their relation to the capsid self-assembly, is debated [63–67].
Despite the diversity of envelope viruses, their matrix proteins are organized in a similar way: they are either helical structures (as in the case of influenza A virus [14, 68, 69], vesicular stomatitis virus [15] and measles virus [16]) or two-dimensional lattices forming perispherical particles (for example, Newcastle disease virus [70] or HIV [71]). Evolutionarily and structurally matrix proteins of different virus families are very similar, such as the polyprotein Gag of the HIV and the M1 protein of the influenza A virus [72]. Such proximity should also indicate common mechanisms of capsid formation and virion budding from the surface of the infected cell.
The relationship between the three-dimensional structure and functional properties of a protein is one of the most important basic concepts in biochemistry. To perform a particular function, a protein should have a specific amino acid sequence and be folded in a certain way [15]. This idea, however, has been reconsidered about two decades ago when many proteins with a partially or completely disordered structure have been found. These proteins, called intrinsically disordered proteins, exist as dynamic ensembles of conformations that do not have a stable folded structure, but nevertheless perform their (often very variable) biological functions [73]. Disordered domains of proteins can promote the rapid response of viruses to changing environmental conditions and, consequently, their survival and development by implementing mechanisms of adaptation and protection [74]. Disorder in protein folding prevents antibody binding that reduces the immune response. This is the key to the extraordinary ability of HIV to evade the immune response, with main contribution of the viral capsid structural features [74].
The process of viral budding is generally similar to the cellular exocytosis in the way of formation a curved structure from the cell membrane. A minimal set of proteins in the viral envelope reduces possible ways the virus can affect the membrane. There are three possible ways of such effect, which are occurring in different viruses separately or in complex combinations. The first mechanism consists in the formation of a virion due to the certain molecular geometry of matrix proteins. It allows proteins to bind the lipid bilayer and form a two-dimensional lattice on the lipid surface. X-ray analysis demonstrates the existence of such mechanism for the Newcastle disease virus [70] and Salmonella infectious anemia virus [75]. In both cases, protein monomers bind in a capsid structure with a certain angle to each other, forming hemispherical viral particles. A nucleation of such particles occurs by the formation of circle protein–lipid domains [76] followed by curving of the lipid membrane. The liquid state of the lipid bilayer implies that transfer of the curvature to the lipid membrane requires the layer of matrix proteins to minimize lipid redistribution in the areas of protein–lipid contacts. Apparently, this should be achieved by hydrophobic interactions, which is the case of the Newcastle disease virus [77]. This mechanism of interaction has been suggested in the early studies of the interaction of the influenza A virus matrix protein with the lipid bilayer [78–81]. However, it has not been proved in later studies [63], which show neither partial integration of the M1 protein into the lipid bilayer, nor its ability to force a certain curvature on the membrane. These studies suggest the predominant electrostatic nature of the interaction of matrix protein M1 with the membrane.
The second possible mechanism is the condensation of lipids under the protein layer due to electrostatic interactions. Such condensation on the inner monolayer of the plasma membrane creates a local imbalance of the membrane monolayer area, leading to the membrane bending and the formation of various outgrowths to relief the excess area [82]. In particular, a formation of filamentous structures close in diameter to viral particles is shown in experiments on negatively charged giant unilamellar vesicles during the adsorption of the influenza C virus matrix protein [83]. Electrostatic interactions with anionic lipids are common to matrix proteins of viruses such as influenza virus [63, 84], Ebola virus [85, 86], and vesicular stomatitis virus [87]. For all these viruses it has been shown that their matrix proteins are able to form virus-like particles [87–90]. At the same time, additional factors contributing to the change in membrane curvature play role in this mechanism of the viral budding. For example, it has been shown that a palmitoylation of the HA of the influenza A virus, that promotes the formation of the correct curvature of the viral particle, enhances the adsorption of protein M1 at the lipid membrane and facilitates the formation of virus-like particles [91].
For some enveloped viruses it was shown that their membrane is more resistant to detergents than the cell plasma membrane [92–96], as it has a raft structure [97]. The presence of lipid rafts in the viral membrane provides another mechanism of vesicle formation and budding: by increasing the linear tension of the raft boundary with their following release from the membrane surface [98]. However, the mechanism of this process has not yet been demonstrated for any enveloped virus. It is supposed that lipid domains can be formed by condensation of negatively charged lipids with fully saturated hydrocarbon chains on the inner monolayer of the plasma membrane, followed by the formation of a sphingomyelin-enriched lipid domain on the outer part of the membrane [99].
Enveloped viruses of different families have different pathways in the cell and differ in the primary sequence of structural proteins, but nevertheless use a number of common physico-chemical mechanisms at different stages of cell infection. The evolutional similarity of the matrix and capsid proteins of different enveloped viruses, as well as the common elements in their tertiary structure, also indicate possible general mechanisms of the capsid self-assembly and disintegration. Viral fusion proteins are able to create a small fusion pore; however, blocking the dissociation of the protein envelope prevents the release of the viral genome into the cell cytoplasm [13, 41]. It is still unclear whether matrix and capsid proteins are active participants in the expansion of the fusion pore or whether they only dissociate from the lipid envelope of the virion. The multifunctionality of viral proteins indicates that matrix proteins can also perform several functions. The effect of these proteins on the cellular transport of viral genetic material has been determined [100–102], but no effect on the release of the viral genome has been demonstrated.
These proteins must also self-assemble to form a viral particle and release it from the infected cell. The formation and budding of the viral particle require extensive topological rearrangements of the cell membranes. This leads to the question of how and which viral proteins can do this. The focus is on matrix proteins, since their amount is the highest for enveloped virus proteins, and they are the ones that establish and support the viral architecture. Therefore, in the process of self-organization, capsid proteins should provide deformations of cell membranes and conditions for the budding or release of virions. Limitations of membrane structure modification strategies imply general functional mechanisms that can be described within the framework of physico-chemical models, but this area is currently less well developed than the issues of virus-induced membrane fusion.
CORONAVIRUSES: A NEW CHALLENGE FOR PHYSICO-CHEMICAL MODELS
Coronaviruses have attracted a great attention as triggers of outbreaks of human respiratory syndromes such as severe acute respiratory syndrome (SARS), Middle East respiratory syndrome (MERS) and new coronavirus infection (COVID-19). These respiratory diseases have arisen from zoonotic transmission from animals to humans, resulting in virus strains that have not been previously circulated in the human population. The SARS-CoV-2 coronavirus, the causative agent of COVID-19, has shown rapid global spread and has led to a pandemic.
Coronaviruses, like the viruses discussed above, belong to the class of enveloped viruses. The coronavirus genome contains four main structural proteins: spike (S), membrane protein (M), envelope protein (E), and nucleocapsid protein (N), which are coded at the 3'-end of the genome [103]. The S protein mediates the viral binding to the surface receptors of the host cell, which leads to the fusion and subsequent entry of the virus. The M protein is the most represented and determines the shape of the viral envelope. The E protein has the lowest amount of the major structural proteins and is involved in viral assembly and budding. The N protein is the only one that binds to the RNA genome and also participates in the assembly and budding. It should be noted that, compared to other enveloped viruses, almost all structural proteins of coronaviruses have transmembrane domains.
Despite their complexity and range of functions [104, 105], structural proteins of coronaviruses cover only about one-third of the genome’s coding part. A larger site of the genome, about two-thirds, located at the 5'-end and codes two long open reading frames 1a and 1b of the viral nonstructural proteins. Each sequence is primarily translated as a polyprotein precursor, pp1a and pp1ab. Polyproteins include several viral proteases that transform pp1a and pp1ab into 16 nonstructural proteins (nsp 1–16) needed at different stages of the viral replication cycle. These proteins are the most conservative proteins of coronaviruses [103]. Many nonstructural proteins interact with membranes as coronavirus replication occurs in dedicated cellular compartments. These are created by viral proteins that modify cell membranes to create sites of viral replication hidden from cell innate immunity [106]. The combination of multiple membrane interacting factors makes coronaviruses one of the most complex models.
Replication of RNA viruses with positive RNA chain, as well as DNA viruses in plant and animal cells causes the formation of a subcellular microenvironment—a replication network—viral factories, or viroplasma [107]. Although the purpose of such membrane formations in the viral life cycle is not entirely known, it is assumed to facilitate the stage of the viral RNA synthesis [108, 109]. The formation of such “mini-organelles” requires a deformation of the host cell membranes and cytoskeleton, causing a cytopathic effect, that has been used as a marker of viral infection to test the potential efficacy of drugs [110]. All positive-chain RNA viruses (+RNA viruses) that infect eukaryotic cells are supposed to form membrane-bound organelles [111]. One of the most common membrane modifications induced by +RNA viruses is the formation of double membranes, two closely spaced lipid bilayers that give a structure for double-membrane vesicles [108]. The widespread of such structures in the replication of +RNA viruses suggests that this is an effective strategy to produce new virions, and that membrane pairing can enhance the competitiveness of these viruses.
The coronavirus family has a highly complex replication membrane network that originates from the endoplasmic reticulum [106], in a similar manner to the Dengue virus [112] and reovirus [113]. Its structure includes many curved membranes and double-membrane vesicles. All of them are interconnected in a continual network with each other and with the endoplasmic reticulum by lipid nanotubes [114]. This restructuring of host cell membranes is considered to be a viral strategy for replication by localizing and concentrating the essential factors and providing protection against the immune response [115]. This hypothesis is proved by the fact that the total level of viral RNA correlates with the number of double-membrane vesicles in the cell [106, 116, 117]. However, there are data that do not confirm the relationship between double-membrane vesicles in the endoplasmic reticulum and viral RNA synthesis in the infected cell [118]. It has been shown that mutations in the viral nonstructural proteins, which prevent them from forming double-membrane vesicles, do not lead to a complete cessation of the viral RNA synthesis. However, in this case the viral replication rate drops considerably.
The mechanism of double-membrane vesicle formation is still unclear. An analysis of the physico-chemical mechanisms of the formation of the double-membrane vesicle from a double-membrane disk is performed in [119]. In this work, the bending energies of the membranes of the double-membrane vesicle and the double-membrane disk are calculated and compared within the framework of the Helfrich elasticity theory [120]. The bending energy of the spherical double-membrane vesicle is independent of its radius, while the bending energy of the double-membrane disk increases linearly as the disk radius increases. At some disc radius, these two energies are compared, and the double-membrane disc can transform into the double-membrane vesicle. This work has not considered the processes of the double-membrane disk formation from a flat membrane or the formation of the double-membrane vesicle from the double-membrane spherical segment. Also, the shape of the membranes is postulated rather than found by elastic energy optimization, and the proteins involved have not been described.
Thus, it is still an open question whether viral replicases are capable to induce membrane curvature on the target organelle. This implies that these proteins must be capable of generating lattice-like structures. Also, they should have lipid specificity since there is no information on other types of specific targeting of these replicases in the endoplasmic reticulum. In hepatitis C, which also forms membrane replication compartments from the endoplasmic reticulum, the NS5A protein stimulates phosphatidylinositol-4-kinase-III activity. This facilitates phosphatidylinositol-4-phosphate production on the cytoplasmic side of the endoplasmic reticulum and hypothetically facilitates RNA polymerase accumulation [121]. It has been shown that enrichment with phosphatidylinositol-4-phosphate is important for the replication of enterovirus and flavivirus RNA.
The fact that the nonstructural proteins of coronaviruses can form scaffolds that contribute to cell membrane deformations makes them similar to the matrix proteins of other enveloped viruses. This suggests the possibility of common mechanisms of protein–lipid and protein–protein interactions. Such double-membrane structures are not found in uninfected cells (except for autophagosomes), indicating that spontaneous “shrinkage” of the endoplasmic reticulum cannot be the driving force of the formation of double-membrane structures. Understanding this formation mechanism is critically important for clarifying the principles of organization of the viral replication compartments.
Another proposed mechanism of double-membrane vesicle formation involves cellular autophagy [122, 123]. It is assumed that the DFCP1 protein (double protein 1 containing the FYVE domain) binds phosphatidylinositol phosphate, thus forming curved regions of the endoplasmic reticulum called omegasomes [124]. Based on the data obtained by some coronaviruses, it has been suggested that their nsp 6 proteins generate autophagosomes [125].
The envelope E proteins of coronavirus play multiple roles during infection, including viral morphogenesis. They are small (74–109 amino acids) hydrophobic viroporins [126]. E proteins consist of two separate structural domains: a hydrophobic domain longer than the thickness of the lipid bilayer and a charged cytoplasmic tail. The role of E proteins in assembly and release is not completely understood. The necessity for them during virus morphogenesis depends on the viral type. Removal of E proteins from transmissible gastroenteritis virus (TGEV) leads to replication of competent but proliferation-deficient viruses [127], as in the case of MERS virus [128]. SARS-CoV demonstrated a 200-fold reduction in virus release in the absence of E protein, which depended on the cell type used for infection [129]. Hence, although the membrane-active proteins of coronaviruses, such as nsp 3, 4, and 6 and E protein, are highly conservative and critical for viral replication, the mechanisms of their functioning and interaction with cell membranes and with each other are still unclear.
The S protein is a glycosylated type I membrane fusion protein that consists of two subunits, S1 and S2. The N-terminal S1 subunit contains a receptor-binding domain (RBD) that mediates binding to the host cell receptor, namely angiotensin converting enzyme 2 (ACE2), for both SARS-CoV and SARS-CoV-2 [130, 131]. Biophysical structural and functional studies of the S protein fusion peptide of SARS-CoV-2 viruses [132], SARS [133], and MERS [134] provided important insights into the functional significance of specific domains of the fusion peptide, such as the large number of conserved amino acid residues in two consecutive fusion peptide fragments named FP1 and FP2 (residues 816–835 and 835–854, respectively, in SARS-CoV-2). These studies showed that collectively the F1 and F2 fragments form a platform of bilateral interactions between membranes, and the amino acid residues in both FP1 and FP2 contribute to membrane binding through their interaction with Ca2+ ions. These data on the functional role of Ca2+ are supported by the results obtained for fusion peptides from other related viruses, such as MERS [134] and Ebola virus [135]. In fact, in the MERS virus fusion peptide, one of these amino acid residues, E891 in the N-terminal (FP1) part (corresponding to E819 in the SARS-CoV-2 fusion peptide numeration), is proved to be crucial for interactions with Ca2+ and providing fusion-related cell membrane deformations [136]. In addition, it is shown that disruption of the lipid bilayer by Ca2+-dependent interactions of the fusion peptide with the membrane affects the organization of the lipid polar groups at the interaction site, but not the central hydrophobic region of the membrane [136]. Nevertheless, the Ca2+ binding sites with the fusion peptide and their role in any specific (but unknown) modes of interaction of the fusion peptide with the membrane remain uncertain. This makes difficult to explain any measurable effects of viral interactions with the membrane and, therefore, any approaches for decreasing infectivity by targeting this important region of the S protein.
As mentioned above, different activation mechanisms of fusion proteins and the depth of the fusion peptide incorporation into the lipid matrix of cell membranes regulate the cooperative action of fusion proteins through deformations of the target cell lipid bilayer rather than through direct protein–protein interactions [62]. A molecular model of the action of influenza A virus and human immunodeficiency virus fusion peptides makes it possible to clarify the role of receptor structure in the cooperativity of the virus fusion proteins. The fact that the initial stages of SARS-CoV-2 coronavirus entry are close to those of HIV (binding to the receptor on the cell surface followed by fusion with the plasma membrane), and the SARS-CoV-2 protein, as the HIV gp41/gp120 protein, refers to type I fusion proteins, suggests that the assumptions of this model are valid for describing the function of the fusion peptide and S protein of the coronavirus.
CONCLUSIONS
The life cycle of all enveloped viruses is a complex multistage process that begins with virus entry into the cell and ends with the release of newly formed virions from its membrane. It involves a precisely determined interaction between the components of the virus and the cell. Disruption of any stage of the virion life cycle prevents the formation of infectiously competent virions and limits the spread of infection.
A key feature of enveloped viruses is the presence of not only the protein shell that protects the viral genome, but also the lipid membrane, which interacts with it and contains the main structural proteins of the virion. Therefore, protein–lipid interactions play one of the defining roles in the processes of viral infection, replication, and release of new viral particles. Such interactions in most cases do not involve the formation of covalent chemical bonds, but are based on electrostatic, hydrophobic, and Van der Waals interactions. Hence, various physico-chemical mechanisms play an important role in the processes of cell infection by enveloped viruses.
Despite the common nature of the described processes for most enveloped viruses, there are still many open questions about the molecular mechanisms of certain stages, about the forces that determine the functioning and interaction of the various viral components and the infected cell. It is obvious that the search for new effective antiviral drugs should be based on the fundamental mechanisms regulating the functioning of viral proteins and determining various stages of viral infection in the cell. Models of physico-chemical mechanisms of viral fusion processes, formation of progeny virions, as well as new challenges related to the replication features of coronaviruses can become the basis for such search. The creation and analysis of such models, initiated by Yu.A. Chizmadzhev and his colleagues in the 1980s, has been successfully developed to this day thanks to his unique scientific school.
REFERENCES
McMahon H.T., Gallop J.L. 2005. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 438, 590–596.
Zimmerberg J., Kozlov M.M. 2006. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 7, 9–19.
Schekman R., Orci L. 1996. Coat proteins and vesicle budding. Science 271, 1526–1533.
Bremser M., Nickel W., Schweikert M., Ravazzola M., Amherdt M., Hughes C.A., Söllner T.H., Rothman J.E., Wieland F.T. 1999. Coupling of coat assembly and vesicle budding to packaging of putative cargo receptors. Cell. 96, 495–506.
Chernomordik L., Kozlov M.M., Zimmerberg J. 1995. Lipids in biological membrane fusion. J. Membr. Biol. 146, 1–14.
Melikyan G.B., Abidor I.G., Chernomordik L.V., Chailakhyan L.M. 1982. Electrically stimulated fusion and fission of bilayer lipid membranes. Dokl. Acad. Sci. USSR (Rus.). 263, 1009–1012.
Kozlov M.M., Markin V.S. 1983. Possible mechanism of membrane fusion. Biofizika (Rus.). 28, 242–247.
Chernomordik L.V., Kozlov M.M., Melikyan G.B., Abidor I.G., Markin V.S., Chizmadzhev Yu.A. 1985. The shape of lipid molecules and monolayer membrane fusion. Biochim. Biophys. Acta BBA – Biomembr. 812, 643–655.
Wang T., Li L., Hong W. 2017. SNARE proteins in membrane trafficking. Traffic. 18, 767–775.
Jahn R., Scheller R.H. 2006. SNAREs – engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643.
Skehel J.J., Wiley D.C. 2000. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 69, 531–569.
Chernomordik L.V., Frolov V.A., Leikina E., Bronk P., Zimmerberg J. 1998. The pathway of membrane fusion catalyzed by influenza hemagglutinin: Restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 140, 1369–1382.
Lee K.K. 2010. Architecture of a nascent viral fusion pore. EMBO J. 29, 1299–1311.
Calder L.J., Wasilewski S., Berriman J.A., Rosenthal P.B. 2010. Structural organization of a filamentous influenza A virus. Proc. Natl. Acad. Sci. USA. 107, 10685–10690.
Ge P., Tsao J., Schein S., Green T.J., Luo M., Zhou Z.H. 2010. Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science. 327, 689–693.
Liljeroos L., Huiskonen J.T., Ora A., Susi P., Butcher S.J. 2011. Electron cryotomography of measles virus reveals how matrix protein coats the ribonucleocapsid within intact virions. Proc. Natl. Acad. Sci. USA. 108, 18085–18090.
Chizmadzhev Y.A. 2004. The mechanisms of lipid–protein rearrangements during viral infection. Bioelectrochemistry. 63, 129–136.
Pettersson R.F. 1991. Protein localization and virus assembly at intracellular membranes. In: Protein traffic in eukaryotic cells. Vol. 170. Current topics in microbiology and immunology. Ed. Compans R.W. Berlin, Heidelberg: Springer, p. 67–106.
White J., Kielian M., Helenius A. 1983. Membrane fusion proteins of enveloped animal viruses. Q. Rev. Biophys. 16, 151–195.
Kielian M., Rey F.A. 2006. Virus membrane-fusion proteins: More than one way to make a hairpin. Nat. Rev. Microbiol. 4, 67–76.
Colman P.M., Lawrence M.C. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4, 309–319.
Poehlmann S., Simmons G. 2013. Viral entry into host cells. New York: Springer Science.
Südhof T.C. 2013. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron. 80, 675–690.
Hernandez L.D., Hoffman L.R., Wolfsberg T.G., White J.M. 1996. Virus–cell and cell–cell fusion. Annu. Rev. Cell Dev. Biol. 12, 627–661.
Duelli D., Lazebnik Y. 2003. Cell fusion: A hidden enemy? Cancer Cell. 3, 445–448.
Chen E.H. 2005. Unveiling the mechanisms of cell–cell fusion. Science. 308, 369–373.
Chen E.H., Grote E., Mohler W., Vignery A. 2007. Cell–cell fusion. FEBS Lett. 581, 2181–2193.
White J. 1992. Membrane fusion. Science. 258, 917–924.
Wickner W., Schekman R. 2008. Membrane fusion. Nat. Struct. Mol. Biol. 15, 658–664.
Brukman N.G., Uygur B., Podbilewicz B., Chernomordik L.V. 2019. How cells fuse. J. Cell Biol. 218, 1436–1451.
Harrison S.C. 2015. Viral membrane fusion. Virology. 479–480, 498–507.
Kuzmin P.I., Zimmerberg J., Chizmadzhev Y.A., Cohen F.S. 2001. A quantitative model for membrane fusion based on low-energy intermediates. Proc. Natl. Acad. Sci. USA. 98, 7235–7240.
Lentz B.R. 2007. PEG as a tool to gain insight into membrane fusion. Eur. Biophys. J. 36, 315–326.
Hong J., Yang H., Pang D., Wei L., Deng C. 2018. Effects of mono- and di-valent metal cations on the morphology of lipid vesicles. Chem. Phys. Lipids. 217, 19–28.
White J.M., Delos S.E., Brecher M., Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43, 189–219.
Benhaim M.A., Lee K.K. 2020. New biophysical approaches reveal the dynamics and mechanics of type I viral fusion machinery and their interplay with membranes. Viruses. 12, 413.
Chlanda P., Mekhedov E., Waters H., Schwartz C.L., Fischer E.R., Ryham R.J., Cohen F.S., Blank P.S., Zimmerberg J. 2016. The hemifusion structure induced by influenza virus haemagglutinin is determined by physical properties of the target membranes. Nat. Microbiol. 1, 16050.
Sammalkorpi M., Lazaridis T. 2007. Configuration of influenza hemagglutinin fusion peptide monomers and oligomers in membranes. Biochim. Biophys. Acta BBA – Biomembr. 1768, 30–38.
Qiang W., Sun Y., Weliky D.P. 2009. A strong correlation between fusogenicity and membrane insertion depth of the HIV fusion peptide. Proc. Natl. Acad. Sci. USA. 106, 15314–15319.
Frolov V.A., Cho M.-S., Bronk P., Reese T.S., Zimmerberg J. 2000. Multiple local contact sites are induced by GPI-linked influenza hemagglutinin during hemifusion and flickering pore formation. Traffic. 1, 622–630.
Markosyan R.M., Cohen F.S., Melikyan G.B. 2000. The lipid-anchored ectodomain of influenza virus hemagglutinin (GPI-HA) is capable of inducing nonenlarging fusion pores. Mol. Biol. Cell. 11, 1143–1152.
Leikin S.L., Kozlov M.M., Chernomordik L.V., Markin V.S., Chizmadzhev Y.A. 1987. Membrane fusion: Overcoming of the hydration barrier and local restructuring. J. Theor. Biol. 129, 411–425.
Helm C., Israelachvili J., McGuiggan P. 1989. Molecular mechanisms and forces involved in the adhesion and fusion of amphiphilic bilayers. Science. 246, 919–922.
Kozlovsky Y., Efrat A., Siegel D.A., Kozlov M.M. 2004. Stalk phase formation: Effects of dehydration and saddle splay modulus. Biophys. J. 87, 2508–2521.
Kozlovsky Y., Chernomordik L.V., Kozlov M.M. 2002. Lipid intermediates in membrane fusion: Formation, structure, and decay of hemifusion diaphragm. Biophys. J. 83, 2634–2651.
Akimov S., Polynkin M.A., Jiménez-Munguía I., Pavlov K.V., Batishchev O.V. 2018. Phosphatidylcholine membrane fusion is pH-dependent. Int. J. Mol. Sci. 19, 1358.
Kozlov M.M., Chernomordik L.V. 1998. A mechanism of protein-mediated fusion: Coupling between refolding of the influenza hemagglutinin and lipid rearrangements. Biophys. J. 75, 1384–1396.
Melikyan G.B., Markosyan R.M., Hemmati H., Delmedico M.K., Lambert D.M., Cohen F.S. 2000. Evidence that the transition of HIV-1 Gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151, 413–424.
Danieli T., Pelletier S.L., Henis Y.I., White J.M. 1996. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J. Cell Biol. 133, 559–569.
Yang X., Kurteva S., Ren X., Lee S., Sodroski J. 2005. Stoichiometry of envelope glycoprotein trimers in the entry of human immunodeficiency virus type 1. J. Virol. 79, 12132–12147.
Kozlovsky Y., Zimmerberg J., Kozlov M.M. 2004. Orientation and interaction of oblique cylindrical inclusions embedded in a lipid monolayer: A theoretical model for viral fusion peptides. Biophys. J. 87, 999–1012.
Pinigin K.V., Kondrashov O.V., Jiménez-Munguía I., Alexandrova V.V., Batishchev O.V., Galimzyanov T.R., Akimov S.A. 2020. Elastic deformations mediate interaction of the raft boundary with membrane inclusions leading to their effective lateral sorting. Sci. Rep. 10, 4087.
Kondrashov O.V., Galimzyanov T.R., Jiménez-Munguía I., Batishchev O.V., Akimov S.A. 2019. Membrane-mediated interaction of amphipathic peptides can be described by a one-dimensional approach. Phys. Rev. E. 99, 022401.
Harrison J.S., Higgins C.D., O’Meara M.J., Koellhoffer J.F., Kuhlman B.A., Lai J.R. 2013. Role of electrostatic repulsion in controlling pH-dependent conformational changes of viral fusion proteins. Structure. 21, 1085–1096.
McMahon H.T., Kozlov M.M., Martens S. 2010. Membrane curvature in synaptic vesicle fusion and beyond. Cell. 140, 601–605.
Kozlov M.M., McMahon H.T., Chernomordik L.V. 2010. Protein-driven membrane stresses in fusion and fission. Trends Biochem. Sci. 35, 699–706.
Chernomordik L. 1996. Non-bilayer lipids and biological fusion intermediates. Chem. Phys. Lipids. 81, 203–213.
Molotkovsky R., Galimzyanov T., Jiménez-Munguía I., Pavlov K., Batishchev O., Akimov S. 2017. Switching between successful and dead-end intermediates in membrane fusion. Int. J. Mol. Sci. 18, 2598.
Akimov S.A., Molotkovsky R.J., Galimzyanov T.R., Radaev A.V., Shilova L.A., Kuzmin P.I., Batishchev O.V., Voronina G.F., Chizmadzhev Yu.A. 2014. Model of membrane fusion: Continuous transition to fusion pore with regard of hydrophobic and hydration interactions. Biochem. (Mosc.) Suppl. Ser. A: Membr. Cell Biol. 8, 153–161.
Merk A., Subramaniam S. 2013. HIV-1 envelope glycoprotein structure. Curr. Opin. Struct. Biol. 23, 268–276.
Martens S., Kozlov M.M., McMahon H.T. 2007. How synaptotagmin promotes membrane fusion. Science. 316, 1205–1208.
Akimov S.A., Kondrashov O.V., Zimmerberg J., Batishchev O.V. 2020. Ectodomain pulling combines with fusion peptide inserting to provide cooperative fusion for influenza virus and HIV. Int. J. Mol. Sci. 21, 5411.
Baudin F., Petit I., Weissenhorn W., Ruigrok R.W.H. 2001. In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology. 281, 102–108.
Leser G.P., Lamb R.A. 2017. Lateral organization of influenza virus proteins in the budozone region of the plasma membrane. J. Virol. 91, e02104-16.
Ruigrok R.W.H., Barge A., Durrer P., Brunner J., Ma K., Whittaker G.R. 2000. Membrane interaction of influenza virus M1 protein. Virology. 267, 289–298.
Ruigrok R.W.H., Schoehn G., Dessen A., Forest E., Volchkov V., Dolnik O., Klenk H.-D., Weissenhorn W. 2000. Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola virus. J. Mol. Biol. 300, 103–112.
Wijesinghe K.J., Urata S., Bhattarai N., Kooijman E.E., Gerstman B.S., Chapagain P.P., Li S., Stahelin R.V. 2017. Detection of lipid-induced structural changes of the Marburg virus matrix protein VP40 using hydrogen/deuterium exchange-mass spectrometry. J. Biol. Chem. 292, 6108–6122.
Shtykova E.V., Dadinova L.A., Fedorova N.V., Golanikov A.E., Bogacheva E.N., Ksenofontov A.L., Baratova L.A., Shilova L.A., Tashkin V.Yu., Galimzyanov T.R., Jeffries C.M., Svergun D.I., Batishchev O.V. 2017. Influenza virus matrix protein M1 preserves its conformation with pH, changing multimerization state at the priming stage due to electrostatics. Sci Rep. 8, e82431.
Peukes J., Xiong X., Erlendsson S., Qu K., Wan W., Calder L.J., Schraidt O., Kummer S., Freund S.M.V., Kräusslich H.G., Briggs J.A.G. 2020. The native structure of the assembled matrix protein 1 of influenza A virus. Nature. 587, 495–498.
Battisti A.J., Meng G., Winkler D.C., McGinnes L.W., Plevka P., Steven A.C., Morrison T.G., Rossmann M.G. 2012. Structure and assembly of a paramyxovirus matrix protein. Proc. Natl. Acad. Sci. USA. 109, 13996–4000.
Briggs J.A.G., Wilk T., Welker R., Kräusslich H.-G., Fuller S.D. 2003. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22, 1707–1715.
Harris A., Sha B., Luo M. 1999. Structural similarities between influenza virus matrix protein M1 and human immunodeficiency virus matrix and capsid proteins: An evolutionary link between negative-stranded RNA viruses and retroviruses. J. Gen. Virol. 80, 863–869.
Dunker A.K., Babu M.M., Barbar E., Blackledge M., Bondos S.E., Dosztányi Z., Dyson H.J., Forman-Kay J., Fuxreiter M., Gsponer J., Han K.H., Jones D.T., Longhi S., Metallo S.J., Nishikawa K., Nussinov R., Obradovic Z., Pappu R.V., Rost B., Selenko P., Subramaniam V., Sussman J.L., Tompa P., Uversky V.N. 2013. What’s in a name? Why these proteins are intrinsically disordered: Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins. 1, e24157.
Goh G., Dunker A.K., Uversky V.N. 2008. A comparative analysis of viral matrix proteins using disorder predictors. Virol. J. 5, 126.
Zhang W., Zheng W., Toh Y., Betancourt-Solis M.A., Tu J., Fan Y., Vakharia V.N., Liu J., McNew J.A., Jin M., Tao Y.J. 2017. Crystal structure of an orthomyxovirus matrix protein reveals mechanisms for self-polymerization and membrane association. Proc. Natl. Acad. Sci. USA. 114, 8550–8555.
Shnyrova A.V., Ayllon J., Mikhalyov I.I., Villar E., Zimmerberg J., Frolov V.A. 2007. Vesicle formation by self-assembly of membrane-bound matrix proteins into a fluidlike budding domain. J. Cell Biol. 179, 627–633.
Faaberg K.S., Peeples M.E. 1988. Association of soluble matrix protein of newcastle disease virus with liposomes is independent of ionic conditions. Virology. 166, 123–132.
Sha B., Luo M. 1997. Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1. Nat. Struct. Biol. 4, 239–244.
Gregoriades A. 1980. Interaction of influenza M protein with viral lipid and phosphatidylcholine vesicles. J. Virol. 36, 470–479.
Gregoriades A., Frangione B. 1981. Insertion of influenza M protein into the viral lipid bilayer and localization of site of insertion. J. Virol. 40, 323–328.
Ye Z.P., Pal R., Fox J.W., Wagner R.R. 1987. Functional and antigenic domains of the matrix (M1) protein of influenza A virus. J. Virol. 61, 239–246.
Shi Z., Baumgart T. 2015. Membrane tension and peripheral protein density mediate membrane shape transitions. Nat. Commun. 6, 5974.
Saletti D., Radzimanowski J., Effantin G., Midtvedt D., Mangenot S., Weissenhorn W., Bassereau P., Bally M. 2017. The Matrix protein M1 from influenza C virus induces tubular membrane invaginations in an in vitro cell membrane model. Sci. Rep. 7, 40801.
Hilsch M., Goldenbogen B., Sieben C., Höfer C.T., Rabe J.P., Klipp E., Herrmann A., Chiantia S. 2014. Influenza A matrix protein M1 multimerizes upon binding to lipid membranes. Biophys. J. 107, 912–923.
Adu-Gyamfi E., Johnson K.A., Fraser M.E., Scott J.L., Soni S.P., Jones K.R., Stahelin R.V. 2015. Host cell plasma membrane phosphatidylserine regulates the assembly and budding of Ebola virus. J. Virol. 89, 9440–9453.
Gc J.B., Gerstman B.S., Stahelin R.V., Chapagain P.P. 2016. The Ebola virus protein VP40 hexamer enhances the clustering of PI(4,5)P2 lipids in the plasma membrane. Phys. Chem. Chem. Phys. 18, 28 409–28 417.
Justice P.A., Sun W., Li Y., Ye Z., Grigera P.R., Wagner R.R. 1995. Membrane vesiculation function and exocytosis of wild-type and mutant matrix proteins of vesicular stomatitis virus. J. Virol. 69, 3156–3160.
Gómez-Puertas P., Albo C., Pérez-Pastrana E., Vivo A., Portela A. 2000. Influenza virus matrix protein is the major driving force in virus budding. J. Virol. 74, 11538–11547.
Latham T., Galarza J.M. 2001. Formation of wild-type and chimeric influenza virus-like particles following simultaneous expression of only four structural proteins. J. Virol. 75, 6154–6165.
Jasenosky L.D., Neumann G., Lukashevich I., Kawaoka Y. 2001. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol. 75, 5205–5214.
Chlanda P., Mekhedov E., Waters H., Sodt A., Schwartz C., Nair V., Blank P.S., Zimmerberg J. 2017. Palmitoylation contributes to membrane curvature in influenza A virus assembly and hemagglutinin-mediated membrane fusion. J. Virol. 91, e00947-17.
Scheiffele P., Rietveld A., Wilk T., Simons K. 1999. Influenza Viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 274, 2038–2044.
Veit M., Thaa B. 2011. Association of influenza virus proteins with membrane rafts. Adv. Virol. 2011, 370606.
Gerl M.J., Sampaio J.L., Urban S., Kalvodova L., Verbavatz J.M., Binnington B., Simons K. 2012. Quantitative analysis of the lipidomes of the influenza virus envelope and MDCK cell apical membrane. J. Cell Biol. 196, 213–221.
Nayak D.P., Balogun R.A., Yamada H., Zhou Z.H., Barman S. 2009. Influenza virus morphogenesis and budding. Virus Res. 143, 147–161.
Pohl C., Duprex W.P., Krohne G., Rima B.K., Schneider-Schaulies S. 2007. Measles virus M and F proteins associate with detergent-resistant membrane fractions and promote formation of virus-like particles. J. Gen. Virol. 88, 1243–1250.
Simons K., Ikonen E. 1997. Functional rafts in cell membranes. Nature. 387, 569–572.
Baumgart T., Hess S.T., Webb W.W. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 425, 821–824.
Bobone S., Hilsch M., Storm J., Dunsing V., Herrmann A., Chiantia S. 2017. Phosphatidylserine lateral organization influences the interaction of influenza virus matrix protein 1 with lipid membranes. J. Virol. 91, e00267-17.
Peeples M.E., Wang C., Gupta K.C., Coleman N. 1992. Nuclear entry and nucleolar localization of the Newcastle disease virus (NDV) matrix protein occur early in infection and do not require other NDV proteins. J. Virol. 66, 3263–3269.
Wang Y.E., Pernet O., Lee B. 2012. Regulation of the nucleocytoplasmic trafficking of viral and cellular proteins by ubiquitin and small ubiquitin-related modifiers. Biol. Cell. 104, 121–138.
Bui M., Wills E.G., Helenius A., Whittaker G.R. 2000. Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J. Virol. 74, 1781–1786.
Masters P.S. 2006. The molecular biology of coronaviruses. Adv. Virus Res. 66, 193–292.
Perlman S., Netland J. 2009. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 7, 439–450.
Reguera J., Mudgal G., Santiago C., Casasnovas J.M. 2014. A structural view of coronavirus-receptor interactions. Virus Res. 194, 3–15.
Ulasli M., Verheije M.H., de Haan C.A.M., Reggiori F. 2010. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 12, 844–861.
Miller S., Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6, 363–374.
Netherton C.L., Wileman T. 2011. Virus factories, double membrane vesicles and viroplasm generated in animal cells. Curr. Opin. Virol. 1, 381–387.
Hsu N.Y., Ilnytska O., Belov G., Santiana M., Chen Y.H., Takvorian P.M., Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 141, 799–811.
de Wilde A.H., Raj V.S., Oudshoorn D., Bestebroer T.M., van Nieuwkoop S., Limpens R.W., van den Hoogen B.G. 2013. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-α treatment. J. Gen. Virol. 94, 1749–1760.
Neuman B.W., Angelini M.M., Buchmeier M.J. 2014. Does form meet function in the coronavirus replicative organelle? Trends Microbiol. 22, 642–647.
Welsch S., Miller S., Romero-Brey I., Merz A., Bleck C.K., Walther P., Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 5, 365–375.
Tenorio R., Fernández de Castro I., Knowlton J.J., Zamora P.F., Lee C.H., Mainou B.A., Dermody T.S., Risco C. 2018. Reovirus σNS and μNS proteins remodel the endoplasmic reticulum to build replication neo-organelles. mBio. 9, e01253-18.
Knoops K., Kikkert M., Worm S.H.E. van den, Zevenhoven-Dobbe J.C., van der Meer Y., Koster A.J., Mommaas A.M., Snijder E.J. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6, e226.
Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. 2002. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 76, 3697–3708.
Verheije M.H., Raaben M., Mari M., Te Lintelo E.G., Reggiori F., van Kuppeveld F.J.M., Rottier P.J.M., de Haan C.A.M. 2008. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLoS Pathog. 4, e1000088.
Stokes H.L., Baliji S., Hui C.G., Sawicki S.G., Baker S.C., Siddell S.G. 2010. A new cistron in the murine hepatitis virus replicase gene. J. Virol. 84, 10148–10158.
Sola I., Almazán F., Zúñiga S., Enjuanes L. 2015. Continuous and discontinuous RNA synthesis in coronaviruses. Annu. Rev. Virol. 2, 265–288.
Knorr R.L., Dimova R., Lipowsky R. 2012. Curvature of double-membrane organelles generated by changes in membrane size and composition. PloS One. 7, e32753.
Helfrich W. 1973. Elastic properties of lipid bilayers: Theory and possible experiments. Z. Für Naturforschung C. 28, 693–703.
Reiss S., Rebhan I., Backes P., Romero-Brey I., Erfle H., Matula P., Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 9, 32–45.
Reggiori F., Monastyrska I., Verheije M.H., Calì T., Ulasli M., Bianchi S., Bernasconi R., de Haan C.A.M., Molinari M. 2010. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 7, 500–508.
Cottam E.M., Maier H.J., Manifava M., Vaux L.C., Chandra-Schoenfelder P., Gerner W., Britton P., Ktistakis N.T., Wileman T. 2011. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 7, 1335–1347.
Axe E.L., Walker S.A., Manifava M., Chandra P., Roderick H.L., Habermann A., Griffiths G., Ktistakis N.T. 2008. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701.
Prentice E., Jerome W.G., Yoshimori T., Mizushima N., Denison M.R. 2004. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 279, 10 136–10 141.
Ruch T.R., Machamer C.E. 2012. The coronavirus E protein: Assembly and beyond. Viruses. 4, 363–382.
Curtis K.M., Yount B., Baric R.S. 2002. Heterologous gene expression from transmissible gastroenteritis virus replicon particles. J. Virol. 76, 1422–1434.
Almazán F., DeDiego M.L., Sola I., Zuñiga S., Nieto-Torres J.L., Marquez-Jurado S., Andrés G., Enjuanes L. 2013. Engineering a replication-competent, propagation-defective Middle East respiratory syndrome coronavirus as a vaccine candidate. mBio. 4, e00650-00613.
DeDiego M.L., Alvarez E., Almazán F., Rejas M.T., Lamirande E., Roberts A., Enjuanes L. 2007. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 81, 1701–1713.
Bosch B.J., van der Zee R., de Haan C.A.M., Rottier P.J.M. 2003. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 77, 8801–8811.
Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Pöhlmann S. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 181, 271–280
Lai A.L., Freed J.H. 2021. SARS-CoV-2 Fusion peptide has a greater membrane perturbating effect than SARS-CoV with highly specific dependence on Ca2+. J. Mol. Biol. 433, 166946.
Lai A.L., Millet J.K., Daniel S., Freed J.H., Whittaker G.R. 2017. The SARS-CoV fusion peptide forms an extended bipartite fusion platform that perturbs membrane order in a calcium-dependent manner. J. Mol. Biol. 429, 3875–3892.
Straus M.R., Tang T., Lai A.L., Flegel A., Bidon M., Freed J.H., Daniel S., Whittaker G.R. 2020. Ca2+ ions promote fusion of middle east respiratory syndrome coronavirus with host cells and increase infectivity. J. Virol. 94, e00426-20.
Nathan L., Lai A.L., Millet J.K., Straus M.R., Freed J.H., Whittaker G.R., Daniel S. 2020. Calcium ions directly interact with the Ebola virus fusion peptide to promote structure-function changes that enhance infection. ACS Infect. Dis. 6, 250–260.
Madu I.G., Belouzard S., Whittaker G.R. 2009. SARS-coronavirus spike S2 domain flanked by cysteine residues C822 and C833 is important for activation of membrane fusion. Virology. 393, 265–271.
ACKNOWLEDGMENTS
The author is grateful to M.M. Popova for technical assistance.
Funding
This work was supported by the Russian Science Foundation (project no. 22-13-00435).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author declares the absence of obvious and potential conflicts of interest related to this publication.
This article does not contain any studies involving animals or humans as participants performed by the author.
Additional information
Translated by M. Popova
Rights and permissions
About this article

Cite this article
Batishchev, O.V. Physico-Chemical Mechanisms of the Functioning of Membrane-Active Proteins of Enveloped Viruses. Biochem. Moscow Suppl. Ser. A 16, 247–260 (2022). https://doi.org/10.1134/S1990747822050038
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1990747822050038