





Abstract
Hazard identification and risk assessment paradigms depend on the presumption of the similarity of rodents to humans, yet species specific responses, and the extrapolation of high-dose effects to low-dose exposures can affect the estimation of human risk from rodent data. As a consequence, a human relevance framework concept was developed by the International Programme on Chemical Safety (IPCS) and International Life Sciences Institute (ILSI) Risk Science Institute (RSI) with the central tenet being the identification of a mode of action (MOA). To perform a MOA analysis, the key biochemical, cellular, and molecular events need to first be established, and the temporal and dose-dependent concordance of each of the key events in the MOA can then be determined. The key events can be used to bridge species and dose for a given MOA. The next step in the MOA analysis is the assessment of biological plausibility for determining the relevance of the specified MOA in an animal model for human cancer risk based on kinetic and dynamic parameters. Using the framework approach, a MOA in animals could not be defined for metal overload. The MOA for phenobarbital (PB)-like P450 inducers was determined to be unlikely in humans after kinetic and dynamic factors were considered. In contrast, after these factors were considered with reference to estrogen, the conclusion was drawn that estrogen-induced tumors were plausible in humans. Finally, it was concluded that the induction of rodent liver tumors by porphyrogenic compounds followed a cytotoxic MOA, and that liver tumors formed as a result of sustained cytotoxicity and regenerative proliferation are considered relevant for evaluating human cancer risk if appropriate metabolism occurs in the animal models and in humans.
INTRODUCTION
Animal cancer bioassays have been used for more than a half century to determine whether pesticides, pharmaceuticals, industrial chemicals, and other products might cause cancer or other health problems in humans. As such, cancer bioassays have become the default for testing the carcinogenic potential of products where human use or exposure is anticipated. Extrapolations (dose and species) are necessary on animal data to predict and estimate human cancer risk. Such extrapolations have been surrounded by intense discussion and debate. Data from molecular and cellular studies have brought together a fuller biological understanding of how chemicals induce neoplasia in animal studies. Such mechanistic work has also raised doubt about the appropriateness of extrapolating some positive rodent tumor data to humans. In particular, the use of rodent liver tumor responses in human cancer risk assessment has often been controversial and has been extensively debated within the scientific community.
From 1989 until 1994, the International Life Sciences Institute (ILSI) Health and Environmental Sciences Institute (HESI) and the U.S. Environmental Protection Agency (EPA) co-organized and co-sponsored a series of five Mouse Liver Tumor Workshops with the ultimate aim being to consider how mouse liver tumor data should be interpreted with respect to assessing human cancer risk. At the conclusion of the last workshop in 1994, the participants considered the following question as central to the debate, “What constitutes a definition of robust evidence for a mode-of-action that would be sufficient to override a default regulatory posture?” (Unpublished observation; but internal reports available from ILSI HESI upon request).
The recommendations from these early HESI–EPA workshops provided a foundation upon which to develop a mode of action (MOA) framework. The International Programme on Chemical Safety (IPCS) defined the criteria for accepting a MOA as adequate for evaluating a specific tumor type in animals (Sonich-Mullin et al., 2001). Subsequent work conducted by the ILSI Risk Science Institute (RSI) determined how MOA studies can be used to establish the relevance of rodent tumors to humans (Cohen et al., 2003; Meek et al., 2003; Cohen et al., 2004).
Scientists representing academia, government, and industry organized a workshop at the 2005 Society of Toxicology annual meeting in New Orleans to determine if this human relevance framework could be used to analyze five MOAs associated with rodent liver tumors. This article summarizes the main points addressed during the Workshop on Mode of Action in Relevance of Rodent Liver Tumors to Human Cancer Risk. Following presentations on the pathogenesis of rodent hepatocarcinogenesis, and an overview of the Framework, several MOAs for rodent liver cancer development were described, including phenobarbital (PB)-like P450 induction, metal overload, porphyrogenicity, hormone perturbation (i.e., estrogen), and cytotoxicity. The MOA for peroxisome proliferator activated receptor alpha (PPARα) agonist-induced hepatocarcinogenesis was not included because it had recently been extensively reviewed, along with an evaluation of its human relevance (Klaunig et al., 2003).
The pathogenesis of rodent hepatocarcinogenesis: potential applications to human cancer risk.
A variety of model systems of hepatocarcinogenesis in the rat and in the mouse have been developed that allow the delineation and characterization of the stages of initiation, promotion, and progression as sequential processes in the pathogenesis of hepatocarcinogenesis (Pitot and Dragan, 2001). Each of these stages has specific characteristics, including mechanism, dose response, reversibility, and implications for human risk assessment. This model has served well for examining theoretical aspects of carcinogenesis, and its main contribution has been to identify a distinction between genotoxic (DNA reactive) and non-genotoxic effects of carcinogens.
In the rat, single hepatocytes expressing the placental form of glutathione-S-transferase (GSTP) have been identified as putative single initiated cells. Single putatively initiated hepatocytes expressing specific protooncogenes have been demonstrated in mice (Satoh et al., 2002). The number of single GSTP-positive hepatocytes exhibits a linear dose response with DNA-reactive chemical carcinogens, and clonal growth of about 1% of such hepatocytes results in altered hepatic foci (AHF), expressing GSTP and other genes in excess or deficient in normal hepatocytes (Pitot, 1993). The dose response for the development of AHF to several promoting agents, including phenobarbital, exhibits a sigmoidal response with a clear no-effect level and hormetic effect (Tsuda et al., 2003). Numerous studies argue strongly that promoting agents selectively (1) enhance cell replication in preneoplastic cells in AHF and (2) inhibit apoptosis in preneoplastic cells (Kolaja et al., 1996; Hikita et al., 1997). Studies with such model systems may have application beyond animals to the human, especially in relation to the importance of classifying a chemical as to whether it is an initiating agent, a promoting agent, or a progressor agent, or whether it is a complete carcinogen capable of all three of these activities (Dragan et al., 1993).
Framework for evaluating the human relevance of carcinogenic modes of action in animals.
Considerable effort has been expended during the past several decades to evaluate the MOA for specific chemicals causing cancer in rodents. However, the key question is the relevance of this MOA to human risk assessment. A framework was developed by an ILSI RSI working group sponsored by the U.S. EPA and Health Canada to address this issue and to provide direction in determining the relevance of rodent tumors to human health (Cohen et al., 2003; Meek et al., 2003; Cohen et al., 2004). This human relevance framework (HRF) describes methods and decision-tree logic to establish a relationship between early cellular events and the development of cancer. Knowledge of key events and the identification of a MOA provide a more rational basis for human hazard and risk assessment. The HRF devised by ILSI RSI evolved from the MOA paradigm in animals established by the IPCS (Sonich-Mullin et al., 2001).
As shown in Figure 1, the process of conducting a MOA analysis is based on three specific questions:
Is the weight of evidence sufficient to establish the MOA in animals? This evaluation is performed based on the framework developed by the IPCS/EPA for determining an animal MOA (Sonich-Mullin et al., 2001). The measurable key events in the MOA are explicitly stated and evaluated. This is not only useful in formulating a MOA, but it identifies any data gaps and uncertainties that remain.
Are key events in the animal MOA plausible in humans? This evaluation is based on a concordance analysis comparing the information known about the specific key events in both animals and humans. This evaluation is primarily a qualitative assessment.
Taking into account kinetic and dynamic factors, are key events in the animal MOA plausible in humans? This is a more quantitative analysis, which addresses the relevance of tumorigenicity to a level of exposure, and again relies on a concordance analysis between the animal model and humans. This approach focuses not only on dose response but also on quantitative differences between species in fundamental biologic processes that can affect exposure.
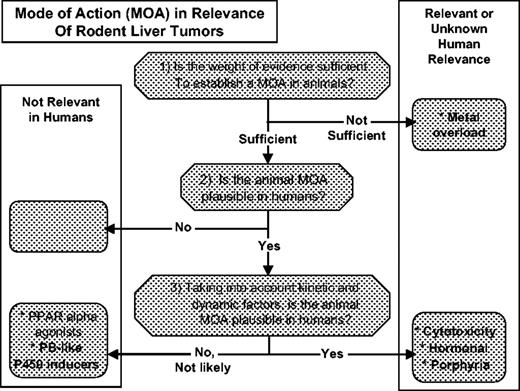
Summary of conclusions for various modes of action (MOA) for rodent liver tumors (RLT) using the human relevance framework (HRF). The workshop considered the following MOA: metal overload, phenobarbital-like P450 inducers, cytotoxicity, hormonal toxicity, and porphyrogenicity. The peroxisome proliferator activated receptor alpha (PPARα) agonists are included for comparative purposes.
For the first two questions, data may or may not be available for the specific chemical in humans, but broad knowledge of processes involved in humans, including anatomy, physiology, biochemistry, metabolism, and the like, are critical. Based on the results of this analysis, a statement of the confidence in the analysis is made, along with implications for carrying forward to the remainder of the risk assessment process. This human relevance framework is focused on hazard identification and evaluation. If the second and third questions are answered in the negative, then there is not a cancer hazard for humans and therefore no cancer risk. Examples based on qualitative differences between species are chemicals producing kidney tumors in male rats by binding to α2u-globulin. Phenobarbital (PB)-induced rat thyroid tumors provide an example where the qualitative processes (PB induction of thyroid hormone metabolism, T3 feedback on TSH, and effects on thyroid proliferation) are the same in rats and humans but where sufficient quantitative differences exist between species to indicate that thyroid tumors would not occur in humans no matter how great the exposure.
Mode of action and human relevance of phenobarbital-like rodent liver carcinogens.
Phenobarbital is the prototype of several rodent hepatocarcinogens (e.g., oxazepam, DDT) that induce tumors by a non-genotoxic mechanism involving liver hyperplasia (Williams and Whysner, 1996). In a wide range of genotoxicity tests, PB is negative and is not cytotoxic (Whysner et al., 1996; IARC, 2001). A diagnostic effect of PB is induction of some P450 enzymes, particularly of the CYP2B family. This effect is due to activation of nuclear receptors, particularly the constitutive androstane receptor (CAR). However, although there is evidence from studies in knockout mice that CAR plays an essential role in the carcinogenicity of PB (Yamamoto et al., 2004), it is uncertain whether CYP induction is a surrogate for a wider pleiotropic response (Ueda et al., 2002) or if P450 itself plays a role, e.g., by generation of active oxygen species (Lehman-McKeeman et al., 1999). Additional PB responses that are key in its tumorigenic effect include increased cell proliferation, inhibition of apoptosis, hypertrophy, and development of altered hepatic foci (Whysner et al., 1996). These effects are all CAR-dependent (Wei et al., 2000; Yamamoto et al., 2004). Hence, it is possible to identify a series of key events for PB-induced liver tumors in rodents that fulfill criteria proposed by Sonich-Mullin et al. (2001) for a MOA.
Although CAR is expressed, and PB induces CYP enzymes in human liver, it may act more through the pregnane X receptor (PXR) than through CAR (Moore et al., 2003). However, there are exceptions such as phenytoin, which has been shown to induce CYP2B6 in humans via CAR (Wang et al., 2004). In addition, there is evidence that human CAR may be activatable by PB, resulting in the induction of non-P450 genes, including UGT1A1 (Sugatani et al., 2001). For example, liver size is increased in humans after prolonged treatment with PB (Pirttiaho et al., 1982). However, limited studies with human hepatocytes indicate that such cells are refractory to the hyperplastic and anti-apoptotic effects of PB (Parzefall et al., 1991; Hasmall and Roberts, 1999). Moreover, although the data for concordance analysis for PB are limited, there are convincing data showing that in patients receiving PB for many years, at doses producing plasma concentrations similar to those following a carcinogenic dose in rodents, there is no evidence of a hepatocarcinogenic effect (IARC, 2001; Lamminpaa et al., 2002). There are a number of data gaps, including the extent to which this MOA extends to other rodent hepatocarcinogens that are P450 inducers, the concordance of key events between rodents and humans, and a more detailed definition of the key events in rodents (e.g., role of CYP2B, reactive oxygen species, DNA-methylation). Nevertheless, the situation with PB affords a somewhat unique opportunity, because extensive epidemiology data from the clinical applications of this drug can be used in a human relevance framework to help bridge some of the data gaps in an inverse direction from the traditional application of the framework. Hence, for those compounds for which there are robust data for a PB-like MOA it can be concluded that the carcinogenic response is not relevant to humans.
Mode of action and human relevance of metal overload and porphyrogenic compounds.
Using the recently developed human relevance framework, the MOA for metal overload (copper and iron) and porphyrinogenic chemical-induced rodent hepatocellular cancer induction were examined. Iron and copper overload in the liver are seen in the human diseases hemochromatosis and Wilson's disease, respectively (Schilsky and Oikonomou, 2005). Both iron and copper overload have been implicated as causal factors in both rodent and human hepatic cancer (Harrison and Bacon, 2005; Kowdley, 2004, Powell, 1994). In evaluating a potential MOA for metal overload in the induction of hepatic neoplasia, multiple references noting an increase in oxidative damage subsequent to metal overload were examined (Bartsch and Nair, 2004; Nair et al., 1999). Oxidative stress has been implicated as an important factor in the carcinogenesis process in both genotoxic and nongenotoxic mechanisms (Klaunig and Kamendulis, 2004). However, using the Framework as depicted in Figure 1, an expert working group concluded that while copper and/or iron overload is associated with the development of rodent and human hepatic neoplasia, evidence that only copper or only iron overload produces liver cancer in rodents or humans was not apparent. It was the conclusion of the working group that additional liver damage, including inflammatory effects, hepatocyte necrosis, cirrhosis, and/or fibrosis were necessary to produce the liver neoplasia (Greaves et al., 2005). Therefore, under the restrictions of the Framework definitions, the working group concluded that without other scientific proof neither metal produces rodent or human liver cancer.
In the discussion of the metal overload data by the working group, it became apparent that in iron overload, a frequently observed pathologic effect was the induction of liver porphyria. Because of this link to metal overload, this discussion led to a consideration of the impact of porphyrinogenic compounds on hepatic rodent neoplasia (Smith et al., 1993). A number of chemical and pharmaceutical agents produce porphyria and hepatocellular cancer in rodents. Using the established porphyrinogenic agent, hexachlorobenzene, as a model (Carthew and Smith, 1994), the expert working group concluded that porphyrinogenic compounds have a definable MOA for rodent hepatic cancer that involves a threshold and a chronic, dose-dependent, persistent hepatocellular injury with resulting persistent compensatory hyperplasia, which is discussed below as a cytotoxic MOA.
Hormonal perturbation as a mode of action for rodent liver tumors.
Because hormonally active agents are believed to contribute to the development of rodent liver tumors, perturbation of hormonal regulation should be considered when determining the MOA of non-genotoxic hepatocarcinogens. Although the liver is not generally considered a target tissue for sex hormones, it is quite responsive to these hormones. As an example, rat liver, but not mouse liver, is responsive to the carcinogenic actions of estrogen. With respect to humans, the mode of action observed in rat, namely hormonal perturbation, is a plausible mechanism for primary liver cancer induction. As summarized by IARC (1999), two case-control studies of benign hepatocellular tumors showed a strong relationship with the duration of use of combined oral contraceptives. Three cohort studies showed no significant association between the use of combined oral contraceptives and the incidence of mortality from liver cancer, but it is important to note that these studies were characterized by low statistical power. Long-term use of combined oral contraceptives was associated with an increase in the risk of hepatocellular carcinoma in all nine case-control studies conducted in populations with low incidences of hepatitis B and C viral infections and chronic liver disease, which are the major causes of human liver cancer, and in analyses in which women with these factors were excluded. Few data are available for the more recent, low-dose contraceptive formulations. In the two case-control studies conducted in populations with a high prevalence of infection with hepatitis viruses, there was no increase in the risk for hepatocellular carcinoma associated with the use of combined oral contraceptives; but there was little information on long-term use. Ultimately, extensive epidemiology has shown a small increased risk of long-term estrogen-containing contraceptives with hepatocellular adenomas and less with carcinomas, although each of these tumors remains rare in the young female populations exposed. Even though there is a differential risk for tumor types associated with estrogen treatment, it does not appear that the adenomas are precursor lesions to the carcinomas.
The key events in rodent liver carcinogenesis following exposure to estrogenic agents are perturbation of hormone level or function, altered cell proliferation to apoptosis balance, and development of altered foci of cellular alteration. Because this MOA is receptor-mediated, the use of the framework approach permits initial development of quantitative risk assessment that can be applied across species. These types of agents typically exhibit a threshold, and their effects are dose level-, dose frequency-, and dose-duration dependent.
Rodent liver tumors: cytotoxicity mode of action.
Cytotoxicity is a generally accepted MOA and has been defined for a number of nongenotoxic rodent carcinogens, including chloroform-induced liver and kidney tumors and melamine-induced bladder tumors (Andersen et al., 1998; Sonich-Mullin et al., 2001). A hepatocyte cytotoxicant would produce continual hepatocyte death, leading to persistent regenerative growth. Such growth results in more opportunities for “spontaneous” DNA mutations, allowing mutated cells to accumulate and proliferate, and giving rise to preneoplastic foci and, ultimately, to tumors via further clonal expansion. To define a cytotoxic MOA in liver, it is critical to ensure that other MOAs do not contribute significantly to hepatocarcinogenesis. For instance, it is important to ensure that DNA reactivity is not the source of the tumor findings. Furthermore, it is important to establish that there are parallel dose responses (not necessarily identical) for the key events (i.e., cytotoxicity and proliferation) and tumors, as well as a specificity of the key events and the tumor response. Hepatotoxicity can be demonstrated with histopathology (necrosis and/or apoptosis), with or without serum enzyme changes. Cell proliferation can be measured by several methods, such as bromodeoxyuridine (BrdU) labeling index and/or cell number, and it may need to be evaluated on the basis of zonal distribution.
Using chloroform as an example, the critical role played by metabolic activation (e.g., CYP 2E1) in the onset of hepatotoxicity is illustrated. As such, liver tumors formed as a result of sustained cytotoxicity and regenerative proliferation are considered relevant for evaluating human cancer risk if appropriate metabolism occurs in the animal model and in humans. A non-linear dose response is expected and will likely entail a threshold effect.
SUMMARY AND CONCLUSIONS
A survey of chemicals tested in chronic rodent bioassays showed that the most common target organ is the liver and the most sensitive species is the mouse (Gold et al., 1991; Gold, 2002). In many cases, the tumor increases were seen only in the mouse liver and were not observed in other species. Interestingly, the incidence of liver tumors in humans (age-adjusted) in the United States is only 2.4/100,000 (El-Serag and Mason, 1999), in comparison to much higher frequency tumors such as prostate, breast, or lung (e.g., 165.4/100,000, 134.1/100,000, and 62.9/100,000, respectively, as summarized by the National Cancer Institute in a review from 1973–1999). These observations can be added to those already discussed above to provide the basis for understanding why the relevance of rodent liver tumors to humans has been so intensely discussed and debated through the years.
In recent years, MOA frameworks were developed through the ILSI RSI (Meek et al., 2003; Cohen et al., 2004), and the IPCS (Sonich-Mullin et al., 2001). The RSI MOA framework was applied to the analysis of the class of PPARα agonists (Klaunig et al., 2003). A summary of the MOA analysis for PPARα agonists is depicted in Figure 1. A MOA was identified (See the first question, above) and determined to be plausible in humans (second question). However, when kinetic and dynamic factors were considered (third question), the plausibility of the animal MOA was determined not to be likely in humans. The primary objective of the present article is to summarize the discussion that took place during a workshop at the 2005 SOT meeting, where additional MOAs for rodent liver tumors were analyzed in terms of the human relevance framework. The analysis of five MOAs are summarized in Figure 1.
With one exception, animal MOAs were identified and determined to be feasible in humans. Using the framework approach, a definable MOA for the formation of hepatic cancer in rodents by either iron or copper (e.g., metal overload) was not achievable in animals based on available scientific evidence.
The situation with PB-like P450 inducers afforded a unique opportunity to capitalize on extensive experience in humans where no incidence of liver tumors has been observed. Therefore, this MOA was determined to be unlikely in humans after considering kinetic and dynamic factors. Once a robust MOA has been established in rodents, and adequate human data show no evidence of a carcinogenic response, these data can be applied to chemicals sharing that MOA. Although a number of compounds appear to be PB-like from the vantage of the pleiotropic gene expression changes induced (e.g., specifically CYP2B induction), it cannot be concluded that this MOA is similar without further data.
Estrogen and related molecules induce liver tumors in rats. Although the MOA for hepatocarcinogenicity is not completely understood, it is accepted that estrogen works in a dose- and duration-dependent manner, through receptors, to change cellular function. As such, the hormonal MOA can be considered a subset of receptor-mediated carcinogenesis. Several examples of receptor-mediated carcinogenesis have already been considered, including the MOA for PPARα agonists and CAR activators. In contrast to these MOA, the conclusion was that after considering kinetic and dynamic factors, estrogen-induced modulation should be considered as plausible in humans. However, because this MOA is receptor-mediated, agents that induce cancer through this MOA typically exhibit a threshold, and their effects are dose level-, dose frequency-, and dose duration-dependent. Also, in humans, estrogens have not been associated with development of malignant hepatocellular carcinomas.
Liver tumors formed as a result of sustained cytotoxicity and regenerative proliferation are considered relevant for evaluating human cancer risk, if appropriate metabolism occurs in the animal model and in humans. However, because of the dose–response relationship for cytoxicity and proliferation that is present with this MOA, a nonlinear model should be used for human health risk assessment, possibly including a threshold. Thus, the relevance of the level of human exposure is critical to the evaluation of potential risk in humans. Using hexachlorobenzene as an example, it was concluded that the induction of rodent liver cancer by porphyrinogenic compounds followed a cytotoxic MOA (Carthew and Smith, 1994).
The SOT workshop provided greater insight into the utility of the MOA-HRF approach as applied to human relevance, and it identified a number of key data gaps. An understanding of the MOA of agents underlying liver tumor induction will place the observation of rodent liver tumors into perspective when human risk assessment is performed, taking appropriate account of dose and exposure duration and frequency.
Disclaimer: The views expressed in this paper are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency, the Food and Drug Administration National Center for Toxicological Research, or other institutions represented by the authors.
References
Andersen, M., Brusick, D., Cohen, S., Dragan, Y., Frederick, C., Goodman, J. I., Hard, G., Meek, B., and O'Flaherty, E. J. (
Bartsch, H., and Nair, J. (
Carthew, P., and Smith, A. G. (
Cohen, S. M., Meek, M. E., Klaunig, J. E., Patton, D. E., and Fenner-Crisp, P. (
Cohen, S. M., Klaunig, J., Meek, M. E., Hill, R. N., Pastoor, T., Lehman-McKeeman, L., Bucher, J., Longfellow, D. G., Seed, J., Dellarco, V., et al. (
Dragan, Y. P., Sargent, L., Xu, Y.-D., Su, Y.-H., and Pitot, H. C. (
El-Serag, H. B., and Mason, A. C. (
Gold, L. S., Slone, T. H., Manley, N. B., and Bernstein, L. (
Greaves, P., Clothier, B., Davies, R., Higginson, F. M., Edwards, R. E., Dalton, T. P., Nebert, D. W., and Smith, A. G. (
Harrison, S. A., and Bacon, B. R. (
Hasmall, S. C., and Roberts, R. A. (
Hikita, H., Vaughan, J., and Pitot, H. C. (
IARC. (
IARC. (
Klaunig, J. E., Babich, M. A., Baetcke, K. P., Cook, J. C., Corton, J. C., David, R. M., DeLuca, J. G., Lai, D. Y., McKee, R. H., Peters, J. M., et al. (
Klaunig, J. E., and Kamendulis, L. M. (
Kolaja, K. L., Stevenson, D. E., Walborg, E. F., Jr., and Klaunig, J. E. (
Kowdley, K. V. (
Lamminpaa, A., Pukkala, E., Teppo, L., and Neuvonen, P. J. (
Lehman-McKeeman, L. D., Caudill, D., Vassallo, J. D., and Fix, A. S. (
Meek, M. E., Bucher, J. R., Cohen, S. M., Dellarco, V., Hill, R. N., Lehman-McKeeman, L. D., Longfellow, D. G., Pastoor, T., Seed, J., and Patton, D. E. (
Moore, J. T., Moore, L. B., Maglich, J. M., and Kliewer, S. A. (
Nair, J., Barbin, A., Velic, I., and Bartsch, H. (
Parzefall, W., Erber, E., Sedivy, R., and Schulte-Hermann, R. (
Pirttiaho, H. I., Sotaniemi, E. A., Pelkonen, R. O., and Pitkanen, U. (
Pitot, H. C. (
Pitot, H. C., III., and Dragan, Y. P. (
Powell, C. J. (
Satoh, K., Itoh, K., Yamamoto, M., Tanaka, M., Hayakari, M., Ookawa, K., Yamazaki, T., Sato, T., Tsuchida, S., and Hatayama, I. (
Schilsky, M. L., and Oikonomou, I. (
Smith, A. G., Carthew, P., Francis, J. E., Cabral, J. R., and Manson, M. M. (
Sonich-Mullin, C., Fielder, R., Wiltse, J., Baetcke, K., Dempsey, J., Fenner-Crisp, P., Grant, D., Hartley, M., Knapp, A., Kroese, D., et al. (
Sugatani, J., Kojima, H., Ueda, A., Kakizaki, S., Yoshinari, K., Gong, Q. H., Owens, I. S., Negishi, M., and Sueyoshi, T. (
Tsuda, H., Fukushima, S., Wanibuchi, H., Morimura, K., Nakae, D., Imaida, K., Tatematsu, M., Hirose, M., Wakabayashi, K., and Moore, M. A. (
Ueda, A., Hamadeh, H. K., Webb, H. K., Yamamoto, Y., Sueyoshi, T., Afshari, C. A., Lehmann, J. M., and Negishi, M. (
Wang, H., Faucette, S., Moore, R., Sueyoshi, T., Negishi, M., and LeCluyse, E. (
Wei, P., Zhang, J., Egan-Hafley, M., Liang, S., and Moore, D. D. (
Whysner, J., Ross, P. M., and Williams, G. M. (
Williams, G. M., and Whysner, J. (
Author notes
*ILSI Health and Environmental Sciences Institute, Washington, DC 20005; †Department of Oncology, McArdle Laboratory, University of Wisconsin, Madison, Wisconsin 53706; ‡Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, Nebraska 68198; §Experimental Medicine & Toxicology, Imperial College London, London, United Kingdom W12 0NN; ¶Indiana University School of Medicine, Indianapolis, Indiana 46202; ‖Syngenta CropScience, Greensboro, North Carolina 27419; |||Office of Pesticides Programs, U.S. Environmental Protection Agency, Washington, DC 20460; and ‖‖National Center for Toxicology Research, Jefferson, Arkansas 72079

{kind=link}
Comments