





Abstract
Candida glabrata cells suspended in water are under hypo-osmotic stress and undergo cell death in 1–2 days, unless they are at a density of more than 105CFU mL−1. The dying cells exhibit FITC-annexin V staining, indicative of programmed cell death (apoptosis). In a higher cell density, cells are protected and survive at least for 4 days. Filtrates from cells at high density can protect those at lower density, indicating that cells release substances, amounting to c. 5 mg L−1 of cell suspension, that protect each other against hypo-osmotic stress. In a concentrated form, the released materials can support growth, indicating that the protective material includes carbon and nitrogen sources, as well as vitamins that are required by C. glabrata for growth. We conclude that cell death from hypo-osmotic stress can be alleviated by small amounts of nutrients.
Introduction
Yeasts are frequently exposed to changes in environmental conditions with some changes, such as osmotic stress, occurring suddenly. A considerable amount of work has been carried out on hyperosmotic stress in yeast (Dickinson & Schweizer, 2004). Less work has been carried out on hypo-osmotic stress, the topic of this paper. Cells suspended in water experience hypo-osmolarity that may result in a rapid inflow of water, cell swelling and increased turgor pressure. Weaknesses in lipid–lipid bonding may also lead to cell lysis due to chaotropic solutes (Cray et al., 2013). However, activation of protective mechanisms, such as release of osmolytes and remodelling of the cell wall, plays an important role in preventing cell rupture (Hohmann, 2002). Cell wall integrity plays an important role in preserving the viability of yeast. Under hypo-osmotic stress, cell wall stress sensors lead to activation of a mitogen-activated protein kinase (MAPK) cascade. MAPK leads to activation of transcription factors that facilitate the biosynthesis of cell wall components, organize actin and control osmolytes. In yeast, hyper- and hypotonic shocks induce two mitogen-activated protein (MAP) kinase signal transduction cascades. While hypertonic shock stimulates the HOG pathway, hypotonic shock stimulates the PKC1 signal transduction cascade (Hohmann, 2002).
Efflux of intracellular electrolytes and small organic solute ‘osmolytes’ is a universal response to cell swelling (Strange & Jackson, 1995). Protozoa and lung cancer cells release amino acids in response to acute hypo-osmotic stress (Kirk, 1997). In yeast, glycerol, arabitol and erythritol are released in response to hypo-osmotic stress (Kayingo et al., 2001; Fuchs & Mylonakis, 2009). In organisms such as the algae Dunaliella tertiolecta, intracellular osmolytes are depleted by metabolism and not released in response to hypo-osmotic stress (Goyal, 1989).
If cellular defence mechanisms fail, cells die by undergoing apoptosis or necrosis. Apoptosis has been defined as programmed cell death, which is an organized energy-dependent process, where the intracellular content (such as DNA) undergoes ordered degradation, exposure or secretion of diverse factors promotes phagocytic cell elimination and the plasma membrane integrity is maintained. An apoptotic ‘dead cell’ will finally suffer from a decline in metabolism which necessarily causes the breakdown of plasma membrane integrity and thus necrotic morphology. This phenomenon is defined as ‘secondary necrosis’. In contrast, necrosis (primary necrosis) is defined as the characterization of cells that die as a result of injury or typically swell and burst, consequently releasing their contents (Kroemer et al., 1998; Eisenberg et al., 2010).
Candida glabrata is considered to be a nondimorphic yeast that forms blastoconidia with cell size ranging from 1 to 4 μm (Bialkova & Subik, 2006). It is a haploid, petite-positive yeast and is a species considered quite similar to Saccharomyces cerevisiae (Barns et al., 1991; Davenport et al., 1995). Candida glabrata is used in the food industry as a starter culture (Tsuyoshi et al., 2005; Aidoo et al., 2006), and it also appears as the second most prevalent Candida species in patients with HIV and patients with advanced cancer (Li et al., 2007). While many studies on the effect of hypo-osmotic stress have been carried out on Saccharomyces cerevisiae (Granot & Snyder, 1991, 1993; Kayingo et al., 2001; Arias et al., 2011), there have been few studies on Candida species.
As hypo-osmolarity may induce the cell wall integrity MAPK pathway, cells in water might be a useful model to study this pathway and the physiology of the cells. Moreover, preparing yeast suspensions in water is a critical step in many experiments, including the preparation of inocula by standard methods to determine antifungal susceptibility. In addition, laboratory studies frequently involve yeast, and frequently C. glabrata, suspended in water to monitor the effects of carbon sources, drugs and peptides such as glucose, dopamine and amyloid beta (Aβ; Granot & Snyder, 1993; Bharadwaj et al., 2008; Macreadie et al., 2010). Here, the effect of cell density on survival after hypo-osmotic stress is shown. Death at low density appears to be through apoptosis. At high cell density, cells are protected through the release of nutrients.
Materials and methods
Viability assays
Candida glabrata cells, subcultured at least five times, were inoculated in 2 mL YEPD medium [1% (w/v) yeast extract (Oxoid), 2% (w/v) bacteriological peptone (Oxoid) and 2% (w/v) dextrose (Oxoid)] and incubated for 24 h with agitation (170 r.p.m.). Cells were collected by centrifugation in a high-speed microcentrifuge (LaboGene ApS, Denmark) at 5250 g for 1 min, washed six times with sterile MilliQ water and suspended in MilliQ water to a specific cell density. Cultures were then incubated at 30 °C with agitation (170 r.p.m.) in 50-ml centrifuge tubes with a working volume of 2 mL. One hundred microlitre aliquots of cells were taken at indicated time points to measure cell viability, as colony-forming units on YEPD solidified with 1.5% (w/v) bacteriological agar (Oxoid).
Propidium iodide staining
Propidium iodide (PI) was purchased from Sigma-Aldrich (Australia). A modified PI staining protocol from Invitrogen was used to measure cell viability. A 3 μM solution of PI was made by diluting 1 mg mL−1 (1.5 mM) stock solution 1 : 500 in phosphate-buffered saline (PBS), which was then filtered through 0.22-μm syringe filters (Millex). One ml samples were centrifuged at 8500 r.p.m. for 1 min, and the pellets were suspended in PBS containing 3 μM PI. The samples were incubated at 30 °C for 30 min before flow cytometry analysis using a FACS Canto II (BD Biosciences).
Bio-protection assays
Materials released by cells were obtained by incubating cells at 106 CFU mL−1 for 1–2 days. Cells were removed by filtration through 0.22-μm syringe filters. Aliquots of filtrates were plated on YEPD to check sterility before further investigations. The filtrates were used either directly, or they were lyophilized and reconstituted as concentrated samples.
For protection studies using dead cells, 104 CFU mL−1 was left in water for 4 days to die naturally (99.7% death). After that, C. glabrata cells, from a YEPD culture grown for 24 h, were washed and mixed with the dead cells at a concentration of 5.7 × 103CFU mL−1. The growth was monitored by counting the colonies. Two independent replicates were performed for this experiment with three replicates each time.
To test whether the effect of hypo-osmolarity on low-density cells can be eliminated by sorbitol, about 2 × 103 CFU mL−1 was suspended in 5 μM, 50 μM and 1 M of sorbitol (Sigma) and incubated for 24 h at 30 °C with shaking (170 r.p.m.). The impact of low nutrients on the survival of low-density cells was tested by suspending the cells in diluted yeast minimal media (0.67% yeast nitrogen base with amino acids (Sigma), d-glucose (2% w/v) as carbon source). Yeast minimal media (YMM) was sterilized by filtration through 0.22-μm syringe filters (Millex). Cells were suspended in diluted YMM as indicated in the Results and Discussion section.
Testing for apoptosis
Due to the difficulty in collection and observation of dead cells in low-density cell suspensions under the microscope, apoptosis was observed by dialysing high-density cell cultures. About 106 CFU mL−1 was washed as described above, and 4 mL of the cell suspension was dialysed in dialysis membranes (12 kDa MWCO), surrounded by 3 L of MilliQ water. The number of colonies was determined after 2 or 3 days at 30 °C.
The annexin V-FITC Apoptosis Detection kit (APOAF-20TST; Sigma-Aldrich) was used for detecting apoptotic cells. Two samples of 1 mL from 3 days dialysed high-density cells (106 CFU mL−1) were centrifuged (8500 r.p.m., 1 min) and resuspended in 1 mL binding buffer (1×). Ten microlitre PI and 5 μL FITC-annexin V solutions were added to each 500 μL dialysed cell suspension and incubated 10 or 30 min at room temperature. After that, cells suspensions were centrifuged at 8500 r.p.m. for 1 min, and the content of the tubes (pellet from 500 μL sample each) were combined in one tube by washing with 500 or 20 μl sterile MilliQ water for flow cytometry or fluorescence microscopy analysis, respectively. Cells grown in YEPD media to exponential phase were used as a negative control. Cells were analysed by FACS Canto II (BD Biosciences) flow cytometer or observed using an epifluorescence microscope (Leica DM 2500) provided with Leica DFC310 FX camera.
For DNA microscopic observation, cells were harvested by centrifugation at 5890 g for 1 min, washed, resuspended in 3 μM DAPI (4′,6-diamidino-2-phenylindole dihydrochloride; Sigma) and incubated for 15 min in room temperature at dark. Then, cells were washed and resuspended in PBS.
For cell cycle analysis by flow cytometry, cell suspensions, of c. 106 cell mL−1 of overnight culture in YEPD, and dialysed cells were fixed and permeabilized with 70% cold ethanol on ice for 5 min, rehydrated in PBS at room temperature for another 5 min and then cells were harvested and stained with DAPI as described above (Li et al., 2006; Liang & Zhou, 2007). Cells were analysed by flow cytometry using a pacific blue filter.
Statistical analysis
For statistical analysis, One-way anova with Dunnett's post-test was performed using GraphPad Prism software. The control groups were the cultures at time zero. Error bars represent the standard error of the mean (SEM). Differences were considered significant for P< 0.05. WEASEL software was used for flow cytometry data analysis.
Results and discussion
Cell viability and cell density
The effect of cell density on viability of C. glabrata in water was examined by plating cells to measure the number of colony-forming units (CFUs). As shown in Fig. 1, cultures with an initial high cell density (more than 8 × 104 CFU mL−1) maintained their viability over 3 days. In contrast, cells at low density (≤ 8 × 104 CFU mL−1) progressively lost their viability. In summary, cells at low density lost viability, while a quorum of more than 105 CFU mL−1 allowed viability to be maintained. This experiment has been performed three times independently with four replicates each time.
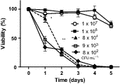
The relation between cell density and cell survival in water. Bars represent SEM. *P < 0.05; **P < 0.003. This experiment was repeated at least three times independently with four replicates each time, the result of one experiment has been shown.
Cell death was also measured using flow cytometry to examine the proportion of cells that could be stained with PI, an indicator of cell death. After 24 h, cells in water at high density, an average of 1.5% of cells, exhibited red fluorescence (Fig. 2a and b), indicative of cell death. Thus, the population had 98.5% viability.
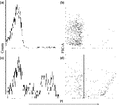
Analysis of cell viability by flow cytometry. (a) Cells at high density (106 CFU mL−1) in water for 24 h without staining with PI. (b) Same cells at A were stained with PI and then analysed for PI staining vs. forward light scatter. (c) Analysis of cells at low density (103 cells mL−1) after 24 h for PI staining, (d) PI staining vs. forward light scatter. This experiment was performed three times independently.
At low cell density (103 CFU mL−1), 44.4% of the population were PI-stained after 24 h (Fig. 2c and d). Of the cells that became stained with PI, populations exhibited either moderate or higher levels of PI staining. Analysis of PI staining vs. forward light scatter (Fig. 2d) indicates that the more highly stained cells were larger and therefore able to incorporate more PI. In previous studies of Saccharomyces cerevisiae, cells suspended at high density (2 × 107 cells mL−1) in water maintained high viability for 20 days (Granot & Snyder, 1991, 1993). Here, we demonstrate that C. glabrata cells at low density in water are sensitive to the hypo-osmolarity and die within 3 or 4 days.
Could cells at high density be protected by materials from dead cells? It has previously been shown that yeast cells can proliferate by utilizing nutrients released from cells where 90–99% of the population has died (Fabrizio et al., 2004). In this study, the percentage of dead cells is very low. To determine whether such a population of dead cells could protect a low-density population, dead cells from an initial population of 104 CFU ml−1 (99.7% dead after 4 days in water) were added to low-density cell suspension (5.5 × 103 CFU mL−1). The suspension of dead cells was incapable of providing protection (data not shown).
Bio-protection
To determine whether a soluble agent(s) could be involved in protection of cells from hypo-osmotic stress, a cell filtrate from high-density cells (3 × 106 CFU mL−1) was added to cells at low cell density (7 × 103 CFU mL−1). Cells at low density, suspended in the filtrate from high-density cells (3 × 106 CFU mL−1), maintained viability (Fig. 3a). In the control (without the filtrate from the high-density suspension), viability decreased to 50% after 24 h. This indicates that the high-density cells release material(s) that protect the population against hypo-osmotic sensitivity and starvation.
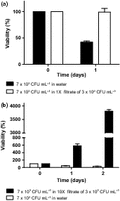
(a) 7 × 103 CFU mL−1 was suspended in the filtrate from 3 × 106 CFU mL−1, obtained after 1 day. (b) The same filtrate was concentrated 10-fold, and cells were added to a density of 7 × 103 CFU mL−1. This experiment was performed three times with independent filtrates. Bars represent SEM of four replicates of one batch of cells.
When this protective filtrate was concentrated 10-fold by lyophilization and added to a suspension of cells at 7 × 103 CFU mL−1, the filtrate enabled cells to multiply 40-fold in 2 days (Fig. 3b). This experiment was performed three times with independent filtrates. As the concentrated filtrate was enough to stimulate divisions, it can be concluded that it contains all necessary nutrients for C. glabrata. Herker et al. (2004) induced growth and survival in aged cells by addition of secretions from high-density, aged yeast cells exhibiting massive cell death. In contrast, in this study, the bioactive materials leading to survival in hypo-osmotic conditions were obtained from populations with high viability (> 97% viable). The dry weight of exuded materials is < 5 mg L−1 of filtrate. Our efforts to identify the protective agent(s) by mass spectroscopy and NMR have been unsuccessful due to these low levels and the diversity of materials in the filtrate.
Many studies use sorbitol as an osmotic stabilizer (de Nobel et al., 2000; Soriano-Carot et al., 2012). Here, we show in Fig. 4 that a low concentration of sorbitol (50 μM) provides protection to cells at low density, but not 5 μM sorbitol. In contrast, the addition of 1 : 100 diluted YMM media induced growth of the cells, whereas a dilution of 1 : 1000 did not contain enough nutrients for growth and did not provide protection. Because protection is provided by < 5 mg of solid matter per litre, the protection is unlikely to be ‘sorbitol-like’ osmotic protection. Moreover, 1 : 100 diluted YMM media is a hypo-osmotic condition (Kayingo et al., 2001), but it was protective. This indicates miniscule of amounts of nutrients in the milieu surrounding cells that appear sufficient to protect against death due to hypo-osmolarity.
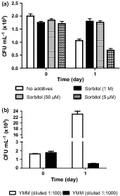
Protection of cells against hypo-osmolarity. (a) By sorbitol, (b) by yeast minimal media (YMM) diluted 1 : 100 and 1 : 1000.
Apoptosis caused by hypo-osmotic treatment
The factors conferring survival were found to pass through 12-kDa MWCO dialysis membranes. After 2 days, c. 80% loss of viability was observed in 7 × 105 CFU mL−1 cultures after dialysis in 12-kDa MWCO dialysis sacs (Fig. 5). This indicates that the bioactive compounds have molecular weights ≤ 12 kDa, and these compounds have been diluted around the cells. As a result, they have lost their resistance to hypo-osmolarity. The loss of viability was similar to death rates seen when the population is at 7 × 103 CFU mL−1 (Fig. 5).
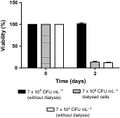
Effect of dialysis on survival. Cells at 7 × 105 CFU mL−1 were dialysed in 14000 MWCO dialysis membranes. Cells from 24-h YEPD liquid media were washed as described and resuspended in water to have 7 × 105 CFU mL−1 and incubated at 30 °C as indicated. Dialysis sacs were dialysed against 100 times the sample volume. This experiment has been performed more than three times with two replicates each time.
Because of the difficulty in examining cells when they are at low density, especially for microscopic observation, dialysed cells at high density were used to study the mechanism of cell death.
The exposure of phosphatidylserine at the outer leaflet of the cytoplasmic membrane is an early morphological marker of apoptosis which is conserved from yeast to mammalian cells (Martin et al., 1995; Madeo et al., 1997). In yeast, phosphatidylserine can be detected by FITC-annexin V staining after digestion of the cell wall. However, in this study, annexin V was applied directly without any pretreatment to remove the cell wall. The analysis of flow cytometric data demonstrate that about one quarter of the cells show FITC-annexin V staining after 3 days dialysis in water (Fig. 6a and b). The cell staining by FITC-annexin V was further observed by epifluorescence microscopy which showed green fluorescence in the periphery of the cytoplasm, indicating phosphatidylserine externalization (Fig. 6c upper right). Unlike other studies of apoptosis in yeast, in this study, the production of protoplasts was not necessary to observe FITC-annexin V staining. In this study, annexin V staining of the phosphatidylserine was probably enabled by the hypo-osmotic conditions leading to increased permeability of the cell wall. PI staining was used in combination with annexin V staining to distinguish between apoptotic and necrotic cells. Around 8% of the collected cells exhibited annexin V and PI staining, indicative of late apoptosis, while 21% of cells exhibit only PI staining, indicative of secondary necrosis (Herker et al., 2004; Allen et al., 2006; Eisenberg et al., 2010).
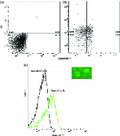
FITC-annexin V staining on high-density cells dialysed for 3 days to examine externalization of phosphatidylserine. (a) Flow cytometry analysis of cells without staining. (b) Flow cytometry analysis of cells in A stained with PI and FITC-annexin V. (c) Flow cytometric analysis of unstained and FITC-annexin V-stained cells. The inset shows a fluorescence microscopy image of FITC-annexin V-stained cells. This analysis was performed more than three times: the result of one experiment has been presented.
In growing cells, DAPI stains the single, large, homogeneous, round-shaped nucleus as well as the peripheral mitochondria (Fig. 7a and b). In contrast, dialysed cells in water show small condensed nuclei or distributed nuclear fragments (Fig. 7c –h), which indicate chromatin condensation, DNA damage and nuclear fragmentation: these are features of apoptosis (Madeo et al., 1997, 1999; Andres et al., 2008). These cells were not fixed to avoid the loss of small DNA fragments, which can occur due to increase in the permeability of fixed cells. Figure 7a–h also shows a difference in cell size. Cells in water are smaller compared to those from growth media. In general, cell shrinkage, dense cytoplasm and tight package of organelles are other indicators of apoptotic death. In contrast, necrotic cells show increased size and swelling (Elmore, 2007).

DAPI staining of growing and dialysed cells. Fluorescence and light microscopy analysis of cells growing in YEPD (a) and cells dialysed in water (c, e and g), after DAPI staining, b, d, f and h are bright field for DAPI staining, respectively. Flow cytometric analysis of DNA content for cells growing in YEPD (i) and cells dialysed for 3 days (j). This experiment has been performed three times independently.
It has been well documented that apoptotic cells show reduced DNA staining with a variety of fluorochromes, including PI and DAPI. A large portion of DNA in apoptotic cells is of low molecular weight due to activation of an endogenous endonucleases which break the linkers between the nucleosomes. These small fragments of DNA can leach out from ethanol-fixed (permeabilized) cells during rehydration and staining process. Consequently, flow cytometric analyses detect cells with lower staining intensity than those at G0/1 phase, a phase when live cells have their minimum content of nonfragmented DNA. These cells can be observed as a ‘sub-G0/1’ peak in a DNA histogram. In contrast, necrotic cells generally do not show an immediate reduction in DNA staining, and sub-G0/1 cannot be discriminated from a histogram DNA content analysis of live cells (Darzynkiewicz et al., 1992, 2010; Dive et al., 1992; Riccardi & Nicoletti, 2006; Calvert et al., 2008). In Fig. 7i–j, about 20% of the population are in sub-G0/1 region and are comprised of apoptotic cells, apoptotic bodies and debris. The accuracy of DNA content measurement was reflected by variation in fluorescence intensity between individual cells with identical DNA content in G0/1 cell population. The coefficient of variation (CV) of the mean value was about 3%, which is at the acceptable range (Darzynkiewicz et al., 2010). We have also used equal number of control and treated cells plus same concentration of DAPI stain to overcome the drawbacks that may consequence on these factors (Haase & Reed, 2002).
Cell death inducers such as H2O2, acetic acid or amphotericin B trigger apoptosis in Saccharomyces cerevisiae when applied at low concentration, but there is necrotic death when used at higher doses (Phillips et al., 2003; Ribeiro et al., 2006; Eisenberg et al., 2010). DNA degradation as indicator of apoptosis in S. cerevisiae also was observed under hyperosmotic stress (60% glucose) by Ribeiro et al. (2006). In this study, we demonstrate preliminary evidence of hypo-osmotic stress-induced apoptosis in C. glabrata, and further investigations to confirm this phenomenon might be required.
In conclusion, this study provides an evidence on the relation between cell number and mechanism of survival for yeast under hypo-osmotic stress. Candida glabrata cells protect each other against hypo-osmotic and starvation-induced death when in a quorum of ≥ 105 CFU mL−1, through the release of materials that enable survival. Moreover, this material is able to induce division once it is concentrated. When this material around is at a low concentration, the cells become damaged and appear to undergo programmed cell death (apoptosis).
Acknowledgement
This work is supported in part by a grant from the Medical Advances Without Animals Trust (MAWA).
References
Author notes
Editor: Richard Calderone

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}