





SUMMARY
Nuclear factor kappa B (NF-κB) is a transcription factor pivotal for the development of inflammation. A dysregulation of NF-κB has been shown to play an important role in many chronic inflammatory diseases including rheumatoid arthritis, inflammatory bowel disease and psoriasis. Although classical NF-κB, a heterodimer composed of the p50 and p65 subunits, has been well studied, little is known about gene regulation by other hetero- and homodimeric forms of NF-κB. While p65 possesses a transactivation domain, p50 does not. Indeed, p50/p50 homodimers have been shown to inhibit transcriptional activity. We have recently shown that Interleukin-10 exerts its anti-inflammatory activity in part through the inhibition of NF-κB by blocking IκB kinase activity and by inhibiting NF-κB already found in the nucleus. Since the inhibition of nuclear NF-κB could not be explained by an increase of nuclear IκB, we sought to further investigate the mechanisms involved in the inhibition of NF-κB by IL-10. We show here that IL-10 selectively induced nuclear translocation and DNA-binding of p50/p50 homodimers in human monocytic cells. TNF-α treatment led to a strong translocation of p65 and p50, whereas pretreatment with IL-10 followed by TNF-α blocked p65 translocation but did not alter the strong translocation of p50. Furthermore, macrophages of p105/p50-deficient mice exhibited a significantly decreased constitutive production of MIP-2α and IL-6 in comparison to wild type controls. Surprisingly, IL-10 inhibited high constitutive levels of these cytokines in wt macrophages but not in p105/p50 deficient cells. Our findings suggest that the selective induction of nuclear translocation and DNA-binding of the repressive p50/p50 homodimer is an important anti-inflammatory mechanism utilized by IL-10 to repress inflammatory gene transcription.
INTRODUCTION
Interleukin-10 (IL-10) is a pleiotropic cytokine produced by many cell types including monocytes/macrophages, cells that play a critical role in the inflammatory process [reviewed in [1]. IL-10 exerts its anti-inflammatory effects by suppressing the production of macrophage inflammatory proteins such as MIP-2α, IL-1β, IL-6, IL-8, IL-12, TNF-α, GM-CSF and G-CSF. Additionally, it inhibits the expression of MHC class II molecules, B7 and intercellular adhesion molecule-1 on antigen presenting cells and diminishes Th1 cell activity by suppressing secretion of IL-2 and IFN-γ in T-cells [reviewed in [1]. The fact that IL-10 deficient mice develop chronic enterocolitis with similarities to inflammatory bowel disease (IBD) is strong evidence for the in vivo role of IL-10 as an important immunoregulator with potent anti-inflammatory and immunosuppressive activities [2]. Treatment with IL-10 is beneficial in models of induced colitis [3–5] and arthritis [4,6], as well as in models of experimental autoimmune encaphalomyelitis, pancreatitis, diabetes mellitus and experimental endotoxemia in vivo[7–10]. Moreover, patients suffering from Crohn's disease [11,12] and psoriasis [13] display clinical improvement following IL-10 treatment.
Many of the pro-inflammatory cytokines and costimulatory proteins demonstrated to be suppressed by IL-10 are known to be regulated by the transcription factor NF-κB [14]. Dysregulation of NF-κB has been implicated in the pathogenesis of a variety of chronic inflammatory diseases including rheumatoid arthritis [15], IBD [16] and psoriasis [17]. Blockade of NF-κB has thus shown beneficial effects in animal models for these chronic inflammatory diseases [18,19]. In mammals the NF-κB family consists of several proteins including NF-κB1 (p50), NF-κB2 (p52), RelA (p65), c-Rel (Rel), and RelB which share the so-called Rel homology domain. The p50 and p52 subunits, which lack transactivation domains, are produced by procession of precursor molecules of 105 kDa and 100 kDa, respectively. p50 may also be processed cotranslationally with p105 [20]. The classical NF-κB heterodimer composed of p50 and p65 subunits is a potent activator of gene expression [21,22]. While p65 possesses a transactivation domain, p50 does not [22]. The classical p65/p50 heterodimer- is sequestered in the cytoplasm as an inactive complex bound to its inhibitor protein IκB [21]. In response to a variety of extracellular stimuli, such as IL-1β, TNF-α, lipopolysaccharide (LPS) or phorbol esters, IκB proteins are rapidly phosphorylated by the IκB kinase (IKK) complex. Phosphorylation targets these inhibitory proteins for rapid polyubiquitination and degradation through the 26S proteasome [23]. Degradation of the inhibitor results in liberation of the classical NF-κB heterodimer p65/p50 from IκB and subsequent translocation of NF-κB to the nucleus where it regulates gene transcription. In contrast, p50/p50 homodimers inhibit transcriptional activation [24]. However, little is known regarding their regulation.
Several mechanisms are currently being discussed for IL-10 mediated cytokine suppression. In human monocytes and neutrophils IL-10 induces the expression of suppressor of cytokine signalling 3 (SOCS-3) [25] which inhibits LPS-induced macrophage activation [26]. Morever, IL-10 has been shown to inhibit the expression of both interferon alpha- and interferon gamma-induced genes by suppressing tyrosine phosphorylation of STAT1 [27]. While the ability of IL-10 to suppress cytokine synthesis apparently does not depend on p38 mitogen-activated protein kinase [28] the involvement of NF-κB in this important anti-inflammatory mechanism has been controversial [29–32]. We have recently demonstrated that the blockade of IKK activation and direct inhibition of DNA-binding of the nuclear p65/p50 heterodimer are targets of IL-10 in the suppression of NF-κB activity [33]. This, however, could not fully explain the inhibitory effects of IL-10 suggesting the existence of further mechanisms. In particular, the mechanisms underlying the ability of IL-10 to inhibit nuclear NF-κB remained unclear. Our data presented here provide evidence that in addition to the inhibition of p65/p50 nuclear translocation, the selective induction and translocation of p50/p50 homodimers is an important mechanism by which IL-10 inhibits NF-κB activity.
MATERIALS AND METHODS
Preparation of PBMC and cell culture
Human PBMC from various healthy donors were isolated from citrated venous blood by density gradient centrifugation using Ficoll-Paque (Pharmacia, Freiburg, Germany). For inhibiting monocyte adherence, culture vessels were coated with poly(2)-hydroxyethylmethacrylate (ICN Biomedicals, Eschwege, Germany) [34]. The human monocytic cell line U937 (CRL-1593) was obtained from the American Type Culture Collection.
Recombinant human (rh)TNF-a was purchased from R & D Systems (Minneapolis, MN, USA). LPS (from E. coli Serotype 055:B5) was purchased from Sigma Chemical Co. (St. Louis, MO, USA) and recombinant human (rh)IL-10 was purchased from R & D Systems.
Western blot analysis
Cytoplasmic (C) and nuclear (N) extracts were prepared as previously described [33]. Cytosolic and nuclear proteins (20–25 µg) were resolved over sodiumdodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). SDS-PAGE were performed according to standard protocols [33]. Equal amounts of extracts were subjected to SDS-PAGE electrophoresis and transferred to nitrocellulose membranes. Specific proteins were visualized by ECL (enhanced chemiluminescence) (Amersham Pharmacia Biotech, Freiburg, Germany). Antibodies (Ab) for p50 (H-119) and p65 (C-20) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies for p65 (N67620) and IκBα (158220) were obtained from Transduction Laboratories (BD Biosciences Pharmingen, Franklin Lakes, NJ, USA). The antibody α-Tubulin (T5168) was obtained from Sigma (Sigma Aldrich, Seelze, Germany).
Nuclear extraction for EMSA
Nuclear extracts (N) were prepared as follows: 1 × 106 cells were treated with 500 µl of lysis buffer (50 mm KCl, 0·5% Nonidet P-40, 25 mm HEPES, 1 mm phenylmethylsulphonylfluoride (PMSF), 10 µg/ml leupeptin, 20 µg/ml aprotinin, 100 µm dithiothreitol (DTT)) and kept on ice for 4 min. Nuclei were pelleted by centrifugation at 25 000 g for 1 min, and supernatants were discarded.
The nuclei were washed one time with lysis buffer not containing Nonidet P-40, and the nuclei were extracted in 300 µl of extraction buffer (500 mm KCl, 10% glycerol, 25 mm Hepes, 1 mm PMSF, 10 µg/ml leupeptin, 20 µg/ml aprotinin, and 100 µm DTT). After centrifugation at 25 000 g for 5 min, the supernatants were harvested. The protein concentration of the resulting nuclear protein extract was determined by the method of Bradford using a Protein Assay Kit (Bio-Rad), and the samples were diluted to 1 µg per µl with extraction buffer.
Electrophoretic mobility shift assays (EMSA)
8 mg of extract were incubated for 20 min in a total of 20 µl containing 1 mg of poly (dI-dC) with 4–6 × 104 cpm of a 32P-labelled oligonucleotide probe containing a κB site from the human class I MHC promoter[5′-CAG GGC TGG GGA TTC CCC ATC TCC ACA GTT TCA CTT C-3′(UV 21)] which binds both p65/p50 heterodimers and p50/p50 homodimers [33] and a κB site from the human TNF-α promotor; 5′-GAT CCA CAG GGG GCT TTC CCT CCA-3′(κB 3) [24]. The final buffer concentration was 10 mm Tris-HCl pH 7·7, 50 mm NaCl, 0·5 mm EDTA, 1 mm DTT, 10% glycerol. Complexes were resolved on a 5% polyacrylamide gel. Dried gels were exposed on film for 15–48 h. For supershift analysis, antibodies against specific NF-κB subunits were added to the nuclear extract and incubated for 15 min prior to the addition of poly (dI-dC) and labelled oligonucleotide probe. The following antibodies were used for supershift analysis: p65 (Rockland, Boyertown, PA, USA), p50 (H-119 from Santa Cruz Biotechnology, CA, USA).
Purification and stimulation of murine peritoneal macrophages and spleen cells
p105/p50 –/– mice (B6; 129P-Nfkb1tm1Bal, aged 8–13 weeks) and wild type controls with the same genetic background (B6; 129PF2/J, age 8–13 weeks) were purchased from Jackson Laboratories (Bar Harbor, ME, USA). All animal studies were approved by the competent authority for labour protection, occupational health and technical safety for the state and city of Berlin, Germany and performed in accordance with Schering AG ethical guidelines. The generation of the p105/p50 –/– mice have been described elsewhere [34]. Peritoneal macrophages were obtained by injection and withdrawal of 7 ml of cold PBS w/o Mg++, Ca++ (Gibco BRL). After plating, the nonadherent cells were removed by washing 3 h later. Spleens were removed and cell suspensions were prepared by homogenization in a tissue grinder. The erythrocytes were lysed by brief incubation in RBC-Lysing-Buffer. The cells were maintained in RPMI 1640 supplemented with 2 mm L-Glutamine, 50 U/ml penicillin, 50 µg/ml streptomycin, and 5% heat inactivated FCS (all from Gibco BRL). The cells were stimulated with recombinant murine (rm)TNF-α (R & D Systems) in the presence or absence of recombinant murine (rm)IL-10 (R & D Systems). Supernatants were collected after incubation for 8–17 h, at 1 × 106 cells per ml.
Cytokine quantification assays
Detection of mIL-6 and mMIP-2α concentrations in murine macrophage and spleen cell culture supernatants was performed by the commercially available Enzyme-linked immunosorbent assay (ELISA) kits from R & D Systems.
Statistics
(where mv is mean value)
In order to test whether the change caused by the treatment is different from zero a 95% confidence interval was calculated under consideration of the variance of observations within the entire experiment. If the interval did not include zero, the hypothesis that there is no change was rejected at the level of α = 0·05.
For the analysis of statistical differences for cytokine secretion levels between the genotypes, the Dunnett Test was used. All data are derived from at least three independent experiments and are expressed as mean + SD.
RESULTS
IL-10 blocks nuclear translocation of p65 but induces nuclear translocation of p50
One of the main points of control for NF-κB activity is the interaction with its inhibitor IκBα, which keeps NF-κB in an inactive state by sequestering it in the cytoplasm. To experimentally address the question whether IL-10 inhibits nuclear translocation of NF-κB by modulating the degradation status of IκBα in U937 cells and PBMC, cells were stimulated with TNF-α or LPS in either the absence or presence of human recombinant IL-10. Cytoplasmic and nuclear proteins were extracted, and Western blot analysis was performed to determine whether addition of IL-10 effected degradation of IκBα and the subcellular localization of the NF-κB subunits p50 and p65. Immunoblot analysis for IκBα demonstrated that this inhibitor protein in comparison to untreated cells (Fig. 1a & 2a, panel 3, lane 1) was unaffected in PBMC and U937 cells treated with IL-10 alone, regardless of variations in dose and duration of IL-10 treatment (Fig. 1a & 2a, panel 3, lanes 2 and 3). Stimulation with TNF-α resulted in the almost complete degradation of IκBα (Fig. 1 & 2a, panel 3, lane 4). However, pretreatment of cells with IL-10 followed by stimulation with a low dose of TNF-α (10 ng/ml, 15 min) delayed degradation of IκBα to some extent (Fig. 1a, panel 3, lane 5). In contrast, IL-10 pretreatment did not visibly delay IκBα degradation when followed by a longer and higher dose of TNF-α stimulation (20 ng/ml, 30 min) (Fig. 2a, panel 3, lane 6). IκBα was not detected in nuclear extracts of cells that were left untreated or had been treated with IL-10 in the absence or presence of TNF-α (Fig. 1b&2b, panel 3, lanes 1–5). Treatment of PBMC with LPS (80 ng/ml, 60 min) resulted in clear degradation of IκBα (Fig. 2a, panel 3, lanes 5 and 7), which was not visibly affected by prior IL-10 treatment. These results confirm earlier results [33] indicating that a single dose of IL-10 pretreatment can at least partially inhibit degradation of IκBα induced by low-dose TNF-α.
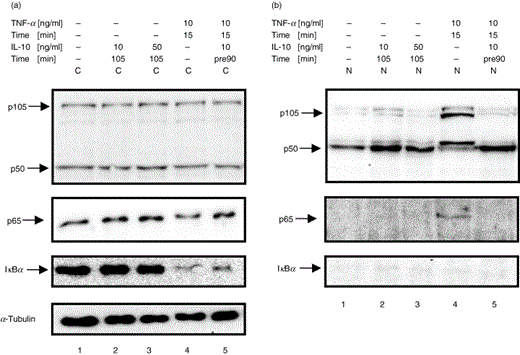
IL-10 blocks nuclear translocation of p65 but induces nuclear translocation of p50 in monocytic U937 cells. U937 cells were left untreated (lane 1) or were treated with different doses of IL-10 alone (lane 2, 10 ng/ml, 105 min; lane 3, 50 ng/ml, 105 min), or treated with TNF-α in the absence (lane 4, 10 ng/ml, 15 min) or presence of increasing doses of IL-10 (lane 5, 10 ng/ml, lane 6, 50 ng/ml; 90 min pretreatment). (a) Cytoplasmic extracts (C) and (b) nuclear extracts (N) were analysed by Western blot using antibodies against p105/p50 (panel 1), p65 (panel 2) and IκBα (panel 3) and were compared for IκBα degradation and p65 and p50 nuclear translocation. Western blotting for α-Tubulin was performed as a loading control (panel 4). One representative of at least three independent experiments with similar results is shown.
IL-10 blocks nuclear translocation of p65 but induces nuclear translocation of p50 in human PBMC. Human PBMC were left untreated (lane 1), were treated with IL-10 alone (lane 2: 10 ng/ml, 35 min; lane 3: 10 ng/ml, 65 min), or treated with TNF-α in the absence (lane 4: 20 ng/ml, 30 min) or presence of IL-10 (lane 5: 10 ng/ml, 5 min pretreatment). (a) Cytosolic extracts (C) and (b) nuclear extracts (N) were extracted and subjected to Western blot analysis and probed for p105/p50 (panel 1), p65 (panel 2), and IκBα (panel 3) and were compared for IκBα degradation and p65 and p50 nuclear translocation. Western blotting for α-Tubulin was performed as a loading control (panel 5). One representative of at least three independent experiments with similar results is shown.
To elucidate whether IL-10 affected the subcellular distribution of NF-κB subunits, Western blot analysis was performed with cytoplasmic and nuclear extracts of U937 cells and PMBCs that had been treated with IL-10 in the absence or presence of TNF-α or LPS. In untreated cells, p65 was predominantly localized in the cytoplasm (Fig. 1a & 2a, panel 2, lane 1 and Fig. 1b&2b, panel 2, lane 1). Cells stimulated with TNF-α (15 and 30 min, respectively) demonstrated an increase in nuclear translocation of p65 and p50 (Fig. 1a & 2 a, panel 1 and 2, lane 4 and Fig. 1b&2b, panel 1 and 2, lane 4). In contrast, while LPS (80 ng/ml, 60 min) alone and in combination with IL-10 clearly augmented nuclear p50, it did not visibly induce nuclear translocation of p65, even though degradation of IκBα had been observed. IL-10 treatment alone did not alter the subcellular distribution of p65 in these cells (Fig. 1a & 2a, panel 2, lanes 2 and 3 and Fig. 1b&2b, panel 2, lanes 2 and 3). In contrast, treatment with IL-10 alone clearly induced nuclear translocation of p50 in U937 cells and PBMC (Fig. 1a and Figure 2a, panel 1, lanes 2 and 3 and Fig. 1b& 2b, panel 1, lanes 2 and 3). The expression of cytoplasmic p105 and p50 was not visibly affected by IL-10, TNF-α or LPS alone or in combination with IL-10 (Fig. 1a & 2a, panel 1). (Note that the nuclear increase of NF-κB is not necessarily accompanied by a visible decrease of cytoplasmic NF-κB, as the translocated protein visualized by Western blot from concentrated nuclear extracts might represent an only small part of the large pool of cytoplasmic NF-κB [33]) Importantly, pretreatment of IL-10 clearly blocked TNF-α-induced nuclear translocation of p65 (Fig. 1a, panel 2, lane 5 and Fig. 2a, panel 2, lane 6 and Fig. 1b, panel 2, lane 5 and Fig. 2b, panel 2, lane 6, respectively), whereas high levels of nuclear p50 remained stable (Fig. 1a, panel 1, lane 5, Fig. 2a, panel 1, lane 6, Fig. 1b, panel 1, lane 5 & Fig. 2b, panel 1, lane 6, respectively). Immunoblotting with α-Tubulin demonstrated equal protein loading of the gels (Fig. 1a, panel 4 & Fig. 2a, panel 4). These data show that IL-10 blocks TNF-α-induced nuclear translocation of p65 while inducing the nuclear accumulation of p50.
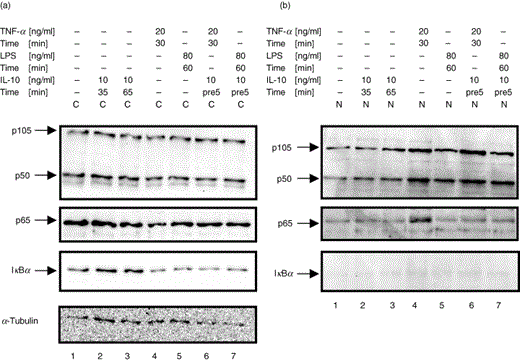
Treatment with IL-10 induces the expression of p105 and p50 in the presence of LPS
Since we had observed that IL-10 induced nuclear translocation of p50 but did not alter the expression of p50 and p105 in the presence of TNF-α, we wanted to further elucidate if treatment with IL-10 would change the synthesis of the p50 and p105 proteins in the presence of a broader, but equally potent stimulus. To address this question, U937 cells were treated with IL-10 in the presence or absence of LPS, and cytoplasmic and nuclear proteins were isolated and subjected to Western blot analysis. Immunoblotting for p105 and p50 demonstrated an approximately 2-fold increase of cytoplasmic protein levels of p105 and p50 upon treatment with a high dose of IL-10 (100 ng/ml) over 120 min (Fig. 3, lane 3). LPS-treatment alone did not visibly change the expression of p105 or p50 (Fig. 3, lane 5). However, in the presence of LPS, IL-10 induced the expression of both p105 and p50 in a dose-dependent fashion (Fig. 3, lanes 7, 9 and 11). Levels of p105 and p50 protein were increased at least 3-fold by high doses of IL-10 (100 ng/ml) in the presence of LPS (Fig. 3, lane 11). In contrast, protein levels of p65 were not affected by IL-10 treatment in the presence or absence of LPS (data not shown).
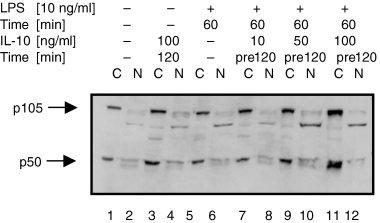
Treatment with IL-10 induces the expression of p105 and p50 in the presence of LPS in monocytic U937 cells. U937 cells were left untreated (lane 1), were treated with IL-10 alone (lane 3 & 4; 100 ng/ml, 120 min), or treated with LPS in the absence (lane 5 & 6; 10 ng/ml, 60 min) or the presence of increasing doses of IL-10 (lane 7 & 8; 10 ng/ml; lane 9 & 10; 50 ng/ml; lane 11 & 12: 100 ng/ml; 120 min pretreatment). (a) Cytoplasmic extracts (C) and (b) nuclear extracts (N) were analysed by Western blot using antibodies against p105/p50 and were compared for p105 and p50 expression levels. Western blotting for α-Tubulin was performed as a loading control (not shown). One experiment of three independent with similar results is shown.
IL-10 inhibits DNA-binding of the classical p65/p50 heterodimer but induces DNA-binding of the repressive p50/p50 homodimer
To assess whether IL-10 inhibited NF-κB-dependent transcription through the suppression of DNA-binding of this transcription factor, EMSAs were performed. For these assays we used UV21, an oligonucleotide probe with binding sites that bind both p65/p50 heterodimers and p50/p50 homodimers [33], and κB3, an oligonucleotide probe with marked preference and high affinity for p50/p50 homodimers compared to p65/p50 [24]. Nuclear extracts were prepared from untreated cells or from cells treated with IL-10 alone, or treated with TNF-α in the presence or absence of an IL-10 pretreatment and nuclear proteins were analysed for the ability to recognize the 32P-labelled UV21 or κB3 probe with an NF-κB consensus site. Gelshift analysis with nuclear extracts of U937 cells using specific DNA-probes for either the p65/p50 heterodimers or the p50/p50 homodimers supported the Western blot analysis data.
Constitutive binding of NF-κB complexes was seen in untreated cells for p50/p50 homodimers and weakly for p65/p50 heterodimers (Fig. 4a,blane 1). IL-10 treatment alone inhibited constitutive NF-κB DNA-binding activity of p65/p50 heterodimers (Fig. 4a, lane 2), but induced NF-κB DNA-binding activity of p50/p50 homodimers (Fig. 4b, lane 2). Stimulation with TNF-α (30 min) strongly induced nuclear translocation and DNA-binding of the classical p65/p50 heterodimer (Fig. 4a, lane 3), while binding of the p50/p50 homodimer was similar to constitutive levels. However, pretreatment with IL-10 followed by TNF-α stimulation significantly inhibited TNF-α-induced DNA-binding of p65/p50 heterodimers (Fig. 4a, lane 4 and 5), whereas DNA-binding activity of p50/p50 homodimers remained at constitutive levels (Fig. 4b, lane 4 and 5). Specificity of binding for NF-κB complexes was demonstrated by competition with 100-fold excess of unlabelled UV21 probe (Fig. 4a, lane 6 and 7). Supershift experiments showed the subunit composition of the p65/p50 heterodimer (Fig. 4a, lanes 9 & 10) and the p50/p50 homodimer.
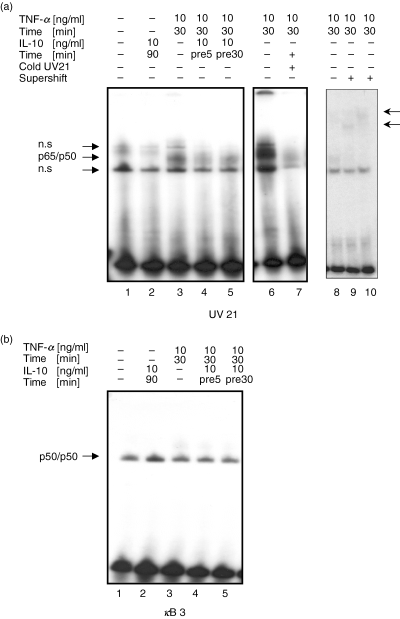
Inhibition of DNA-binding of the classical p65/p50 heterodimer but induction of DNA-binding of the repressive p50/p50 homodimer by IL-10 in U937 cells. U937 cells were left untreated (lane 1), were treated with IL-10 alone (lane 2: 10 ng/ml, 90 min) or were stimulated with TNF-α (10 ng/ml, 30 min) in the absence (lane 3 and 8) or presence of IL-10 (lane 4 & 5, 10 ng/ml, 5 and 30 min pretreatment, respectively). Nuclear extracts were prepared and subjected to EMSA. Supershift experiments used antibodies to p50 (lane 9) and to both p50 and p65 (lane 10). (a) DNA-binding activity of nuclear extracts were analysed by using the UV21 oligonucleotide specific for p65/p50 complexes or (b) the κB3 oligonucleotide which is specific for p50-p50 complexes. Specificity for NF-κB binding was shown with cold competition of 100-fold excess of unlabelled UV21 oligonucleotide probe (a, lane 6 & 7). One representative of three independent experiments with similar results is shown.
Similar effects of IL-10 treatment were observed in human PBMC (data not shown).
Cells deficient of p105/p50 exhibit very low constitutive and induced inflammatory cytokine/chemokine secretion levels – IL-10 looses its ability to inhibit constitutive cytokine/chemokine secretion in these cells
Since our results from the Western blot and gelshift analyses with U937 cells and PBMC were pointing towards a role for p50 in the IL-10-mediated inhibition of NF-κB, we further wanted to elucidate the ex vivo effects of IL-10 and TNF-α on the expression and secretion of IL-6 and MIP-2α, cytokines/chemokines that are central in the inflammatory response and tightly regulated by NF-κB [35,36], in the presence or absence of p50 expression. In order to experimentally address this question, macrophages and spleen cells isolated from p105/p50 sufficient or deficient mice were treated with TNF-α in the absence or presence of IL-10, and supernatants were analysed for the secretion of the inflammatory cytokine/chemokine IL-6 and MIP-2α by ELISA.
Unstimulated macrophages isolated from p105/p50 sufficient mice showed considerable constitutive secretion levels for IL-6 and MIP-2α(Fig. 5a,b, left panel, group a). IL-10 (10 ng/ml, 8 h) treatment alone inhibited these constitutive IL-6 and MIP-2α levels by 38% and 42%, respectively (Fig. 5a,b, left panel, group b). TNF-α (20 ng/ml, 8 h) treatment in the absence of IL-10 increased IL-6 and MIP-2α levels by 31% and 24%, respectively (Fig. 5a,b, left panel, group c). Importantly, IL-10 treatment (10 ng/ml, 8 h) followed by immediate stimulation with TNF-α (20 ng/ml, 8 h) significantly inhibited IL-6 and MIP-2α levels with a maximum of 40% and 42%, respectively (Fig. 5a,b, left panel, groups d–f). However, no dose-dependency of the inhibitory effect on IL-6 and MIP-2α secretion could be demonstrated with increasing doses of IL-10 (Fig. 5a,b, left panel). Maximal inhibition was already reached with the lowest dose of this ant-inflammatory cytokine (Fig. 5a,b, left panel, group d).
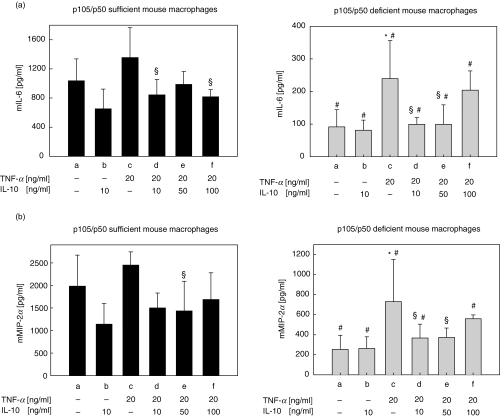
IL-10 mediated effects on NF-κB-regulated inflammatory cytokine secretion in p105/p50 sufficient and deficient mouse macrophages. Peritoneal macrophages were isolated from (a) p105/p50 sufficient mice or (b) p105/p50 deficient mice as described in Materials and Methods. Cells were incubated with medium alone or were incubated with TNF-α in the absence or presence of IL-10 and supernatants were assayed for mIL-6 and mMIP-1α secretion by ELISA as described in Materials and Methods: (a) culture medium only; (b) IL-10 only (10 ng/ml, 8 h); (c) TNF-α only (20 ng/ml, 8 h); (d) IL-10 (10 ng/ml, 8 h) + TNF-α (20 ng/ml, 8 h); (e) IL-10 (50 ng/ml, 8 h) + TNF-α (20 ng/ml, 8 h); (f) IL-10 (100 ng/ml, 8 h) + TNF-α (20 ng/ml, 8 h). *P < 0·05 versus control (a); §P < 0·05 versus TNF-α (c); ♯P < 0·05 versus p105/p50 sufficient cells. Results represent the mean values of three independent experiments; standard deviations are indicated by error bars.
Remarkably, unstimulated macrophages of p105/p50-deficient mice exhibited a significantly decreased constitutive production of IL-6 and MIP-2α (11- and 8-fold decrease, respectively) in comparison to wt controls (Fig. 5a,b, right panel, group a). Importantly, IL-10 treatment alone (10 ng/ml, 8 h) did not further inhibit these low constitutive levels of both inflammatory proteins (Fig. 5a,b, right panel, group b).
TNF-α-stimulated IL-6 and MIP-2α secretion in p105/p50 deficient cells was significantly lower in p105/p50 deficient macrophages in comparison to p105/p50 sufficient macrophages, although in p105/p50 deficient cells the relative increase compared to unstimulated cells was about 2-fold higher (Fig. 5a,b, right panel, group c). Importantly, IL-10 (10–100 ng/ml) inhibited TNF-α-induced IL-6 and MIP-2α production in p105/p50 deficient macrophages with a maximum of 59% and 50%, respectively (Fig. 5a,b, right panel, groups d–f). However, as for the p105/p50 sufficient cells, no dose-dependency was seen for IL-10-mediated inhibition of TNF-α-induced inflammatory cytokine secretion in p105/p50 deficient cells. In contrast, the inhibitory effect was partially lost with a high-dose of IL-10 (Fig. 5a,b, right panel, group f). Remarkably, the inhibitory effect of IL-10-mediated suppression of TNF-α-induced IL-6 and MIP-2α secretion was even more pronounced in p105/p50 deficient cells in comparison to p105/p50 sufficient cells (Fig. 5a,b, groups d–f). Similar data was obtained with p105/p50 sufficient and deficient spleen cells (data not shown). These results indicate that while IL-10 looses its ability to inhibit constitutive inflammatory cytokine secretion in the absence of p50, inducibility of NF-κB-regulated cytokine secretion by stimuli like TNF-α is fully maintained or even increased. Moreover, IL-10 fully maintains its ability to inhibit TNF-α-induced cytokines in the absence of p105/p50.
DISCUSSION
It is well established that IL-10 controls inflammatory processes by suppressing the expression of pro-inflammatory cytokines, chemokines, adhesion molecules, as well as antigen-presenting and costimulatory molecules in monocytes/macrophages, neutrophils and T-cells [reviewed in [1]. Different mechanisms have been discussed that mediate IL-10's cytokine inhibiting function. These mechanisms include the up-regulation of SOCS3 [25,26], the inhibition of tyrosine phosphorylation of STAT1 [27] but apparently do not involve the p38 pathway [28]. Since an abundance of the inflammatory proteins suppressed by IL-10 are transcriptionally controlled by NF-κB it was suggested that IL-10 may exert a significant part of its anti-inflammatory properties through inhibiting this transcription factor. We have recently shown that IL-10 inhibits NF-κB activity by dual mechanisms. Since the inhibition of nuclear NF-κB could not be explained by an increase of nuclear levels of its inhibitor IκB [33] we sought to further investigate the mechanisms underlying this observation.
We show here for the first time that IL-10 selectively induced p50/p50 homodimer translocation into the nucleus and that IL-10 pretreatment followed by TNF-α stimulation blocked the nuclear translocation of the p65 subunit without affecting the nuclear translocation of p50.
We further demonstrate that high doses of IL-10 significantly increased both p105 and p50 protein levels in the absence or presence of low-dose LPS stimulation in monocytic cells (Fig. 3). This observation is supported by reports of p105/p50 up-regulation in the presence of LPS tolerance that can only be induced with low-dose LPS [37]. Since we observed differential effects of IL-10 on p105 and p50, possible mechanisms of interference with p105/p50 generation have to be considered. Two different models of p105/p50 generation are presently discussed. The model proposed by Heissmeyer et al. [38] describes the inducible phosphorylation by IKKα and IKKβ and the subsequent degradation of p105 leading to the liberation of sequestered subunits including the processing product p50. The other model proposed by Lin et al. [20] describes cotranslational processing which allows the production of both p50 and p105 from one single mRNA. The fact that TNF-α-induced activity of the IKK complex can be inhibited by IL-10 [33] suggests that IL-10 treatment in our cell models inhibited the IKK-dependent post-translational processing of p105.
Interestingly, increased expression of p105 has also been described in LPS-tolerant cells [37] and IL-10 itself has been implicated to play a role in the process of LPS tolerance induction [39]. Both observations, the IL-10-induced expression of p105/p50 and the selective nuclear translocation of p50, could be explained by a putative IL-10-mediated induction of the cotranslational processing of p105 and p50 that would substitute for the putatively blocked post-translational processing of p105. Furthermore, several studies have provided evidence indicating that the C-terminal region of p105 has a functional role similar to the IκB protein, as p105 can form heterodimers and sequester the p50 subunit or other Rel proteins in the cytoplasm [40–45]. Considering the IκB-like function of p105, it might also bind p65 to sequester this subunit in the cytoplasm. IL-10-mediated up-regulation of p105 may thus additionally contribute to the inhibition of TNF-α-induced nuclear translocation of p65.
Importantly, IL-10 treatment alone inhibited high constitutive secretion levels of IL-6 and of MIP-2α in p105/p50 sufficient but not in deficient macrophages. NF-κB is the exclusive transcription factor for induction of IL-6 in response to TNF-α[35] and also MIP-2α is tightly regulated by NF-κB activity [36]. Based on our in vitro experiments, this inhibition of constitutive cytokine/chemokine secretion in cells expressing p105/p50 could thus be explained by the selective induction of nuclear translocation of repressive p50/p50 homodimers that would bind to DNA and inhibit NF-κB activity.
IL-10 inhibited TNF-α-induced IL-6 and MIP-2α secretion both in p105/p50 sufficient and deficient murine macrophages and spleen cells (Fig. 5 and data not shown). Importantly, constitutive secretion levels of the NF-κB-controlled cytokines IL-6 and MIP-2α were significantly decreased in p105/p50 deficient when compared to p105/p50 sufficient macrophages. These results suggest that p50 is required in a complex with p65 for the transcriptional activation of constitutive cytokine/chemokine synthesis. In summary, we demonstrate here that IL-10, selectively induced the expression of p105/p50 as well as the nuclear translocation and DNA-binding of the repressive p50/p50 homodimer (Fig. 6). Furthermore, pretreatment with IL-10 followed by TNF-α inhibited nuclear translocation of the p65/p50 heterodimer without affecting strong translocation of p50. Additionally, IL-10 lost its ability to inhibit constitutive cytokine/chemokine secretion in p105/p50 deficient cells suggesting that the selective induction of p50/p50 homodimer nuclear translocation is an important mechanism employed by IL-10 to mainly inhibit constitutive NF-κB activity in immune cells. Thus, by specifically inducing nuclear translocation of the repressive p50/p50 homodimer while inhibiting translocation of the classical p65/p50 heterodimer, IL-10 exerts a dual inhibitory function in the NF-κB pathway. This study broadens our understanding of the mechanisms used by IL-10 to control inflammatory processes and might thus contribute to the development of new therapeutic avenues in the management of chronic inflammatory diseases.
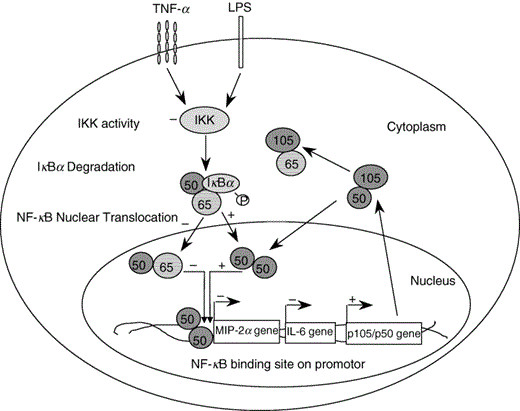
Scheme representing the molecular mechanisms employed by IL-10 to inhibit NF-κB activity. In the absence of an activating stimulus such as TNF-α, IL-10 specifically induces the nuclear translocation of repressive p50/p50 homodimers, which compete with pro-inflammatory p65/p50 heterodimers for DNA-binding to NF-κB promotor sites on inflammatory genes such as IL-6 or MIP-2α. In the presence of a stimulus such as TNF-α, IL-10 can suppress nuclear translocation and DNA-binding of p65/p50 heterodimers by inhibiting IKK activity and thus delaying degradation of IκBα. Conserved levels of IκBα will sequester p65 in the cytoplasm, while p105/p50 expression is up-regulated and p50 free to translocate to the nucleus to form homodimers. Up-regulated p105 may also additionally sequester p65 in the cytoplasm. In the absence of the p105/p50 gene, IL-10 looses its ability to suppress constitutive NF-κB activity. Upon activation of p105/p50 deficient cells, p65 may recruit a different Rel protein to form transcriptionally active heterodimers, which can still be inhibited by IL-10.
ACKNOWLEDGEMENTS
The authors wish to thank Sandy Westerheide, Chapel Hill for critical reading of the manuscript and Detlef Opitz, Berlin for his help in performing statistics.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}