


Abstract
The GPR40/FFA1 receptor is a G-protein-coupled receptor expressed in the pancreatic islets and enteroendocrine cells. Here, we report the pharmacological profiles of (3S)-3-cyclopropyl-3-{2-[(1-{2-[(2,2-dimethylpropyl)(6-methylpyridin-2-yl)carbamoyl]-5-methoxyphenyl}piperidin-4-yl)methoxy]pyridin-4-yl}propanoic acid (SCO-267), a novel full agonist of GPR40. Ca2+ signaling and insulin and glucagon-like peptide-1 (GLP-1) secretion were evaluated in GPR40-expressing CHO, MIN6, and GLUTag cells. Hormone secretions and effects on fasting glucose were tested in rats. Single or repeated dosing effects were evaluated in neonatally streptozotocin-induced diabetic rats (N-STZ-1.5 rats), diet-induced obese (DIO) rats, and GPR40-knockout (Ffar1–/–) mice. Treatment with SCO-267 activated Gq signaling in both high- and low-FFAR1–expressing CHO cells, stimulated insulin secretion in MIN6 cells, and induced GLP-1 release in GLUTag cells. When administered to normal rats, SCO-267 increased insulin, glucagon, GLP-1, glucose-dependent insulinotropic peptide, and peptide YY (PYY) secretions under nonfasting conditions. These results show the full agonistic property of SCO-267 against GPR40. Hypoglycemia was not induced in SCO-267–treated rats during the fasting condition. In diabetic N-STZ-1.5 rats, SCO-267 was highly effective in improving glucose tolerance in single and 2-week dosing studies. DIO rats treated with SCO-267 for 2 weeks showed elevated plasma GLP-1 and PYY levels, reduced food intake, and decreased body weight. In wild-type mice, SCO-267 induced GLP-1 secretion, food intake inhibition, and body weight reduction; however, these effects were abolished in Ffar1–/– mice, indicating a GPR40-dependent mechanism. In conclusion, SCO-267 stimulated islet and gut hormone secretion, improved glycemic control in diabetic rats, and decreased body weight in obese rats. These data suggest the therapeutic potential of SCO-267 for the treatment of diabetes and obesity.
Introduction
The GPR40/FFA1 receptor, a G-protein-coupled receptor (GPCR), couples predominantly with the Gq/11 protein, promoting phospholipase C–dependent hydrolysis of phosphatidylinositol 4,5-bisphosphate into diacylglycerol and inositol 1,4,5-triphosphate (Ghislain and Poitout, 2017). This, in turn, increases intracellular Ca2+ concentrations (Li et al., 2018). Medium-to-long chain fatty acids activate intracellular Ca2+ responses in GPR40-expressing cells, demonstrating that they are natural GPR40 ligands (Briscoe et al., 2003; Itoh et al., 2003; Kotarsky et al., 2003). GPR40 is expressed in pancreatic β-cells and intestinal endocrine cells, and its activation stimulates insulin and incretin secretion (Mancini and Poitout, 2013; Pais et al., 2016). Since insulin and incretin are pivotal for glycemic control (Nauck and Meier, 2018), GPR40 activation is considered a novel option for treating diabetes (Eleazu et al., 2018).
To date, several synthetic compounds targeting GPR40 have been developed, and the structure-activity relationships of these agonists have been investigated (Defossa and Wagner, 2014). Importantly, fasiglifam (Negoro et al., 2010; Tsujihata et al., 2011; Ito et al., 2013), a GPR40 agonist, has been shown to significantly improve glycemic control in patients with type 2 diabetes (Burant et al., 2012; Kaku et al., 2013, 2016); however, fasiglifam testing was voluntarily terminated in phase 3 clinical trials because of possible adverse effects on the liver (Wolenski et al., 2017). Proof of concept of a GPR40 agonist to treat diabetes has attracted considerable attention owing to its potential as a valuable drug target. Concurrently, fasiglifam was demonstrated as a GPR40 partial agonist based on Ca2+ mobilization in GPR40-expressing cells (Yabuki et al., 2013). Additional studies identified a novel series of GPR40 agonists with different profiles from partial agonists. A seminal study demonstrated that (3S)-3-cyclopropyl-3-(3-{[3-(5,5-dimethylcyclopenten-1-yl)-4-(2-fluoro-5-methoxyphenyl)phenyl]methoxy}phenyl)propanoic acid (AM-1638) has full agonistic activity toward GPR40, and was more effective in stimulating insulin secretion and improving glucose-lowering efficacy in vivo than partial agonists (Brown et al., 2012; Luo et al., 2012).
Importantly, GPR40 full agonists can activate the enteroendocrine system while stimulating insulin secretion (Luo et al., 2012). Such mechanisms seem highly valuable in developing a novel strategy for treating diabetes. Indeed, glucagon-like peptide-1 (GLP-1) signaling activation has been proven to have beneficial effects, including improved long-term glucose control and decreased incidence of cardiovascular death, in patients with type 2 diabetes mellitus (Nauck et al., 2017). Since GPR40 partial agonists improve glycemic control in patients with diabetes, GPR40 full agonists may provide superior efficacy and additional benefits in patients with metabolic diseases. To date, there has been no report on the clinical efficacy of the GPR40 full agonist. Thus, preclinical efficacy studies including fasiglifam as a comparator would provide important information to develop novel GPR40 full agonists.
Here, we report the pharmacological profiles and efficacy of (3S)-3-cyclopropyl-3-{2-[(1-{2-[(2,2-dimethylpropyl)(6-methylpyridin-2-yl)carbamoyl]-5-methoxyphenyl}piperidin-4-yl)methoxy]pyridin-4-yl}propanoic acid (SCO-267), a novel GPR40 full agonist (Aida et al., 2015). Its in vitro efficacy was evaluated using cell models, and its effect on islet, gut hormones, and glycemic control was evaluated in rat models. SCO-267’s contribution to body weight reduction was evaluated in obese rats and GPR40 knockout mice. Finally, we compared some key effects induced by SCO-267 to fasiglifam.
Materials and Methods
Materials.
All reagents were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan), Sigma-Aldrich (Tokyo, Japan), or Cayman Chemical (Michigan) unless otherwise indicated. SCO-267, fasiglifam, and AM-1638 were obtained from SCOHIA PHARMA, Inc., and Takeda Pharmaceutical Company Limited. Glimepiride was purchased from FUJIFILM Wako Pure Chemical Corporation. For in vitro studies, compounds were dissolved in dimethylsulfoxide, and for in vivo studies, compounds were suspended in 0.5% methylcellulose solution (FUJIFILM Wako Pure Chemical Corporation). The dose of each compound was expressed as the free base form.
FFAR1-Expressing CHO Cell Assay.
CHO dihydrofolate reductase-deficient cells (clones 104 and 2) stably expressing human FFAR1 (Yabuki et al., 2013) were cultured with minimum essential medium-α (FUJIFILM Wako Pure Chemical Corporation) containing 10% FBS, 10 mmol/l HEPES (Thermo Fisher Scientific, Waltham, MA), 100 IU/ml penicillin, and 100 µg/ml streptomycin in 5% CO2 at 37°C. The cells (1 × 104 cells/well) were seeded in 384-well (black-walled clear-bottom) culture plates and incubated overnight in 5% CO2 at 37°C. After removing the medium, the cells were incubated in 30 µl of loading buffer (Hanks’ balanced salt solution; Thermo Fisher Scientific) containing 20 mmol/l HEPES, 0.1% fatty acid-free bovine serum albumin (BSA), 0.08% Pluronic F127 (CSK-01F; Dojindo, Kumamoto, Japan), 2.5 mmol/l probenecid (CSK-03F; Dojindo), and 2.5 µg/ml Fluo4 (F311; Dojindo) for 60 minutes in 5% CO2 at 37°C. Test compounds of various concentrations were added to the cells and the increase in intracellular Ca2+ concentration was monitored for 180 seconds using the FLIPR Tetra System (Molecular Devices, Tokyo, Japan).
MIN6 Cell Assay.
The pancreatic β-cell line, MIN6, displays features of glucose metabolism and glucose-induced insulin secretion similar to those of normal islets (Miyazaki et al., 1990). MIN6 cells were seeded at 5 × 104 cells/well in 96-well plates and cultured as described previously (Miyazaki et al., 1990). After discarding the medium, the cells were preincubated for 2 hours at 37°C with 100 μl of Krebs-Ringer bicarbonate/HEPES buffer (116 mmol/l NaCl, 4.7 mmol/l KCl, 1.17 mmol/l KH2PO4, 1.17 mmol/l MgSO4, 25 mmol/l NaHCO3, 2.52 mmol/l CaCl2, and 24 mmol/l HEPES) containing 0.2% fatty acid–free BSA and 1 mmol/l glucose. After discarding the buffer, Krebs-Ringer bicarbonate/HEPES buffer containing 1 or 16 mmol/l glucose, 0.2% fatty acid–free BSA, and test materials at indicated concentrations was added and the plate was further incubated for 2 hours at 37°C. After incubation, supernatants were collected from all wells and the insulin concentrations were determined.
GLUTag Cell Assay.
GLUTag cells are stable, immortalized, and relatively differentiated murine enteroendocrine cells (Lee et al., 1992), and in this study these cells were seeded at 1 × 104 cells/well in 96-well poly-l-lysine–coated plates. Cells were cultured in Dulbecco’s modified Eagle’s medium with 10% FBS, 100 IU/ml penicillin, 100 µg/ml streptomycin, and 25 mmol/l glucose. The culture medium was replaced with medium containing 10% FBS, 100 IU/ml penicillin, 100 µg/ml streptomycin, and 5.5 mmol/l glucose prior to overnight incubation. After discarding the medium, Krebs-Ringer bicarbonate/HEPES buffer containing 10 mmol/l glucose, 0.2% fatty acid–free BSA, and the test materials at indicated concentrations was added and the plate was further incubated for 2 hours at 37°C. The supernatants were collected from all wells and the GLP-1 concentrations were determined.
Plasma Protein Binding.
Plasma samples of rats, dogs, and humans were purchased from Charles River Laboratories Japan, Inc. (Kanagawa, Japan), Kitayama Labes Co. Ltd. (Nagano, Japan), and Cosmo Bio (Tokyo, Japan), respectively. In vitro plasma protein binding of SCO-267 in rats, dogs, and humans was evaluated by using liquid chromatography–tandem mass spectrometry following ultracentrifugation.
Animals.
All animals were housed in a room with controlled temperature (23°C), humidity (55%), and lighting (lights on between 7:00 AM and 7:00 PM). All animals were allowed free access to standard laboratory chow diet (CE-2; CLEA Japan, Inc., Tokyo, Japan) and tap water. The care and use of animals and the experimental protocols were approved by the Experimental Animal Care and Use Committee of Takeda Pharmaceutical Company Limited and SCOHIA PHARMA, Inc. All experiments were performed according to the guidelines and regulations of Takeda Pharmaceutical Company Limited and Shonan Health Innovation Park. For animal studies, 0.5% methylcellulose was used as vehicle. All blood samples used in the present study were obtained via the tail vein of animals.
Sprague-Dawley Rat Study.
Male Sprague-Dawley rats were obtained from CLEA Japan, Inc. For the hormone secretion experiment in the nonfasted state, 6-week-old rats were randomized into groups (n = 6) based on body weight. To evaluate the compound’s effect on fasting plasma glucose levels, 8-week-old rats were fasted overnight. Rats were randomized into groups (n = 6) based on body weight and plasma glucose. The test materials were orally administered at the indicated doses. Blood samples were collected at indicated time points, and plasma parameters were determined. A fasting plasma glucose range below 70 mg/dl was considered hypoglycemic (American Diabetes Association, 2018). A pharmacokinetic study was performed using nonfasted 6-week-old male Sprague-Dawley rats (n = 3) obtained from Charles River Laboratories Japan Inc.
Neonatally Streptozotocin-Induced Diabetic Rat Study.
Male neonatally streptozotocin-induced (N-STZ) diabetic rats were developed via subcutaneous administration of 120 mg/kg streptozotocin (STZ) to Wistar Kyoto rats (RABICS, LTD. Kanagawa, Japan) at 1.5 days after birth (N-STZ-1.5 rats). Neonatally streptozotocin-induced rats were reported to display defects in insulin secretion and action, which in many ways resemble those observed in human diabetic patients (Portha et al., 2003). To evaluate the efficacy of single dosing, 25-week-old N-STZ-1.5 rats were subjected to overnight fasting and randomized into groups (n = 6) based on fasting plasma glucose, triglyceride levels, and body weight. The test materials were orally administered 60 minutes before oral glucose loading (1.5 g/kg). The effect of an oral dose of AM-1638 on glucose tolerance and its plasma concentration was compared with SCO-267 in a similarly designed experiment using male N-STZ-1.5 rats [32- and 18-week-old for glucose tolerance test (n = 6) and pharmacokinetic study (n = 3), respectively]. Then, to evaluate the effects of repeated-dosing, 27-week-old N-STZ-1.5 rats (baseline body weight, 373 g) were randomized into groups (n = 6) based on glycosylated hemoglobin level, plasma glucose concentration, body weight, and food intake. Each group was treated with the indicated test materials once daily for 2 weeks. Following the 2-week treatment period, the rats were fasted overnight, and the test materials were orally administered at indicated doses 60 minutes before an oral glucose load (1.5 g/kg). Blood samples were collected at indicated time points and plasma parameters were determined. Following the oral glucose load test, a pharmacokinetic study was performed with the same nonfasted rats. A pharmacokinetic study of 0.3 mg/kg SCO-267 and 3 mg/kg fasiglifam was conducted under the same experimental conditions in 20-week-old male N-STZ-1.5 rats (n = 6).
Diet-Induced Obese Rat Study.
Male F344 rats aged 29 weeks were obtained from CLEA Japan, Inc., and fed a high-fat diet (D12451M, 45 kcal%; Research Diets, Inc., New Brunswick, NJ) to induce obesity. At 49 weeks, the diet-induced obese (DIO) rats (baseline body weight, 487 g) were randomized into groups (n = 6) based on body fat mass, daily food intake, and body weight. Each group was treated with the indicated test materials once daily for 2 weeks. Blood samples were collected at indicated time points and plasma parameters were determined. Body compositions were determined after 2 weeks of treatment. GLP-1 and peptide YY (PYY) levels were measured 16 hours after the 15th dosing and a pharmacokinetic study was performed with nonfasted DIO rats after the 20th drug dosing.
Ffar1–/– Mouse Study.
Male Ffar1–/– and their wild-type (WT) littermates were obtained from RABICS, LTD. (Matsuda-Nagasumi et al., 2013). Ffar1–/– and WT mice were fed a high-fat diet (D12451; Research Diets, Inc.) from the age of 21 weeks. At 29 weeks of age, mice from each strain were randomized into groups based on food intake and body weight (baseline body weight, 40 g for WT and 41 g for Ffar1–/–, n = 6). The test materials were orally administered once daily. After 3 days of dosing, food intake and body weight changes were calculated. GLP-1 levels were measured 1 hour after the fourth dosing of the test materials. A pharmacokinetic study was performed using 10-week-old male nonfasted C57BL/6J mice (n = 3) obtained from CLEA Japan, Inc.
Measurements.
Plasma metabolic parameters were measured with an Autoanalyzer 7180 (Hitachi, Tokyo, Japan). Insulin levels were measured using a radioimmunoassay kit (RI-13K; Merck Millipore, Burlington, MA), ELISA kit (MS301; Takara, Shiga, Japan), or alphaLISA (PerkinElmer, Waltham, MA). Plasma total glucose-dependent insulinotropic peptide (GIP) levels were determined using an ELISA kit (EZRMGIP-55K; Merck Millipore). Plasma glucagon was measured using a radioimmunoassay kit (GL-32K; Merck Millipore) or ELISA kit (10-1271-01; Mercodia, Uppsala, Sweden). Plasma total GLP-1 was determined by using an ELISA established by Takeda Pharmaceutical Company Limited or a commercially available ELISA kit (299-75501; FUJIFILM Wako Pure Chemical Corporation). Plasma total PYY levels were determined using ELISA (291-73501; FUJIFILM Wako Pure Chemical Corporation) or Rat/Mouse PYY RIA (RMPYY-68HK; Merck Millipore). Body composition was quantified by magnetic resonance imaging to directly measure total body fat mass and total body lean mass of rats that were not anesthetized, at indicated ages (EchoMRI-900; Hitachi).
ELISA for GLP-1.
Anti-GLP-1 mAbs (GLIT2-863.35-7 and GLIT4-1448.7) were generated from Balb/c mice immunized with GLP-1-BSA. These mAb pairs were screened for specificity to total GLP-1 in plasma and serum. Conventional sandwich ELISAs were established in a 96-well plate using these mAb pairs. GLP-1 (7-36) amide (PEPTIDE INSTITUTE, INC., Osaka, Japan) was used as a reference standard for the assay. The detection limit of this assay was 0.3–300 pmol/l; intra-assay and interassay variations were ≤15%.
Statistical Analysis.
Statistical significance was first analyzed using Bartlett’s test for homogeneity of variances, followed by the Williams’ test (P > 0.05) and Shirley-Williams test (P ≤ 0.05) for dose-dependent studies, and Dunnett’s test (P > 0.05) and Steel test (P ≤ 0.05) for multiple comparisons. Alternatively, statistical significance was analyzed using the F test for homogeneity of variances, followed by Student’s t test (P > 0.2) or the Aspin-Welch test (P ≤ 0.2). The Williams’ and Shirley-Williams tests were conducted using a one-tailed significance level of 2.5% (0.025). Other tests were conducted using a two-tailed significance level of 5% (0.05). The EC50 and maximum effect (Emax) values were calculated using the four-parameter logistic equation in Prism 7 software (GraphPad Software, San Diego, CA). All data are presented as mean ± S.D. (for in vivo experiments) or mean ± S.E.M. (for in vitro experiments).
Results
SCO-267 Is a GPR40 Full Agonist that Stimulates Insulin and GLP-1 Secretions in Cells.
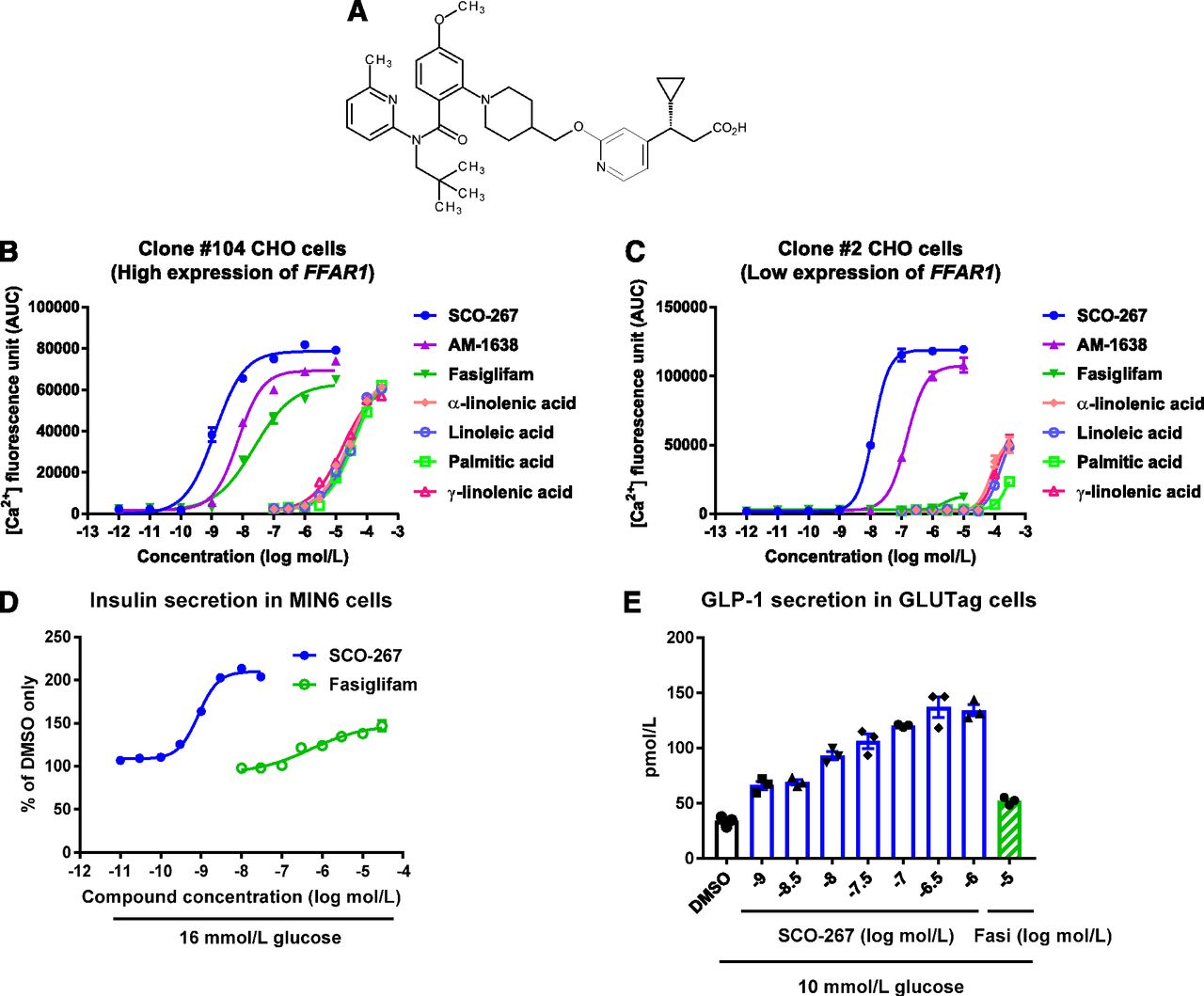
We identified SCO-267 as a new GPR40 agonist (Fig. 1A), and compound profiles were determined by in vitro experiments. We used CHO cells expressing high (clone 104) or low (clone 2) levels of human FFAR1 to evaluate the agonistic activity of SCO-267 on Ca2+ response. SCO-267 showed EC50 values of 1.3 nmol/l (clone 104) and 12 nmol/l (clone 2) and Emax values (percentage of γ-linolenic acid, an endogenous ligand) of 125% (clone 104) and 201% (clone 2) (Fig. 1, B and C; Table 1). AM-1638 (Brown et al., 2012), a well-known GPR40 full agonist, had EC50 values of 7.1 nmol/l (clone 104) and 150 nmol/l (clone 2) and Emax values (percentage of γ-linolenic acid) of 110% (clone 104) and 182% (clone 2) (Fig. 1, B and C; Table 1). The insulinotropic effect of SCO-267 was examined in mouse insulinoma MIN6 cells. SCO-267 effectively stimulated insulin secretion under the high-glucose condition, unlike fasiglifam [EC50 = 0.85 nmol/l for SCO-267 and EC50 = 530 nmol/l for fasiglifam; Emax (percentage of fasiglifam), 142% for SCO-267] (Fig. 1D). When tested in mouse enteroendocrine GLUTag cells, SCO-267 stimulated GLP-1 secretion by more than 4-fold (EC50 = 8.5 nmol/l) the level observed upon treatment with dimethylsulfoxide. SCO-267 was also more effective than 10 μmol/l fasiglifam (Fig. 1E). These data indicate that SCO-267 has full agonistic activity against GPR40. SCO-267 showed 99.6%–99.7% plasma protein binding ability in rats, dogs, and humans (Table 2).

Effects of SCO-267 on Ca2+ concentration in FFAR1-expressing CHO cells, insulin secretion in MIN6 cells, and GLP-1 secretion in GLUTag cells. (A) Chemical structure of SCO-267. Intracellular Ca2+ responses against SCO-267 and other agonists in CHO cells expressing high [(B), clone 104] and low [(C), clone 2] levels of human FFAR1 (mean ± S.E.M. of quadruplicate wells; similar results were obtained in an independent study). (D) Concentration-dependent insulin secretion response of SCO-267 in MIN6 cells with 16 mmol/l glucose condition (mean ± S.E.M. of triplicate wells). (E) Stimulation of GLP-1 secretion in GLUTag cells with 10 mmol/l glucose condition (mean ± S.E.M. and individual data of triplicate wells). SCO-267 activated an intracellular Ca2+ response in CHO cells expressing high and low levels of human FFAR1, showing full agonistic potency for GPR40. SCO-267 stimulated insulin secretion from MIN6 β cells and GLP-1 secretion from GLUTag enteroendocrine cells. Fasi, fasiglifam.
Ca2+ influx activity in human FFAR1-expressing CHO cells
In vitro plasma protein binding ratios of SCO-267 in rats, dogs, and humans
The mean of three samples is presented. Matrices shown are pooled plasma samples of 20 male Sprague-Dawley rats (8 weeks old), pooled plasma samples of 10 male beagles (10 months old), and pooled plasma samples of 10 male and 10 female humans.
SCO-267 Stimulates Secretion of Islet and Enteroendocrine Hormones and Does Not Induce Hypoglycemia in Normal Rats.
In the nonfasting condition, SCO-267 (0.3–10 mg/kg) administration stimulated the secretion of insulin, glucagon, GLP-1, GIP, and PYY (Fig. 2, A–E). Fasiglifam (10 mg/kg) was not effective in inducing these hormonal changes (Fig. 2, A–E). Administration of glimepiride, a sulfonylurea class antidiabetic drug (Korytkowski, 2004), at 10 mg/kg induced overt hypoglycemia, with plasma glucose concentrations below 70 mg/dl, in fasting rats. Although SCO-267 slightly decreased fasting plasma glucose levels, it did not induce hypoglycemia in normoglycemic rats (Fig. 2F). Pharmacokinetics analysis showed that a 0.3 mg/kg oral dose of SCO-267 resulted in a Cmax value of 5.34 ng/ml, time to reach maximum plasma concentration of 4.0 hours, area under the curve from zero to 24 hours (AUC0–24 h) of 59.4 ng·h/ml, and bioavailability of 24.8% in normal rats (Supplemental Fig. 1).

Effect of a single dosing of SCO-267 on hormone secretion and fasting glucose in normal rats. Plasma levels of insulin (A), glucagon (B), GLP-1 (C), GIP (D), and PYY (E) in SCO-267–administered nonfasting normal rats. (F) Effects on fasting plasma glucose levels in overnight-fasted normal rats. SCO-267 increased insulin, glucagon, GLP-1, GIP, and PYY levels in nonfasting normal rats. When administered to fasted normal rats, SCO-267 did not induce hypoglycemia, which was defined as plasma glucose levels <70 mg/dl in this experiment. *P < 0.025 and #P < 0.025 vs. vehicle by one-tailed Williams’ test and Shirley-Williams test, respectively. Values are presented as mean ± S.D. (n = 6). Fasi, fasiglifam.
SCO-267 Improves Glucose Tolerance and Its Efficacy Is Durable in Diabetic Rats.
A single oral administration of SCO-267 (0.1, 0.3, 1, and 3 mg/kg) increased plasma insulin and GLP-1 levels and improved glucose tolerance after an oral glucose load in N-STZ-1.5 rats in a dose-dependent manner (Fig. 3, A–C). SCO-267 (0.3 mg/kg) and fasiglifam (3 mg/kg) had Cmax values of 22.7 ng/ml and 6.17 μg/ml, respectively, in N-STZ-1.5 rats. The efficacy of SCO-267 was also compared with AM-1638 (Luo et al., 2012), a well-studied GPR40 full agonist, in N-STZ-1.5 rats. As shown in Supplemental Fig. 2, a lower plasma exposure of SCO-267 was more effective in enhancing insulin secretion and improving glucose tolerance compared with those by AM-1638 in N-STZ-1.5 rats. To test the efficacy and durability, SCO-267 (1 mg/kg) was repeatedly administered to N-STZ-1.5 rats for 2 weeks. Consistent with the single dosing efficacy, SCO-267 increased insulin secretion and improved glucose tolerance in N-STZ-1.5 rats after 2 weeks of repeated dosing (Fig. 3, D and E). At the end of the study, SCO-267 (1 mg/kg) had a Cmax value of 139 ng/ml and AUC0–24 h of 626 ng·h/ml, and fasiglifam (10 mg/kg) had a Cmax value of 39.8 μg/ml and AUC0–24 h of 255 μg·h/ml in N-STZ-1.5 rats (Supplemental Fig. 3). Food intake and body weight were unaltered in SCO-267–administered N-STZ-1.5 rats (Supplemental Fig. 4).

Effects of single and repeated administration of SCO-267 on hormone secretion and glucose control in diabetic N-STZ-1.5 rats. Plasma insulin (A), GLP-1 (B), and glucose levels (C) in a single-dosing study with N-STZ-1.5 rats. Plasma levels of insulin (D) and glucose (E) after 2 weeks of repeated-dosing with SCO-267 in N-STZ-1.5 rats. In a single dosing study, SCO-267 increased insulin levels, stimulated GLP-1 secretion, and improved glucose tolerance. Enhanced insulin secretion and improved glucose tolerance were maintained after the 2-week repeated administration of SCO-267. *P < 0.025 and #P < 0.025 vs. vehicle by one-tailed Williams’ test and one-tailed Shirley-Williams test, respectively. †P < 0.05 vs. vehicle by Student’s t test. §P < 0.05 and ¶P < 0.05 vs. vehicle by Dunnett’s test and Steel test, respectively. Values are presented as mean ± S.D. (n = 6). Fasi, fasiglifam.
SCO-267 Elevates Circulating Gut Hormone and Decreases Body Weight in DIO Rats.
The chronic effect of SCO-267 (0.3–3 mg/kg) was evaluated in obese rats that were fed a high-fat diet. After chronic dosing, GLP-1 and PYY levels were elevated after 16 hours of SCO-267 dosing (Fig. 4, A and B). During the experimental period, food intake levels were lower and total food intake was decreased in SCO-267–treated rats (Fig. 4, C and D). Consistent with the reduction in food intake, body weight and fat mass were decreased in SCO-267–treated rats (Fig. 4, E and F). The lean mass of SCO-267–treated DIO rats did not change. Plasma levels of glucose, triglycerides, alanine aminotransferase, and aspartate aminotransferase remained the same, whereas total cholesterol was reduced in SCO-267–treated DIO rats (Table 3). Pharmacokinetic analysis confirmed that sustained exposure to SCO-267 was effective in inducing these efficacies in DIO rats (Supplemental Fig. 5).

Effects of SCO-267 on gut hormone secretion and body weight control in DIO rats. Plasma levels of GLP-1 (A), PYY (B), daily food intake (C), total food intake (D), body weight change (E), and fat mass change (F) in DIO rats. SCO-267 increased plasma GLP-1 and PYY after 2 weeks of treatment. Food intake was inhibited, and body weight was decreased by SCO-267 treatment. Fat mass was decreased in SCO-267–administered rats. *P < 0.025 vs. vehicle by one-tailed Williams’ test. Values are presented as mean ± S.D. (n = 6).
Plasma metabolic parameters in SCO-267–treated DIO rats
*P < 0.025 vs. vehicle by one-tailed Williams’ test. n = 6, mean ± S.D.
SCO-267 Induces GLP-1 Secretion and Food Intake Inhibition in a GPR40-Dependent Manner.
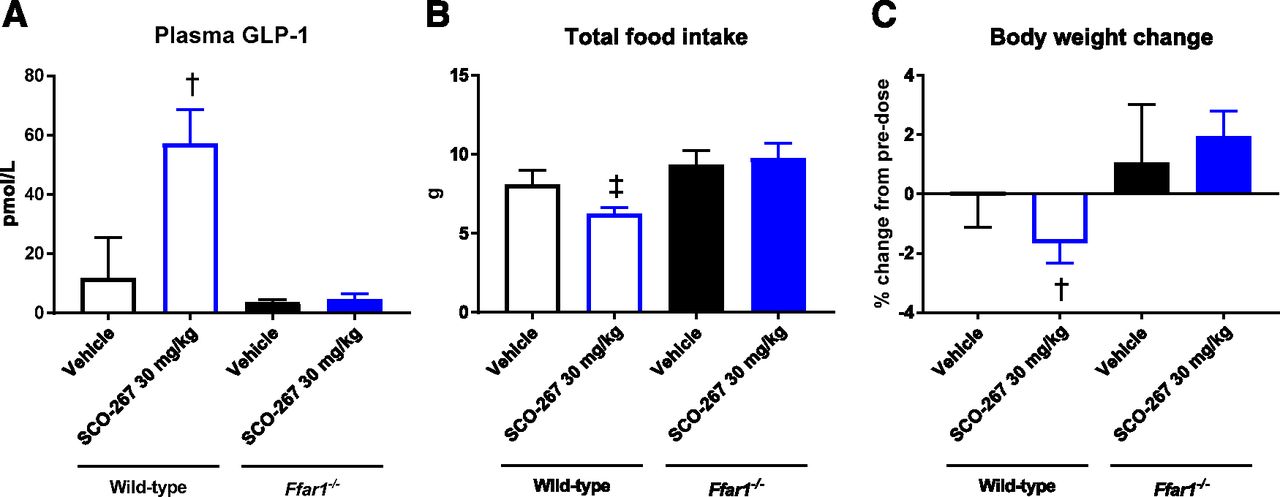
Based on the pharmacokinetic data of the relatively short half-life of SCO-267 in mice (Supplemental Fig. 6), we used 30 mg/kg as the oral SCO-267 dose in a mouse study. In WT mice, SCO-267 stimulated GLP-1 secretion, inhibited food intake, and reduced body weight (Fig. 5). In contrast, SCO-267 did not induce GLP-1 secretion, food intake inhibition, and body weight reduction in Ffar1–/– mice (Fig. 5).

Effects of SCO-267 on GLP-1, food intake, and body weight in Ffar1–/– mice. GLP-1 levels (A), food intake (B), and body weight change (C). WT and Ffar1–/– mice were treated with either vehicle or SCO-267 (30 mg/kg) for 3 to 4 days. SCO-267 increased plasma GLP-1, decreased total food intake, and decreased body weight in WT mice, whereas these effects were abolished in Ffar1–/– mice. †P < 0.05 and ‡P < 0.05 vs. vehicle-treated WT mice by Student’s t test and the Aspin-Welch test, respectively. Values are presented as mean ± S.D. (n = 6).
Discussion
In this study, we demonstrated that SCO-267, a new GPR40 agonist, was highly effective in activating GPR40 and improving glucose control in vivo. The in vitro experiments confirmed the stimulatory effect of SCO-267 on Ca2+ signaling, insulin release, and GLP-1 secretion in FFAR1-expressing CHO, MIN6, and GLUTag cells, respectively. When tested in normal rats, SCO-267 stimulated the secretion of hormones from the islet and gut. These results demonstrate the full agonistic property of SCO-267 against GPR40. Hypoglycemia was not induced in rats during the fasting condition. Studies in N-STZ-1.5 rats with diabetes confirmed the efficacy of SCO-267 in improving glucose control, and the durability of this effect. SCO-267 was also effective in decreasing body weight in DIO rats, which may have been via a GPR40-dependent mechanism. The data suggest that SCO-267 has potential as a novel therapeutic agent for treating diabetes and obesity.
In a single dosing study of N-STZ-1.5 rats, 0.3 mg/kg SCO-267 (Cmax = 22.7 ng/ml) had a glucose-lowering efficacy comparable to that of 3 mg/kg fasiglifam (Cmax = 6.17 μg/ml). This indicated that a substantially lower compound exposure is sufficient for inducing the efficacy of SCO-267. In a repeated-dosing study of N-STZ-1.5 rats, 1 mg/kg SCO-267 (Cmax = 139 ng/ml; AUC0–24 h = 626 ng·h/ml) was more effective in improving glucose tolerance than 10 mg/kg fasiglifam (Cmax = 39.8 μg/ml; AUC0–24 h = 255 μg·h/ml). Plasma protein binding of SCO-267 was similar across species (99.6%–99.7%), and fasiglifam showed similar plasma protein binding across species (>99.4% for fasiglifam (Kogame et al., 2019)). With the clinically effective exposure of 50 mg fasiglifam (Cmax = 5.3 μg/ml; AUC0–24 h = 100.3 μg·h/ml) (Leifke et al., 2012), SCO-267 may be effective in improving glucose control in patients with type 2 diabetes. Additionally, the lower plasma exposure to SCO-267 may be a safety advantage compared with exposure to fasiglifam, which was voluntarily terminated in phase 3 due to possible adverse effects on the liver.
SCO-267 robustly stimulated insulin secretion in dysfunctional β-cells in N-STZ-1.5 rats. In addition, SCO-267 stimulated the secretion of GLP-1 and GIP, both of which are incretin hormones having insulinotropic action (Nauck and Meier, 2018). Taken together with the direct insulinotropic effect on β-cells expressing Ffar1, increased secretion of GLP-1 and GIP may have contributed to the enhanced insulin secretion observed in SCO-267–administered rats.
We compared the efficacy of SCO-267 to AM-1638, a well-studied GPR40 full agonist. The in vitro experiment showed that Ca2+ influx activity of SCO-267 was 5.5- to 12.5-fold more potent compared with AM-1638 in human FFAR1-expressing CHO cells. When each compound was orally dosed at the same dosage levels, SCO-267 was more effective in improving glucose tolerance compared with AM-1638 in N-STZ-1.5 rats. These results indicate that SCO-267 is a potent GPR40 full agonist. Considering the lower plasma exposure of SCO-267 in inducing efficacy and good pharmacokinetic profiles, SCO-267 is likely a good candidate for testing in clinical trials.
When a GPCR is exposed to its agonist over a period of minutes to hours or days, the response is significantly reduced and is associated with decreased receptor expression at the plasma membrane. This process is referred to as downregulation (Zhang et al., 2014; Rajagopal and Shenoy, 2018). Some GPCRs undergo receptor desensitization after repeated stimulation, thus decreasing and in some cases abolishing the biologic response to a drug (Zhang et al., 2014; Rajagopal and Shenoy, 2018). In diabetes and obesity, sustained efficacy is important for a drug targeting GPCRs to induce therapeutic benefits. In the present study, 2 weeks of repeated dosing of SCO-267 resulted in sustained glucose lowering, and the efficacy was much better than that of fasiglifam in N-STZ-1.5 rats. This suggests that the efficacy of SCO-267 is strong and durable. Although the agonism of fasiglifam is partial, it has shown effectiveness in decreasing HbA1c levels in a 52-week study of patients with type 2 diabetes (Kaku et al., 2016). Therefore, SCO-267 may induce similar durability and better efficacy in humans.
Food intake was diminished, and body weight was lowered in SCO-267–treated DIO rats. Plasma GLP-1 and PYY levels remained high 16 hours after the final dose was administered to DIO rats. Considering the elevated levels of GLP-1 and PYY, both of which are physiologic hormones regulating satiety and body weight (Moran, 2006; Troke et al., 2014), these hormones may have contributed to the body weight reduction observed in SCO-267–administered DIO rats. In addition, sustained plasma exposure of SCO-267 was confirmed in DIO rats with reduced body weight, suggesting that the efficacy is durable. SCO-267–induced GLP-1 secretion and food intake inhibition were observed in WT mice and were abolished in Ffar1–/– mice. This suggests that the aforementioned processes are controlled by GPR40-dependent mechanisms. Although a further study is essential to elucidate the mechanism by which GPR40 agonism regulates food intake and body weight, this property of SCO-267 may be beneficial in patients who are overweight or obese and have type 2 diabetes.
SCO-267 stimulated glucagon secretion in rats, which is consistent with a previous study using a GPR40 full agonist (Pachanski et al., 2017). This suggests that GPR40 full agonists exhibit a direct targeted effect on glucagon secretion. Glucagon has pleiotropic physiologic effects in the body as evidenced by the high expression of its receptors in the liver and low expression in the kidneys, adipose tissue, lymphoblasts, spleen, pancreas, brain, adrenal glands, and gastrointestinal tract (Svoboda et al., 1994). In addition to its pivotal role in hepatic glucose metabolism, glucagon regulates hepatic fat metabolism promoting lipid oxidation and lowering lipid synthesis (Seghieri et al., 2018). It is well established that nonalcoholic fatty liver disease is common among individuals with type 2 diabetes, and increases the risk of diabetes and diabetic complications (Hazlehurst et al., 2016). Therefore, the activation of liver glucagon signaling, which decreases hepatic lipid, may improve hepatocyte function. Furthermore, a study suggested that glucagon receptor signaling is essential in the control of murine hepatocyte survival (Sinclair et al., 2008). Glucagon signaling activation has also been suggested to exert antiapoptotic actions in the liver (Sinclair et al., 2008). Therefore, glucagon signaling activation may be beneficial in treating liver diseases such as nonalcoholic fatty liver disease and more advanced nonalcoholic steatohepatitis. In fact, the addition of glucagon agonism to GLP-1 receptor agonism improved lipid metabolism and hepatic steatosis when compared with GLP-1 receptor agonism alone in rodents (Day et al., 2009). However, the contribution of SCO-267–induced glucagon stimulation to liver health requires further studies.
In conclusion, the GPR40 full agonist, SCO-267, stimulated insulin, glucagon, GLP-1, GIP, and PYY secretion. Furthermore, SCO-267 effectively improved glucose control and exerted strong efficacy in rats with diabetes. In addition, body weight loss was observed in obese rats. Thus, SCO-267 is effective in improving diabetes and obesity in rats and may induce similar favorable effects in patients with diabetes and obesity.
Acknowledgments
MIN6 murine pancreatic β-cell cells and GLUTag intestinal L-cells were kindly provided by Jun-ichi Miyazaki (Osaka University) and Daniel J. Drucker (University of Toronto), respectively. We thank Ikumi Chisaki for measuring the pharmacokinetics parameters and Seigo Izumo for valuable discussions and helpful suggestions. We gratefully acknowledge Tomofumi Kurokawa, Yukio Toyoda, Yoshitaka Yasuhara, Naoyoshi Noguchi, Toshitake Kobayashi, Toshikazu Ando, and Masahiro Ide for support.
Authorship Contributions
Participated in research design: Ueno, Ito, Abe, Ookawara, Miyashita, Ogino, Miyamoto, Yoshihara, Kobayashi, Tsujihata, Takeuchi, Watanabe, Yamada, Maekawa, Nishigaki, Moritoh.
Conducted experiments: Ueno, Ito, Abe, Ookawara, Miyashita, Ogino, Kobayashi, Moritoh.
Contributed new reagents or analytic tools: Miyamoto, Yoshihara, Maekawa.
Performed data analysis: Ueno, Ito, Abe, Ookawara, Miyashita, Ogino, Miyamoto, Yoshihara, Kobayashi, Tsujihata, Takeuchi, Watanabe, Yamada, Maekawa, Nishigaki, Moritoh.
Wrote or contributed to the writing of the manuscript: Ueno, Nishigaki, Moritoh.
Footnotes
- Received December 26, 2018.
- Accepted April 1, 2019.
The present study was conducted with financial support from Takeda Pharmaceutical Company Limited and SCOHIA PHARMA Inc.
H.U., R.I., H.M., H.O., Y.Mi., T.Y., Y.T., K.T., and N.N. are/were employees of Takeda Pharmaceutical Company Limited. S.A., M.O., A.K., M.W., Y.Y. T.M., and Y.Mo. are employees of SCOHIA PHARMA, Inc.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- AUC0–24 h
- area under the curve from zero to 24 hours
- BSA
- bovine serum albumin
- DIO
- diet-induced obese
- Emax
- maximum effect
- GIP
- glucose-dependent insulinotropic peptide
- GLP-1
- glucagon-like peptide-1
- GPCR
- G-protein-coupled receptor
- N-STZ
- neonatally streptozotocin-induced
- PYY
- peptide YY
- WT
- wild type
- Copyright © 2019 The Author(s).
This is an open access article distributed under the CC BY Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}