





Abstract
Inflammatory bowel diseases (IBDs) such as Crohn's disease and ulcerative colitis are characterized by recurrent inflammation in the gastrointestinal tract. Infiltration of CD4+ lymphocytes and neutrophils is one of the predominant features of IBD.
Recently, interleukin (IL)-23 and the downstream T cell-derived cytokine IL-17 have been found to be elevated in intestinal tissue and serum of IBD patients. However, the role of IL-17 and IL-17R signaling in gut inflammation is unknown. To examine this role, we investigated gut inflammation in wild-type or IL-17R knockout mice.
Using a model of acute trinitrobenzenesulfonic acid (TNBS)-induced colitis, we found that IL-17 was produced in colon tissue at 24 and 48 hours and that IL-17R knockout mice were significantly protected against TNBS-induced weight loss, IL-6 production, colonic inflammation, and local macrophage inflammatory protein-2 induction. This protection occurred in the presence of equivalent induction of local IL-23 and higher levels of IL-12p70 and interferon-γ in IL-17R knockout mice compared with wild-type mice. Moreover, IL-17R knockout mice showed reduced tissue myeloperoxidase activity. Furthermore, overexpression of an IL-17R IgG1 fusion protein significantly attenuated colonic inflammation after acute TNBS.
These results demonstrate that IL-17R signaling plays a critical role in the development of TNBS-induced colitis and may represent a target for therapeutic intervention for IBD.
Crohn's disease and ulcerative colitis, collectively referred to as inflammatory bowel disease (IBD), are unique gastrointestinal disorders characterized by chronic relapsing inflammation.1,2 The histology associated with IBD includes inflammation of the intestinal mucosa with neutrophil and other inflammatory cell infiltration.3,4 Although the origin of IBD is complex and mutifactorial, it is believed that dysregulated host immunity in response to gut bacterial flora is involved in IBD pathogenesis.1,2 Recent studies have demonstrated that CD4+ lymphocytes play an important role in the development of IBD.5,6 CD4+ lymphocytes are increased and activated in intestinal lamina propria of IBD patients and in several colitis models.7 Furthermore, reconstitution of severe combined immunodeficient mice with functional CD4+ lymphocytes has been shown to lead to a lethal inflammatory colitis.7 The dysregulated T cell response in IBD is echoed by alterations in the mucosal cytokine expression,5 and Crohn's disease is well characterized for its TH1 cytokine pattern.8
In light of the critical role of TH1 CD4+ lymphocytes in IBD, further elucidation of CD4+ cell-derived cytokine regulatory networks will greatly facilitate the understanding of the disease and the development of novel therapeutic strategies.7,9 Recently, it has been shown that infusion of monoclonal antibody against interleukin (IL)-12, a critical regulator of TH1 cell development, is efficacious in Crohn's disease.9 However, this antibody is against the IL-12p40 subunit, which is shared by IL-23.10 Thus, neutralization of IL-23 could in part be responsible for the therapeutic effect of this reagent. In fact, the other subunit of IL-23, IL-23p19, has been shown to be elevated in intestinal tissue of patients with IBD.11 Our laboratory and others have shown that IL-23 is produced after Toll-like receptor 4 stimulation and is critical for CD4+ T cell IL-17 production12,13; and recently it was demonstrated that IL-23 specifically expands a population of CD4+T cells called ThIL-17 cells, which produce IL-17A, IL-17F, IL-6, and tumor necrosis factor-α.14,–17 These IL-23-driven CD4+ T cells are highly pathogenic and elicit IL-17-dependent inflammation, including experimental autoimmune encephalitis and collagen-induced arthritis.14
IL-17 is produced mainly by activated CD4+ T cells18 although CD8+ T cells also have been reported to produce IL-17.12 IL-17 induces neutrophilia19 and upregulates numerous inflammatory mediators, including inducible nitric oxide synthase, IL-6, and the CXC chemokines IL-8 and GCP-2,20 and the mouse homologues macrophage inflammatory protein-2 (MIP-2)21,22and LIX.23 Recently, IL-17 expression has been found to be increased in the serum and intestinal mucosa of IBD patients.24 From these data, we hypothesized that IL-17 and IL-17R signaling plays a key proinflammatory role in the early phase of IBD pathogenesis, leading to the observed leukocyte infiltration. To test this, we investigated the role of IL-17R signaling in an acute trinitrobenzenesulfonic acid (TNBS)-induced model of colitis.
Materials and Methods
Animals
All mice were housed in an AAALAC-approved specific pathogen-free vivarium at Louisiana State University Health Sciences Center or Children's Hospital of Pittsburgh. Colonies of IL-17 receptor knockout mice (IL-17R KO) with a C57BL/6 genetic background were established from breeder pairs developed at Immunex Corp (Seattle, Wash).22 Control C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, Me). Six-week-old male mice were randomized into control and experimental groups (n = 6–10).
Induction of Acute Colitis
Mice were anesthetized with intraperitoneal injection of ketamine (100 mg/kg). A rubber catheter was inserted into the rectum of mice, and 2 mg of TNBS in 50% ethanol (vol/vol) or vehicle alone was introduced into the colon through the catheter inserted 5 cm distal to the anus. Then, 24 or 48 hours after the challenge, the mice were killed, and the colons were removed for macroscopic scoring of inflammation according to the following semiquantitative criteria: 0 = no visible edema or hemorrhage; 1 = focal hyperemia or bowel wall edema and no hemorrhage; 2 = focal hyperemia or bowel wall edema and hemorrhage at 1 site; 3 = extended hyperemia or bowel wall edema and hemorrhage at>1 site; and 4 = extended hyperemia or bowel wall edema, hemorrhage at>1 site, and perforation. After scoring, the tissue was processed for histology, tissue myeloperoxidase (MPO) assay, and cytokine analysis.
Adenovirus Vectors
To antagonize IL-17R signaling in established TNBS colitis, we administered a control adenovirus encoding firefly luciferase (AdLuc) or a vector encoding a soluble murine IL-17F:Fc fusion protein (AdIL-17R:Fc) that neutralizes IL-17 bioactivity22 12 hours after TNBS administration, and mice were killed at 48 hours for tissue and cytokine analyses. The construction of these vectors has been described.22
Histology and Ribonuclease Protection Assays
Harvested colons were fixed in 10% neutral formalin, dehydrated, embedded in paraffin, and sectioned. The sections were stained with hematoxylin and eosin and evaluated under light microscopy. Total RNA was extracted from colonic tissue with Tri-Zol (Invitrogen) and hybridized to 32p-labeled ribonuclease protection assay probes using the MCK-5 RiboQunat kit (BD BioSciences, San Jose, Calif).
Enzyme-linked Immunosorbent Assay of IL-17, IL-6, and MIP-2
IL-17, IL-6, and MIP-2 were measured with an enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's protocol (R & D Systems, Minneapolis, Minn). Murine IL-23 was measured by ELISA (E-Bioscience, San Diego, Calif). To prepare ELISA samples from colon tissues, the colons in each treatment group were collected and homogenized with 0.5 mL of phosphate-buffered saline containing 1% Triton X-100 with proteinase inhibitor cocktail (Amersham, Piscataway, NJ). The homogenates were centrifuged at 12,000g for 15 min, and the supernatants were collected for ELISA. IL-6 was assayed in serum.
MPO Assay
MPO is an enzyme localized in azurophilic granules of neutrophils. MPO activity can be used as an index of neutrophil infiltration. To measure MPO, the tissue was collected and homogenized as described above, and the supernatants were assayed for MPO using the published protocols as previously described.22
Statistical Analysis
Statistical comparisons were made between experimental groups, and when differences were noted among treatment responses, multiple-comparison tests were conducted using the ANOVA f test at the 0.05 significance level.
Results
Induction of IL-17 Production in TNBS-treated Colon
To investigate the role of IL-17 in inflammatory colitis, we first examined whether TNBS treatment induced the local production of IL-17. At both 24 and 48 hours after treatment, the colons were harvested, and IL-17 in the tissue homogenates and serum was measured by ELISA. Compared with control mice receiving vehicle alone (Fig. 1), TNBS treatment increased detectable IL-17 by ≈3-fold. The production of IL-17 was compartmentalized to the colon because IL-17 was not detected in the serum of either group (data not shown). Tissue examination at 48 hours after administration of TNBS revealed increased numbers of tissue neutrophils (Fig. 2). Moreover, using a ribonuclease protection assay, we observed the induction of transcripts for a number of chemokines involved in neutrophil recruitment (MIP-1α, MIP-1β, MIP-2), T cells, eosinophils (RANTES), and macrophages (MCP-1).
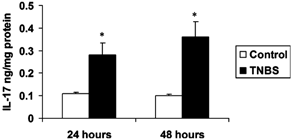
IL-17 levels in colonic tissues from wild-type (WT) mice treated with TNBS or vehicle control. Colons from mice were collected at 24 or 48 hours after intrarectal administration of TNBS, and the tissue homogenates were prepared and compared with control mice. Results represent mean + SEM of 5 mice. *P < 0.05 vs control.
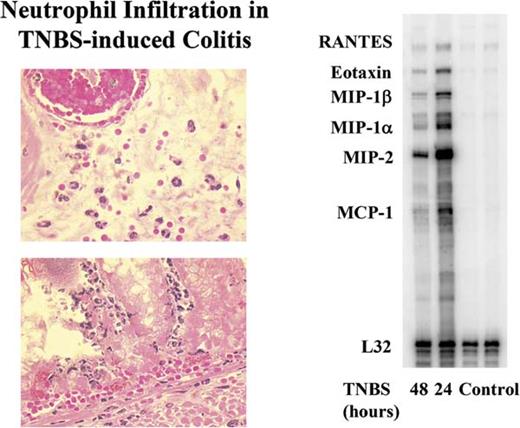
Neutrophilic inflammation and increased transcripts for MIP-2 in acute TNBS-induced colitis. Representative hematoxylin and eosin-stained section of colonic tissue from C57BL/6 mice 48 hours after TNBS. Note the presence of neutrophils in the tissue and within the vasculature. Right, Representative ribonuclease protection assay analysis demonstrating the induction of transcripts for MIP-2 in colonic tissue of TNBS-treated mice at 24 and 48 hours.
TNBS-induced Colonic Inflammation Is Less Severe in IL-17R KO Mice
As a result of the extensive induction of IL-17 protein and MIP-2 message, a gene known to be regulated by IL-17R signaling,22 and an increase in the influx of neutrophils into the colon, we investigated whether IL-17 and IL-17R signaling was critical for these events. We treated WT and IL-17R KO mice with TNBS or vehicle and assessed the tissue at 48 hours. A subgroup was followed up for 9 days for daily weight determinations. The colons from WT and IL-17R KO mice displayed normal histology (Fig. 3, A and B). TNBS treatment in WT mice resulted in severe colitis characterized by extensive leukocyte infiltration, significant hemorrhage, marked villi destruction, and edema (Fig. 3C) In contrast, TNBS caused only mild inflammation in the colon of IL-17R KO mice (Fig. 3D). Moreover, compared with WT mice, IL-17R KO mice exhibited significantly less neutrophil infiltration in the colon after TNBS challenge (Fig. 3). Histological inflammation also was measured with a 4-point macroscopic scoring based on the following parameters of inflammation: severity and size of hemorrhage, hyperemia, and edema (see Materials and Methods). This analysis also demonstrated a critical role of IL-17R signaling in regulating tissue inflammation in response to acute TNBS (Fig. 3E). Moreover, IL-17R KO showed significantly less weight loss during the acute phase of TNBS-induced colitis (Fig. 3F).
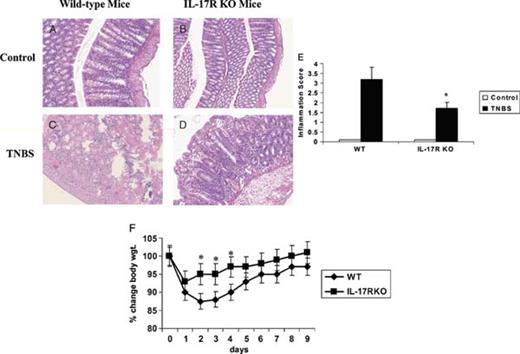
Reduced tissue destruction in IL-17R KO mice after induction of TNBS colitis. Representative hematoxylin and eosin-stained paraffin sections of untreated colons show normal tissue architecture in both WT (A) and IL-17R KO mice (B). C, Representative section of colon from a WT mouse 48 hours after intrarectal TNBS treatment. D, Section of colon of IL-17R KO mouse 48 hours after intrarectal TNBS treatment. E, Colon inflammation was graded on the basis of the criteria listed in Table 1. Results represent mean + SEM of 5 mice. F, Changes in body weight. WT mice or IL-17R KO mice received intrarectal TNBS and weighed daily (n = 8). *P < 0.05 vs control).

Because IL-17 has been shown to be positively regulated by IL-2312,13 and IL-1525 and negatively regulated by IL-12p70,13 we assayed levels of these cytokines in colonic tissue 24 and 48 hours after TNBS. IL-23 was induced in the colon by TNBS to an equivalent extent in WT and IL-17R KO mice at 24 hours (Fig. 4A). Similar levels also were observed at 48 hours (data not shown). IL-15 was undetectable in colonic tissues at 24 or 48 hours, but serum levels increased in TNBS-treated mice at 48 hours from 24 + 8.6 to 30.3 + 11.2 pg/mL in WT mice (P < 0.05; n = 4–6) and 30.9 + 10.1 to 43.2 + 11.8 pg/mL in IL-17R KO mice (P < 0.05; n = 4–6). Serum levels of IL-12p70 also increased in TNBS-treated mice at 48 hours from 70.2 + 35 to 169 + 56 pg/mL in WT mice (P < 0.05; n = 4–6) and 101 + 39.3 to 347.5 + 47.8 pg/mL in IL-17R KO mice (P < 0.05; n = 4–6). Colonic tissue levels of IL-112p70 could not be detected in WT mice but were detectable in 3 of 6 IL-17R KO mice at 48 hours (mean value of 15.45 + 2.69 pg/mg protein, P = NS vs WT controls). Taken together, these data suggest that the proximal regulators of IL-17 are induced in both WT and IL-17R KO mice; in fact, IL-17R KO mice had higher levels of IL-12 p70 despite less tissue inflammation and weight loss. In support of the higher IL-12p70 levels in IL-17R KO mice, we also observed higher levels of interferon-γ in tissues from TNBS-treated IL-17R KO mice compared with WT mice (Fig. 4B).
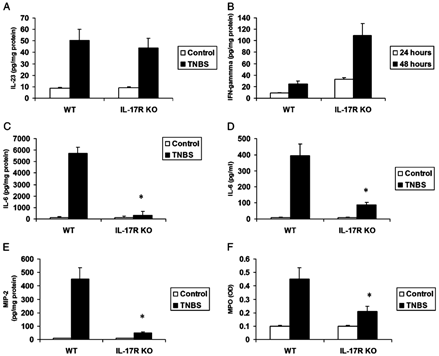
Critical role of IL-17R signaling for tissue neutrophil recruitment and tissue IL-6 and MIP-2 induction. A, Tissue IL-23 levels in WT and IL-17R KO mice in response to intrarectal TNBS (24 hours). Results represent mean + SEM of 4–6 mice. B, Tissue interferon-γ levels in WT and IL-17R KO mice in response to intrarectal TNBS (24 hours). C, Tissue IL-6 levels in WT and IL-17R KO mice in response to intrarectal TNBS (48 hours). D, Serum IL-6 levels in WT and IL-17 R KO mice in response to intrarectal TNBS (48 hours). E, Tissue MIP-2 levels in WT and IL-17R KO mice in response to intrarectal TNBS (48 hours). Results represent mean + SEM of 5 mice. F, Tissue MPO levels in WT and IL-17R KO mice in response to intrarectal TNBS (48 hours). Results represent mean + SEM of 4–7 mice. *P < 0.05 vs control.
IL-17 has been shown to be a potent inducer of IL-6 and the CXC chemokine MIP-2 in synoviocytes26 and fibroblasts; thus, we hypothesized that IL-17R signaling also regulates IL-6 and MIP-2 during acute colitis. To this end, we measured IL-6 and MIP-2 in the colons and IL-6 in the serum of WT mice and IL-17R KO mice 48 hours after vehicle or acute TNBS administration. Compared with the control group, TNBS treatment significantly increased IL-6 in colonic tissue (Fig. 4C) and serum (Fig. 4D) and local levels of MIP-2 (Fig. 4E) protein levels in the colon of WT mice. However, there was significantly less colonic IL-6 and MIP-2 production and lower serum IL-6 levels in IL-17R KO mice in response to TNBS treatment (Fig. 4, C-E). These results suggest that the local cellular response to IL-17 is critical for the production of systemic IL-6 and local CXC chemokine MIP-2, which ultimately leads to augmented neutrophil infiltration. Because neutrophils are the predominant inflammatory cell type in acute TNBS-induced inflammation, we assayed MPO levels as a biomarker of neutrophilic inflammation. In support of this MPO activity, a measure of neutrophil infiltration was significantly attenuated in the colons of IL-17R KO mice challenged with TNBS (Fig. 4F).
To confirm that IL-17R signaling is critical for acute TNBS colitis, we treated WT mice with a control adenovirus (AdLuc) or an Ad vector encoding a soluble IL-17 receptor IgG fusion22 12 hours after administration of TNBS. Mice treated with the AdIL-17R:Fc fusion protein showed significantly reduced tissue MPO and MIP-2 levels as reduced serum IL-6 (Fig. 5) at 48 hours compared with AdLuc control mice, supporting the critical role of IL-17R signaling in acute TNBS-induced colitis.
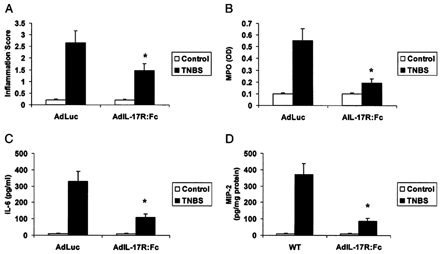
Antagonizing IL-17R signaling with a soluble IL-17R:Fc fusion protein antagonizes colitis, tissue neutrophil recruitment, and tissue IL-6 and MIP-2 induction. Mice were treated with 109 pfu of AdLuc or AdIL-17R:Fc 12 hours after administration of TNBS. Mice were killed at 48 hours. A, Tissue inflammation score in AdLuc- or AdIL-17R:Fc-treated mice in response to intrarectal TNBS. Results represent mean + SEM of 5–6 mice. B, Tissue MPO levels in AdLuc- or AdIL-17R:Fc-treated mice in response to intrarectal TNBS. Results represent mean + SEM of 4–6 mice. C, Serum IL-6 levels in AdLuc- or AdIL-17R:Fc-treated mice in response to intrarectal TNBS. Results represent mean + SEM of 4–6 mice. D, Tissue MIP-2 levels in AdLuc- or AdIL-17R:Fc-treated mice in response to intrarectal TNBS (48 hours). Results represent mean + SEM of 4–6 mice. *P < 0.05 versus control.
Discussion
TNBS has long been used as a valuable model to investigate the pathophysiological mechanism of IBD. Histological changes in TNBS-induced colitis resemble that of IBD in terms of ulceration, inflammation, and leukocyte infiltration. Although the caustic property of TNBS is partly responsible for the mucosal injury, TNBS has been shown to elicit intestinal protein antigenicity by functioning as a hapten,27 thereby developing an immune response and inflammation. The early acute phase of TNBS-induced inflammation is likely essential to establish the continued progression of the colitis that is mediated by T cell-dependent immunity. Because we have previously shown that IL-23-dependent IL-17 production by T cells occurs within 12 to 24 hours in a model of experimental gram-negative infection, we focused on early time points after induction of TNBS-induced colitis.
IL-17 is a T cell-derived cytokine produced by memory CD4+ and CD8+T cells and regulates tissue inflammation in the joint space,26 lung,21 and central nervous system.14 Recently, IL-17 also has been shown to be elaborated by intestinal γδ T cells.28 IL-17 has been shown to trigger the NF-κB signaling cascade, leading to the transcription of several cytokines and chemokines.29 Moreover, serum IL-17 levels have been reported to correlate with severity of Crohn's disease in patients.24 The present study demonstrates for the first time that TNBS increases local IL-17 production in the colon, which we postulate is produced locally by activated T cells. The time course of induction in the colon is similar to what is observed in lipopolysaccharide-induced T cell IL-17 responses in the lung compartment.20 Moreover, in this study, IL-17R KO mice developed less severe inflammation in response to acute TNBS treatment, despite higher levels of IL-12p70 and interferon-γ, which strongly indicates a pathophysiological role of IL-17 in colitis.
Previous studies in our laboratory found that systemic overexpression of IL-17 caused neutrophilia in mice.19 Furthermore, in response to a bacterial challenge in the mouse lung, IL-17 has been shown to be involved in MIP-2 production and influx of neutrophils.22 Thus, IL-17-stimulated production of chemokines may be a key element in the inflammatory cascade in a variety of pathological conditions.
Several studies report that the CXC chemokine IL-8 is elevated in the colons of IBD patients, which may explain the observed neutrophil infiltration.30,31 Our study focused on IL-17, a more proximal mediator of inflammation that may contribute to the early phase of TNBS-induced colitis. We have demonstrated that an increase in IL-17 after TNBS coincides with the production of MIP-2 and neutrophil infiltration. Several cellular sources of MIP-2 are found in the intestinal mucosa, including macrophages, endothelial cells, and fibroblasts.29,32 We postulate that the cellular target of IL-17 signaling is intestinal epithelial cells and/or fibroblasts that produce MIP-2 because these cell populations express IL-17R to a greater extent than tissue macrophages.21 It is important to note that in a prior study, using an anti-IL-17 antibody in model of dextran sulfate sodium colitis in BALB/c mice exacerbated colitis. The difference in outcome of that study compared with our study could be caused by several factors. One is the mouse strain; our study was performed in C57BL/6 mice. Second, the models of colitis are different, and we focused on an acute colitis in this article. Third, the dextran sulfate sodium study used a monoclonal antibody specific to IL-17 that does not cross-react with IL-17F (unpublished observations). Although we did not assess IL-17F in this model, IL-17-producing T cells often also express IL-17F.14,16,17,21 Thus, it is possible that the colitis observed in the dextran sulfate sodium study was caused by unopposed IL-17F signaling, whereas IL-17R KO mice do not show IL-6 elaboration to mouse IL-17A or IL-17F.21
Although our report does not delineate cellular sources of cytokine and chemokine production in the colons of TNBS-treated mice, our studies using IL-17R KO mice suggest that IL-17 enhances MIP-2 expression and subsequent sequestration of neutrophils. The results of our study using an animal model of colitis and a recent report of elevated IL-17 levels in patients with IBD24 warrant further studies to explore the role of IL-17 in the pathogenesis of IBD. Of note, anti-IL-12/23 p40 antibodies show early efficacy in Crohn's disease9,33 and have been associated with a reduction in serum IL-17 levels in these patients.34 Taken together, these data support ongoing investigation of potential IL-17-based therapies in IBD.
Acknowledgment
The authors would like to thank Dr Jacques Peschon for providing the IL-17R KO mice and reviewing this manuscript.
References
Author notes
Reprints: Dr Jay K. Kolls, MD, University of Pittsburgh, Children's Hospital of Pittsburgh, Suite 3765, 3705 Fifth Avenue, Pittsburgh, PA 15213 (e-mail: jay.kolls@chp.edu)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}