





Objective. IL-26 has been shown to have high expression in RA. However, the effects of IL-26 on bone destruction in RA have not been evaluated. The aim of this study was to investigate the effects and mechanisms of IL-26 on RANK ligand (RANKL)-induced osteoclastogenesis.
Methods. We treated cells with IL-26 in RANKL-induced oseteoclastogenesis to monitor osteoclast formation by tartrate-resistant acid phosphatase (TRAP) staining. Osteoclast activity was assessed by pit formation assay and F-actin ring formation. The mechanism of the inhibition was studied by biochemical analyses such as RT-PCR, immunofluorescence staining and immunoblotting. In addition, cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
Results. IL-26 inhibited RANKL-induced TRAP-positive multinucleated cells and inhibited RANKL-induced nuclear factor κB (NF-κB) activation and nuclear factor of activated T cells, cytoplasmic 1 (NFATc1) nuclear translocation in RAW264.7 cells. Also, IL-26 significantly inhibited the bone-resorbing activity and F-actin ring formation ability of mature osteoclasts. Moreover, IL-26 suppressed RANKL-induced mitogen-activated protein kinase activation and NFATc1 downstream gene expression.
Conclusion. We suggest that the inhibitory activity of IL-26 on osteoclastogenesis is via down-regulation of RANKL-induced NF-κB and NFATc1 expression. Our results suggest IL-26 as a possible new remedy against osteolytic bone destruction.
Rheumatology key messages
IL-26 reduced osteoclastogenic activities in receptor activator of nuclear factor κB ligand stimulated monocytes or macrophages in RA.
Modulation of IL-26 may represent a new approach against inflammatory osteolytic diseases like RA.
Introduction
RA is characterized by the presence of synovitis concomitant with pannus formation, cartilage destruction and bone erosion [1–3]. Evidence has demonstrated that osteoclasts are highly activated in the bone resorption site in inflammatory joint diseases. Furthermore, it has been reported that mice lacking osteoclasts were resistant to arthritis-induced bone erosion [4]. Recently, Th17 cells, a subset of T helper cells, were considered to be culprits in the disease progression of RA [5]. Also, IL-26, a newly identified cytokine that is produced by Th17 cells, was shown to be highly expressed in RA [6, 7].
IL-26, also named AK155, was first identified by transformation of T lymphocytes with the monkey herpesvirus saimiri. It belongs to the IL-10 cytokine family with 47% amino acid similarity and 24.7% amino acid identity. The IL-26 gene is located on chromosome 12q14 near the IFN-λ and IL-22 genes. IL-26 is similar to IL-10 in the conformational form of a homodimer [8]. Previous studies show that IL-26 mRNA is not detected in basal type of monocytes, NK, B and T cells, but IL-26 can be induced by IL-2 and IL-12 in NK cells and anti-CD3 mAb in T cells [9]. Also, using IL-23 to polarize T cells toward Th17 cells will induce IL-26 expression [10]. IL-26 signals through a membrane receptor complex, IL-20 receptor 1 (IL-20R1) and IL-10 receptor 2 (IL-10R2), and triggers its intracellular signals through signal transducer and activator of transcription (STAT) 1, STAT3, extracellular signal-regulated protein kinases (ERK), c-Jun N-terminal kinase (JNK) and protein kinase B (AKT) [11, 12].
Recently, IL-26 was founded to be a modulator in innate immunity with antimicrobial effects in neutrophil-infiltrated lung diseases [13]. IL-26 expression in monocytes could be suppressed by Mycobacterium tuberculosis infection [14]. However, IL-26 is encoded by a unique human gene without a murine homologue and this limits the experimental opportunities to study the phenotypes in vivo. Moreover, there is no study published on the effects of IL-26 on monocyte/macrophage differentiation as in RA-induced activation of osteoclastogenesis. Therefore to understand the role of IL-26 in the development of RA-induced osteoclast activation appears to be crucial for clarifying its pathophysiology.
Methods
Cell line and reagents
Murine RAW 264.7 cells were purchased from the Food Industry Research and Development Institute (Hsinchu, Taiwan). Recombinant human IL-26 protein was purchased from MyBiosource (San Diego, CA, USA) and R&D Systems (Minneapolis, MN, USA). Human or mouse recombinant RANK ligand (RANKL) and M-CSF were purchased from Peprotech (London, UK). Nuclear factor-κB (NF-κB) pathway, phospho-mitogen-activated protein kinases (MAPK) and total MAPK Family Antibody Sampler Kit were obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-α-tubulin came from Abcam Inc. (Cambridge, MA, USA). Anti-TATA-box-binding protein (anti-TBP) came from Millipore (Carrigtwohill, Co. Cork, Ireland). Anti-NFATc1 and anti-RANK antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). All other reagents were purchased from Sigma-Aldrich (St Louis, MO, USA).
Cell culture and osteoclast differentiation
The methods of maintaining RAW264.7 cells and further differentiating them into osteoclasts were descripted in our previous publication [15]. In brief, RAW264.7 cells were seeded at 5 × 103 cells/well in a 96-well plate, and cultured with 50 ng/ml soluble murine RANKL plus IL-26 or IgG control for 5 days. Human peripheral blood mononuclear cells (PBMCs) were obtained from healthy adult volunteers, with consent and ethical approval (Tri-Service General Hospital, 1-1-4-05-128), and isolated by density centrifugation using lymphocyte separation medium according to the manufacturer’s instructions (Lonza, Walkersville, MD, USA). Moreover, PBMCs were resuspended in alpha minimal essential medium (α-MEM) (Gibco BRL/Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 30 ng/ml macrophage colony-stimulating factor (M-CSF), penicillin (100 U/ml) and streptomycin (100 mg/ml), and plated in 96-well plates at a density of 4 × 105 cells/well for osteoclast differentiation as previously described [16]. All differentiation media were replaced on days 2–3.
IL-26 plasmid transfection
The human IL-26 gene was isolated from Jurkat T cell by using reverse transcription PCR with forward primer 5′-GCGGCCGCGTGAGTGACACACGCTGA-3′ and reverse primer 5′-GCGGCCGCGAGAAGGAAACCCAATTTAT-3′. Then, the PCR product was digested by NotI restriction enzyme and inserted into pCMV-hDCR3 backbone vector at the NotI site to form the pCMV-hIL-26 construct. RAW264.7 cells were transfected with IL-26 plasmid or empty vector by using Lipofectamine 2000 reagent according to the manufacturer’s instructions (Invitrogen/Thermo Fisher Scientific). Briefly, plasmid was formulated with reagent and diluted in α-MEM, then incubated with RAW264.7 cells. After a 6 h incubation, the mixture was replaced by osteoclastic differentiation medium. Differentiation media were also replaced on days 2–3.
Tartrate-resistant acid phosphatase staining
Cells were washed with PBS and fixed with 3.7% formaldehyde for 30 min. After washing with PBS, cells were incubated at 37 °C in a humid and light-protected incubator for 1 h in the reaction mixture of the Leucocyte Acid Phosphatase Assay kit (Cat. no. 387, Sigma-Aldrich), as directed by the manufacturer. Cells were washed three times with distilled water, and tartrate-resistant acid phosphatase (TRAP)-positive multinucleated cells containing three or more nuclei were counted under a light microscope and photographed.
Resorption assay
RAW264.7 cells were seeded onto 20 mm2 dentine slices (Cat. no. 3988, Corning, NY, USA) in 24-well plates at a density of 2 ×105 cells/well to test osteoclast resorption activity. Then, the cells were washed off the bottom of the plates with a 5% hypochlorite solution. Further, the wells were washed twice with distilled water and allowed to dry at room temperature for 5 h. The ratios of the resorbed area to the total area were measured in four optical fields on a slice using NIH ImageJ software at 100-fold magnification.
Quantitative RT-PCR analysis
Total RNA was extracted by NucleoSpin RNA KIT (Macherey-Nagel GmbH & Co. KG, Düren, Germany). One microgram of total RNA was reversed transcribed to cDNA by using SuperScript III reverse transcriptase (Invitrogen/Thermo Fisher Scientific). Real-time PCR was performed on the LightCycler 480 II system in a total volume of 20 μl using a white LightCycler 480 Multiwell Plate 96 covered with adhesive sealing foil (Roche, Mannheim, Germany). The PCR primers used were previously described [15]. Quantitative thermal cycling parameters were 95 °C for 15 min, followed by 40 cycles for 30 s at 95 °C, 30 s at 60 °C and 1 min at 72 °C, and 10 min at 72 °C for the final elongation. The ratios of each gene were normalized to the corresponding values for GAPDH.
Immunoblotting analysis
Whole cell extract and nuclear extracts were prepared according to our previous study [15]. In brief, RAW264.7 cells treated with IL-26 or IgG control in the absence or presence of 50 ng/ml RANKL were harvested and suspended in radioimmunoprecipitation assay (RIPA) lysis buffer for whole cell extract; for nuclear extracts cells were suspended in 200 μl of buffer A (10 mM HEPES-KOH [pH 7.8], 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 1mM dithiothreitol, 0.1 mM PMSF) then incubated on ice for 15 min. Further, nuclei were pelleted by centrifugation for 5 min at 18 000 g, and the supernatant was collected as the cytoplasmic fraction. The nuclei were resuspended in 40 μl of buffer C (50 mM HEPES-KOH [pH 7.8], 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 1 mM DTT, 0.1 mM PMSF, 25% glycerol [vol/vol]), incubated on ice for 20 min, and centrifuged for 5 min at 18 000 g at 4°C. The supernatant was used as the nuclear extract. Equivalent amounts of protein were loaded for SDS-PAGE, and immunoblotting was performed using specific antibodies for p38, phospho-p38, ERK, phospho-ERK, JNK, phospho-JNK, NF-κB p65, nuclear factor of activated T cells, cytoplasmic 1 (NFATc1), inhibitor of NF-κB α (IκBα), phospho-IκBα, TBP, STAT1, phospho-STAT1, STAT3, phospho-STAT3 and α-tubulin.
NFATc1 and F-actin immunofluorescence staining
RAW264.7 cells were seeded onto glass coverslips and then incubated with 50 ng/ml RANKL plus IL-26 or IgG control. The distribution of NFATc1 protein 24 h after stimulation was assessed according to previously published protocols [15]. F-actin rings were detected after 5 days’ culture. Coverslips were removed, washed in PBS, fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, incubated with 5% BSA and incubated with a specific anti-NFATc1 mAb (1:50) at 4 °C overnight. Cells were washed in PBS, and incubated for 2 h with FITC-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). After the immunostaining procedures, cells were nuclear-stained with 4',6-diamidino-2-phenylindole (Sigma-Aldrich). Fluorescence was visualized using an Olympus microscope with SPOT software. The percentage of cells displaying nuclear staining was then quantified; 100 cells per group were measured from three separate coverslips per group.
MTT assay
Cells (1 × 104/well) were seeded in a 96-well plate with medium supplemented with 10% FBS and treated with various concentrations of IL-26 for 48 h, and then washed three times with PBS and treated with medium containing 500 µg/ml of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide for 30 min at 37°C. Cells were then washed with PBS and solubilized in 100 µl of dimethyl sulfoxide. The intracellular purple formazan concentrations were determined at 550 nm in an ELISA plate reader.
Statistical analysis
Data are shown as means (s.d.) and were analysed using one-way ANOVA with Newman–Keuls multiple comparisons post hoc tests. P < 0.05 was considered to be statistically significant.
Results
Effects of IL-26 on RANKL-induced osteoclast differentiation
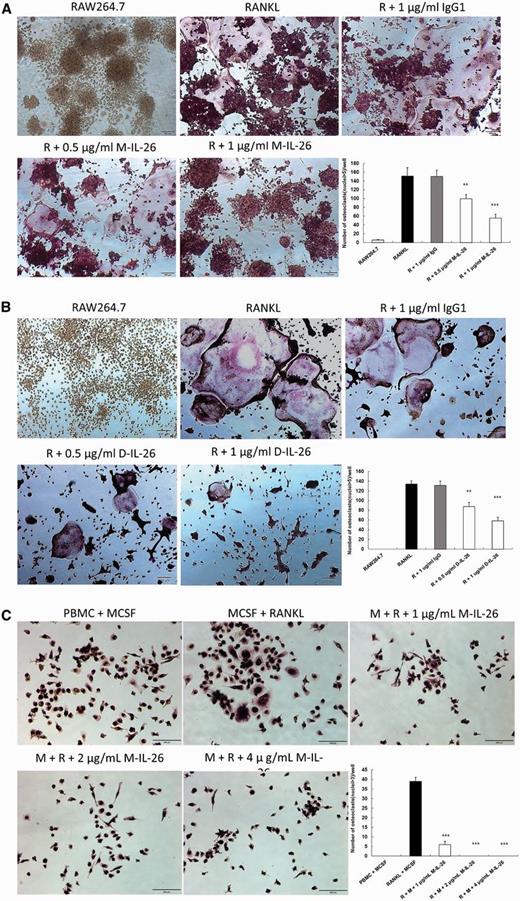
Effects of IL-26 on RANKL-induced osteoclast differentiation of RAW264.7 cells and PBMCs
(A) and (B) RAW264.7 cells were treated with serial dilution of IL-26 (0.5 and 1 μg/ml; A, monomer; B, dimer) or IgG control (1 μg/ml) in the presence of RANKL (50 ng/ml) for 5 days. (C) PBMCs were treated with various concentrations of monomer IL-26 in the presence of RANKL (50 ng/ml) and M-CSF (30 ng/ml) for 5 days. After incubation, the cells were fixed and stained for TRAP, and TRAP+ multinucleated cells containing more than three nuclei were counted as multinucleated osteoclasts. The data represent the means (s.d.) of more than three cultures (**P < 0.01, ***P < 0.001). M-CSF: macrophage colony-stimulating factor; TRAP: tartrate-resistant acid phosphatase.
Effects of IL-26 on RANKL-induced osteoclast function
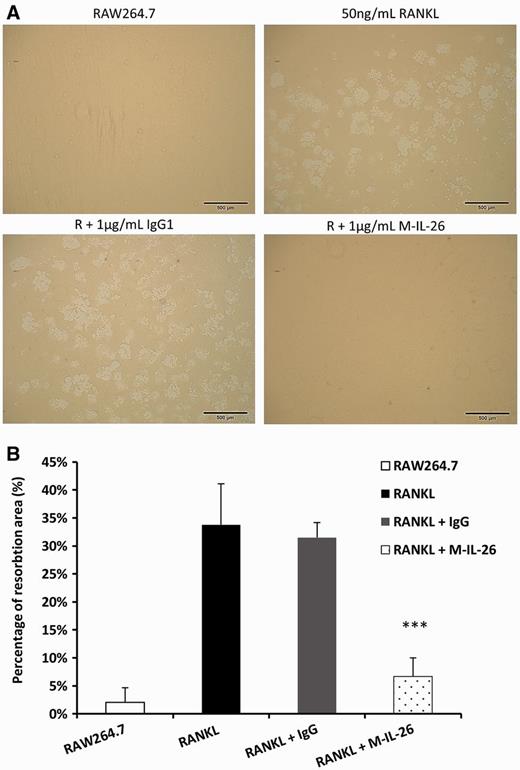
Effects of IL-26 on resorption pit formation
(A) RAW264.7 cells were seeded on bone slices with 1 μg/ml of monomer IL-26 or IgG control in the presence of RANKL (50 ng/ml). After incubating for 5 days, the dentine slices were recovered from the culture and resorption pits were visualized. (B) Percentages of the resorbed areas were determined using the NIH ImageJ software. The data represent the means (s.d.) of more than four slices. (***P < 0.001).
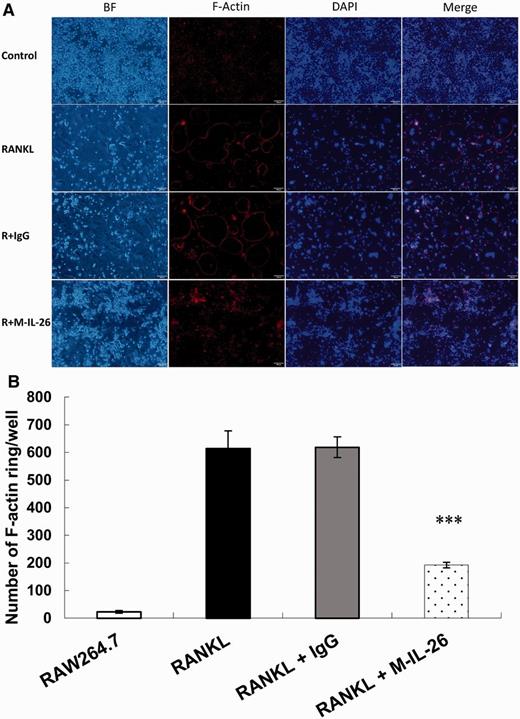
Effects of IL-26 on actin ring formation
(A) RAW264.7 cells were seeded at a density of 2 × 104 cells/well in 24-well plates and treated with 1 μg/ml of monomer IL-26 or IgG control in the presence of RANKL (50 ng/ml). After incubating for 5 days, the cells were fixed by 4% formaldehyde and stained by anti-F-actin antibody for immunofluorescence staining. (B) F-actin-positive rings were counted in each group. The data represent the means (s.d.) of more than three cultures (***P < 0.001).
Effects of IL-26 on NFATc1-regulated gene expression during RANKL-induced osteoclast development
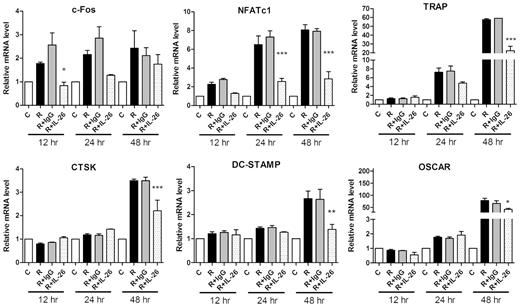
Effect of IL-26 on osteoclastogenic mRNA expression
RAW264.7 cells were treated for 12, 24 and 48 h with 1 μg/ml monomer IL-26 or IgG control in the present of RANKL (50 ng/ml). Total RNA was isolated and 1 μg of total RNA was used to transcribe cDNA. cDNA was used as a template for PCR with mouse-specific primers. Expression of osteoclastogenic genes for c-Fos, NFATc1, TRAP, CTSK, DC-STAMP and OSCAR was detected by real-time PCR. A representative result of at least three independent experiments is shown. (C: RAW264.7 cells alone; R: RANKL treatment; R + G: RANKL + IgG; R + IL-26: RANKL + IL-26.) The data represent the means (s.d.) of more than three cultures (*P < 0.05, **P < 0.01, ***P < 0.001). CTSK: cathepsin K; DC-STAMP: dendrocyte expressed seven transmembrane protein; NFATc1: nuclear factor of activated T cells, cytoplasmic 1; OSCAR: osteoclast-associated receptor; TRAP: tartrate-resistant acid phosphatase.
Effects of IL-26 on RANKL-induced activation of MAPKs in osteoclasts
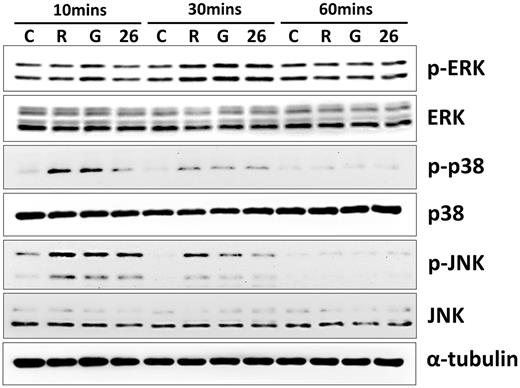
Effects of IL-26 on the activation of MAPKs in RAW264.7 cells
RAW264.7 cells were serum-starved for 16 h and treated with 1 μg/ml monomer IL-26 or IgG in the present of RANKL (50 ng/ml) stimulation for 10, 30 or 60 min. Cell extracts were analysed by western blot using antibodies specifically directed against the phosphorylated forms of MAPKs, and compared with data obtained with antibodies directed against the unphosphorylated states of the kinases. Equal amounts of protein were loaded in each lane as demonstrated by the level of α-tubulin. A representative result of at least three independent experiments is shown. (C: RAW264.7 cells; R: RANKL; G: RANKL + IgG; 26: RANKL + IL-26.).
Effects of IL-26 on RANKL-induced nuclear translocation and expression of NF-kB and NFATc1
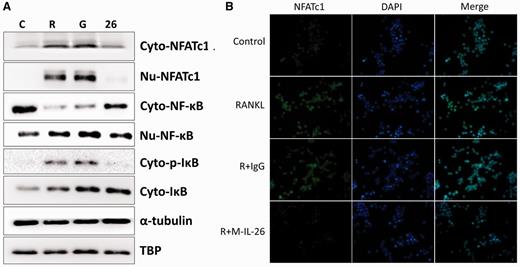
Effects of IL-26 on RANKL-induced nuclear translocation of NF-κB-p65 and NFATc1
(A) Western blot analysis. RAW264.7 cells were serum-starved overnight and then incubated with RANKL (50 ng/ml) for 24 h in the absence or presence of IL-26. Cytoplasmic extracts (Cyto) and nuclear extracts (Nu) were analysed by western blot using antibody specifically directed against NF-κB p65 and NFATc1 protein. Equal amounts of protein were loaded in each lane as demonstrated by the level of TBP (Nu) and α-tubulin (Cyto). (B) Immunofluorescence analysis. RAW264.7 cells were treated with or without IL-26 after RANKL (50 ng/ml) stimulation for 24 h. After the time indicated, the cells were stained with anti-NFATc1 antibody (green) and DAPI (nuclear staining, blue). The nuclear localization of NFATc1 was confirmed in merged images of RAW264.7 cells treated with RANKL. DAPI: 4',6-diamidino-2-phenylindole; NFATc1: nuclear factor of activated T cells, cytoplasmic 1.
Discussion
In the present study we are the first group to have found that IL-26, a newly identified cytokine, will suppress RANKL-induced osteoclastogenesis both in monomer and dimer form. The inhibitory effect of IL-26 was reinforced by IκB-α binding with NF-κB p65 and restricted NFATc1 nuclear translocation. Previous studies majorly focused on the IL-26 cytokine in Th17 or NK cells. For example, Oral et al. [23] reported that IL-26 was without any effect on treated naïve T cells with Th1 or Th2 cytokine polarization. Manel et al. [24] reported that for Th17 cell differentiation, the transcription factor RORγtT would induced chemokine receptor CCR6, IL-23 receptor, IL-17F and IL-26 gene expression. Also, a human NK cell subset, NK-22, secreted IL-22, IL-26 and leukaemia inhibitory factor [25]. IL-26 was overexpressed in chronically HCV-infected patients and enhanced TNF-related apoptosis-inducing ligand (TRAIL)-mediated cytotoxicity and interferon production by NK cells [26]. These findings suggested that IL-26 was not a direct inducer of T helper or NK cell differentiation but seemed to be a response element secreted by NK or Th17 cells. Furthermore, many scientists regard the function of IL-26 as a part of skin immunity. The abnormal keratinocytes expressing IL-10RB and IL-20RA seemed to be targets for IL-26 in the pathogenesis of psoriasis [27, 28]. Also, IL-26 did not induce biological activities in the reconstituted human epidermis system [29]. All evidence pointed to IL-26 as a response element, elevated against foreign pathogens and functioning in innate immunity regulation. Here we found that IL-26 could directly affect macrophages and inhibit osteoclast differentiation.
In many inflammatory or osteolytic diseases, like RA, hyper-activation of immune function or osteoclast function promotes disease pathogenesis and progression. RANKL–RANK was the major ligand–receptor pathway for monocytes to differentiate to osteoclasts. Binding of RANKL on RANK dominantly activated transcription factor NF-κB to translocate into the nucleus and subsequently enhance expression of downstream target genes [30]. Also, RANKL–RANK transduced a series of intracellular signalling pathways such as phosphorylation of JNK, ERK and p38 MAPK and activation of AP-1 (c-Fos) and NFATc1 transcription factors. Moreover, NFATc1 has been demonstrated to be a key mediator of osteoclastogenesis by auto-amplifying and conducting the expression of osteoclast-specific genes including TRAP, DC-STAMP, OSCAR and CTSK [31, 32]. In this study, we found that IL-26 partially decreased the phosphorylation of ERK, JNK and p38 MAPK in response to RANKL. Also, downstream genes, AP-1 (c-Fos) and NFATc1 transcription factors, were suppressed in the early stage (12 or 24 h). TRAP, DC-STAMP, OSCAR and CTSK were suppressed in the late stage (48 h). We speculated that the high level of IL-26 in RA patients might account for compensatory effects on osteoclastogenesis by potent cytokines, such as IL-1 and TNF-α.
Previous studies showed that IL-26 functions by strongly binding with heterodimeric IL-10R2 and IL-20R1 receptor [11], and the intracellular signalling pathways were stimulating STAT1 and STAT3 activation [33]. However, monocytes did not express IL-20R1 receptor in the cell surface [28]. Blockade of IL-22 or IL-26 with sIL-10R2 protein did not decrease the activation of STAT3 in the COLO-205 cell line [34]. In our study, we found that monocytes stimulated with or without IL-26 would not enhance IL-20R1 or IL-10R2 expression (supplementary Fig. S4, available at Rheumatology Online). Also, the STAT1 and STAT3 intracellular signalling pathways could not be activated by IL-26 treatment (supplementary Fig. S3, available at Rheumatology Online). Therefore, we postulated that IL-26 may contain other binding factors, like heparin or other receptors in monocytes. Also, polymorphism in the IL-26 gene region contributing to sex bias in susceptibility to rheumatoid arthritis has been demonstrated [35]. Sex hormone or endocrine system regulation by IL-26 cytokine expression may be another target to investigate in RA.
In conclusion, we have provided the first evidence of IL-26 exhibiting inhibitory effects on RANKL-stimulated osteoclastogenesis as well as functioning in pit formation on dentine slices and F-actin ring formation. We have also provided molecular mechanisms for this inhibition, involving MAPKs and transcription factors like NF-κB and NFATc1, which modulate genes involved in osteoclastogenesis. Our study showed that IL-26 blocked the activation of MAPKs and NF-κB/IκB signalling pathways under RANKL stimulation, and subsequently suppressed NFATc1 nuclear translocation. Finally, IL-26 inhibited osteoclastogenesis via down-regulation of NFATc1-acting osteoclastic gene expression. Taken together, our data may decipher the possible molecular mechanisms of IL-26 in osteoclastogenesis and may make IL-26 a potential therapeutic agent for bone disorders such as RA or osteoporosis by reducing osteoclast formation and functioning to promote bone health.
Acknowledgements
This work was supported in part by grants from the Ministry of National Defence Medical Affairs Bureau (MAB-105-062 to C.P.C. and 104-C10-35 to Y.J.P.). The authors acknowledge the technical supports provided by Instrument Center of National Defence Medical Center.
Funding: This work was funded by the Ministry of Science and Technology, Taiwan, Republic of China (MOST 104-2320-B-016-019) and Tri-Service General Hospital, National Defence Medical Center, Taipei, Taiwan (TSGH-C104-074).
Disclosure statement: The authors have declared no conflicts of interest.
Supplementary data
Supplementary data are available at Rheumatology Online.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments