





Abstract
Children with chronic inflammatory diseases, such as juvenile idiopathic arthritis (JIA), suffer from a variety of growth disorders. These range from general growth retardation to local acceleration of growth in the affected limb. These disorders are associated with the increased production of proinflammatory cytokines, which may influence growth through a local effect in the growth plates of long bones and/or systemic effects throughout the whole body. In this article we review these aspects and also discuss the evidence for interaction between the inflammatory cytokine and growth-signalling pathways.
Child growth is multifactorial, and requires the tightly controlled mobilization and utilization of energy, coordinated by several biochemical and physiological regulatory mechanisms. Impaired linear growth is commonly encountered in children suffering from chronic inflammatory diseases such as juvenile idiopathic arthritis (JIA), both at disease presentation and following treatment with glucocorticoids. In these children, maintenance of growth is a complex process that is influenced by a number of different mechanisms, including not only the steroid therapy, but also other factors such as the disease process, nutritional status, endocrine status and the response of the body to inflammatory mediators. The growth abnormalities observed in children with inflammatory diseases are often associated with delayed onset of sexual maturation and altered bone development. Evidence of an imbalance of proinflammatory cytokines in patients with inflammatory diseases includes the positive correlation of serum and synovial cytokine concentrations with JIA disease activity [1, 2], an increase in antagonists or soluble receptors with a flare of arthritis [3] and the effectiveness of JIA therapies that involve cytokine modulation [4, 5]. The proinflammatory cytokines that have been reported to play a major role in JIA include interleukin 1β (IL-1β), tumour necrosis factor α (TNF-α) and interleukin 6 (IL-6) [6–8]. These proinflammatory cytokines may influence child growth through systemic effects and/or a local effect at the level of the growth plate of long bones.
The growth plate and how bones grow
The articular chondrocyte is well known to the rheumatologist. Its function is to synthesize and maintain an extracellular matrix that is able to withstand physical deformation and facilitate joint articulation. Articular cartilages persist and survive. This is in contrast to the growth cartilage produced at the epiphyseal growth plate, which is progressively synthesized and replaced by bone with accompanying longitudinal (endochondral) bone growth [9].
The growth plate is a thin layer of cartilage found near the ends of long bones and vertebrae [10]. It comprises both chondrocytes and their extracellular matrix. A characteristic of endochondral bone growth is the precise temporal and spatial organization of chondrocytes within the growth plate, where they differentiate through a series of maturational stages whilst remaining in a spatially fixed location throughout its existence [11]. Undifferentiated stem cell progenitors differentiate into chondrocytes and progress through a proliferative phase. Immediately following cessation of cell division, the cells undergo terminal differentiation into hypertrophic chondrocytes [12]; the chondrocytes become more voluminous with increases in rough endoplasmic reticulum and Golgi apparatus, reflecting their increased matrix production [13]. The rate of longitudinal bone growth is determined by a complex interplay of proliferative kinetics, size of the proliferative pool, matrix synthesis and hypertrophic chondrocyte enlargement [14]. The precise control of these processes is still a matter of debate and any perturbation of these synchronized variables may underlie the growth modulatory effects of external agents, such as inflammatory cytokines (Fig. 1).
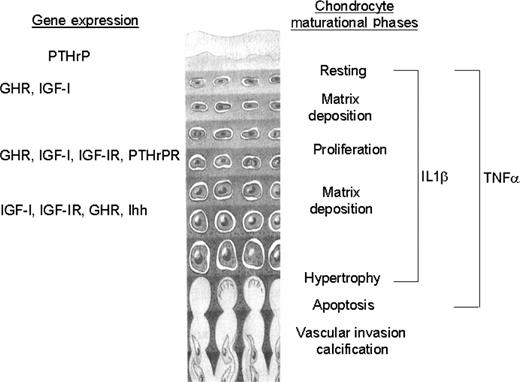
Growth plate gene expression and the chondrocyte maturational phases that may be modulated by the proinflammatory cytokines TNF-α and IL-1β.
Histologically, the chondrocytes are arranged in columns that parallel the longitudinal axis of the bone. Each column and each chondrocyte within a column are respectively separated by longitudinal and transverse septae made up of a collagenous and proteoglycan-rich extracellular matrix. The extracellular matrix of the epiphysis determines the mechanical properties of the tissue and contributes to the structural arrangement of the growth plate by providing a scaffold for chondrocyte attachment and migration [15]. During terminal differentiation, the collagenous matrix mineralizes and it functionally changes to an environment allowing vascular invasion from the marrow of the metaphysis, thereby providing a conduit for the recruitment of osteoclasts and differentiating osteoblasts that remodel the newly formed cartilage into bone tissue [16]. During the growth period the rate of cartilage addition and replacement are coupled, so that the width of the growth plate remains constant. From animal studies it has been calculated that eight hypertrophic chondrocytes (including its associated matrix) are eliminated by apoptosis from each column of cells every day [17]. However, towards the end of the growth period the growth plate narrows and finally disappears. It has been widely accepted that growth cessation is a result of systemic control and the fusion of the epiphysis with the metaphysis. However, it is now believed that regulation is intrinsic to the growth plate and that growth plate fusion does not precede, but follows, the cessation of growth [18, 19].
In children, maintenance of growth is a complex process that is influenced by a number of systemic and local autocrine/paracrine mechanisms. This list includes vitamin D metabolites, androgens, fibroblast growth factor and bone morphogenic proteins together with other members of the transforming growth superfamily. Two of the most important and widely studied regulators of postnatal bone growth are growth hormone (GH) and insulin-like growth factor 1 (IGF-1). The dual effector theory of GH/IGF-1 action at the growth plate proposes that GH acts directly on germinal zone precursors of the growth plate to stimulate the differentiation of chondrocytes and then amplify local IGF-1 synthesis, which in turn induces the clonal expansion of chondrocyte columns in an autocrine/paracrine manner [20, 21]. IGF-1 is, however, expressed by chondrocytes situated in all maturational zones of the growth plate, with IGF-1 mRNA expression mainly restricted to the hypertrophic zone. Infusion of hypophysectomized rats with IGF-1 stimulated growth plate chondrocytes at all stages of differentiation, including those in the hypertrophic zone [22–24]. Although liver-derived IGF-1 is the main determinant of serum IGF-1 levels, it is not as important for postnatal growth as locally derived IGF-1 [25, 26].
Recent investigations have indicated that the local control of cellular function by parathyroid hormone-related peptide (PTHrP) extends to cells of the skeleton and in particular to the growth plate chondrocytes. Mice lacking the PTH/PTHrP receptor gene have a growth plate morphology similar to that of mice that are homozygous for the ablation of the PTHrP gene [27]. There is widespread accelerated differentiation of chondrocytes and premature mineralization, resulting in a narrow growth plate. In contrast, the phenotype of mice in which the PTHrP gene is overexpressed is characterized by a dramatic slowing down of the differentiation of chondrocytes and a wider growth plate [28]. These and other experiments have led to the acceptance that PTHrP, together with the morphogen Indian hedgehog (Ihh), is one of the major influences on the endochondral growth process [10].
Sex steroids are of crucial importance in the control of longitudinal growth, and exert direct effects on the growth plate. A number of studies have demonstrated the presence of the androgen receptor and both oestrogen receptors, ERα and ERβ, in growth plate tissue at the mRNA and protein level in several species, including the rat, rabbit and human [29], indicating that androgens and oestrogens directly regulate processes in the growth plate. Furthermore, the growth plate possesses the ability for steroidogenesis as well as aromatization [30]. However, it has been difficult to prove whether androgens have direct effects on growth plate cartilage. Non-aromatizable androgens, such as dihydrotestosterone, have been shown to regulate both proliferation and differentiation of cultured human epiphyseal chondrocytes, probably by promoting local IGF-1 synthesis and increasing IGF-1 receptor expression [31, 32].
Oestrogen is the critical hormone in controlling growth plate acceleration and fusion in both females and males [33, 34]. In vitro studies have shown that oestrogen alters alkaline phosphatase activity, cell proliferation and proteoglycan synthesis [35, 36]. Indeed, oestrogen has a biphasic effect on proliferation, which is stimulated by low levels and inhibited by high levels of oestrogen [37].
Linear growth in children with JIA
Maintenance of growth in children with JIA is a complex process that is influenced by a number of different mechanisms, including not only drugs but also other factors, such as the disease process [38]. Estimates of significant short stature (final height standard deviation score (SDS) of less than –2) in children with JIA range from 11% of all patients [39] to 41% of patients with systemic forms of JIA [40]. Clinical studies by our own group and others have shown that growth and skeletal development are reversibly impaired during periods of intensive therapy, especially during treatment with prednisolone and dexamethasone [41]. However, 87% of patients in the retrospective study of 24 children with systemic JIA reported by Simon et al. had a final height below their target height [40]. In this study, the authors also noted that, after remission of the disease and discontinuation of glucocorticoid therapy, 30% of the patients did not show any catch-up growth. The patients who showed no catch-up were more likely to be shorter at diagnosis and had a lower target height.
As a group, children with JIA have a unique pattern of growth disturbance; systemic JIA is often associated with general growth retardation whereas oligoarticular JIA is associated with local excess growth. This discrepancy manifests itself as increased growth in the affected limb in young children and premature fusion of the epiphyses in the older child with resultant limb shortening [42]. In the young child, early use of intra-articular steroids has been reported to prevent the leg length discrepancy, but some recent evidence suggests that intra-articular steroids may depress the growth of the contralateral leg [43, 44]. Although hyperaemia to the juxtaposed growth plates is thought to be the mechanism for overgrowth in young children, there is no clear scientific explanation for this phenomenon, especially as the reverse seems to happen in the adolescent child.
Children with JIA and severe growth retardation may have normal pulsatile GH secretion and are reported to have reduced IGF-1 levels, suggestive of GH resistance [45]. After monitoring height velocity for 1 yr, Davies et al. [45] treated 20 children for the following year with either 12 IU/m2 per week or 24 IU/m2 per week of recombinant human GH (rhGH). There was a significant increase in height velocity in almost all children during the treatment period. Children with mild to moderate disease activity grew at a better rate than those with very active disease and those with polyarticular disease responded better than those with systemic JIA. In addition, those children receiving high-dose rhGH grew significantly more than those on the low-dose regimen. Touati et al. [46] and Bechtold et al. [47] have also shown that treatment with a relatively high dose of rhGH (0.35–0.46 mg/kg per week) in children with systemic or polyarticular JIA for a year led to an improvement in height velocity. Overall, these studies suggest that the adverse effects of glucocorticoids and chronic inflammatory disease on growth and skeletal development in children with JIA may be halted, but not reversed, by treatment with recombinant GH.
A number of clinical studies suggest a direct link between factors produced during chronic inflammation and growth failure [45, 48, 49]. In systemic JIA, impairment of linear growth is seen during periods of disease activity, with subsequent normalization of growth rate during remission [48, 49]. In patients with systemic JIA and growth defect treated with GH, growth velocity during treatment appears inversely correlated with the intensity of inflammation [45].
It is likely that cytokines may influence the development of these abnormal growth patterns in children with inflammatory diseases, through both systemic effects and local effects at the level of the growth plate.
Systemic effects of cytokines on the GH/IGF-1 axis
Studies using transgenic murine models have examined in detail the mechanisms by which IL-6 induces systemic effects on growth retardation. The murine model NSE/hIL-6 overexpresses IL-6, leading to high levels of circulating IL-6 and reduced growth rate [50]. The growth defect is completely abolished by neutralization of IL-6 [51]. The mice show reduced circulating IGF-1 levels, while GH production remains unaltered [50]. This mirrors the observations in patients with systemic JIA [52, 53], and demonstrates that the effect on IGF-1 levels is not mediated indirectly via an effect on GH production.
A relevant portion of circulating IGF-1 is carried in a ternary complex with IGF binding protein (IGFBP)-3 and an acid-labile subunit. This complex prolongs the half-life of circulating IGF-1. Decreased levels of IGFBP3 are observed in NSE/hIL-6 mice and wild-type mice treated with IL-6 [52]. Patients with systemic JIA [52–54] also show markedly reduced levels of IGFBP-3.
The decrease in IGFBP-3 levels in NSE/hIL-6 mice is associated with impaired formation of the ternary complex. This is likely to be, at least in part, due to IGFBP-3 proteolysis, which was also observed in NSE/hIL-6 mice [52]. Proteolytic degradation of IGFBP-3 has also been demonstrated in patients with systemic JIA [52]. It has therefore been proposed that IL-6 decreases IGF-1 levels by increased clearance, as association of IGF-1 in the circulating ternary complex prolongs the half-life of IGF-1 from less than 10 min to 16 h [52].
Excessive production of TNF-α also causes growth failure in TNF-transgenic mice [55, 56]. However, no studies have specifically examined alterations in the IGF-1/GH axis in these models.
Il-1β has also been shown to reduce plasma concentrations of both IGF-1 [57] and the acid-labile subunit [58, 59]. Increased levels of IGFBP1 inhibit IGF-1 bioactivity, as phosphorylated IGFBP1 has a higher affinity for IGF-1 than the IGF-1 receptor [60, 61], and therefore complexes with IGF-1 and prevents IGF-1 receptor binding. Hepatic IGFBP-1 expression is elevated in septic rats; this can be completely prevented by treatment with an IL-1 receptor antagonist [61]. IL-1β also directly stimulates IGFBP-1 protein and mRNA synthesis in the HepG2 hepatoma cell line [62–64].
Local effects of cytokines on the growth plate
The cytokines that have been studied most extensively for their ability to regulate bone and cartilage function in cell cultures of chondrocytes are IL-1β, IL-6 and TNF-α. However, whilst many studies have examined the effects of cytokines on articular chondrocytes, relatively few have investigated growth plate chondrocytes.
Local destruction of the growth plate has been observed following inflammatory synovitis, denoted by elevated synovial TNF-α, IL-1β and IL-6 [65]. This suggests that the proinflammatory cytokines present in the fluid can reach the growth plate from the adjacent synovial space.
IL-1β and TNF-α inhibit the expression of a number of genes encoding chondrocyte-specific matrix molecules, including collagen types IX and XI and aggrecan [66–69]. In cultures of rabbit growth plate chondrocytes, IL-1β decreased alkaline phosphatase activity during hypertrophy and suppressed increases in cell size and type X collagen expression, suggesting inhibition of chondrocyte differentiation [70]. IL-1β has been shown to induce a dose-dependent rise in rat growth plate chondrocyte DNA synthesis, which may explain the increased longitudinal bone growth seen in affected limbs of children with arthritis [71]. TNF-α has also been reported to stimulate DNA synthesis in cultured rat [72] and rabbit [73] costal chondrocytes. TNF-α induces apoptosis in chick chondrocyte cultures [74] and suppresses cartilaginous nodule formation and the accumulation of cartilage-specific proteoglycan reduction in the ATDC5 cell line [75]. TNF-α has also been shown to reduce proteoglycan synthesis in fetal mouse metatarsals [76]. Furthermore, this study demonstrated that IL-17 synergizes with TNF-α to further reduce proteoglycan production. IL-1β has also been shown to synergize with TNF-α to inhibit longitudinal growth in fetal rat metatarsal bones [77].
IL-6 belongs to a cytokine subfamily whose members share a common signal-transducing molecule, gp130, in their respective complexes [78]. In articular chondrocytes, contradictory results have been reported on the effects of IL-6 on proteoglycan synthesis. It must be noted that, in most of the related studies, the IL-6 effect was investigated in connection with that of other cytokines, such as IGF-1 and IL-1. For example, very high doses of IL-6 were found to decrease the enhancing effect of IGF-1 on proteoglycan synthesis [79]. It was also shown that IL-6 is required for the inhibition of proteoglycan synthesis by IL-1 in human articular chondrocytes [80], but these latter results have not been reproduced by others [81]. The contradictory results may be because the IL-6 effects were investigated in the absence of soluble IL-6 receptor. Indeed, it has been shown that the levels of membrane-anchored IL-6 receptor on chondrocytes are lower than those on other cell types, such as hepatocytes, and that in vitro addition of soluble IL-6 receptor to IL-6 is required to observe the full inhibitory effect of IL-6 on proteoglycan synthesis [82]. IL-6 in the presence of additional soluble IL-6 receptor also markedly down-regulates the expression of cartilage-specific matrix genes, including type II collagen, aggrecan and link proteins in bovine articular chondrocytes [83]. IL-6 has been reported to have no effect on growth plate chondrocyte dynamics [84]; however, these studies were undertaken in the absence of soluble IL-6 receptor.
Oncostatin M (OSM) is a cytokine that also belongs to the IL-6 family [78] and has been detected in JIA synovial fluid [65]. Additionally, the injection of an adenovirus vector that expresses murine OSM induces damage to the growth plate cartilage, including proteoglycan depletion and loss of matrix integrity [65]. This damage was shown to be dependent on endogenous IL-1. The authors proposed that OSM could either enhance or modify the autocrine effects of IL-1 on growth plate chondrocytes, thereby leading to growth plate proteoglycan loss, disorganization and finally growth abnormalities.
Underlying mechanisms
The cellular machinery by which cytokines may act on the growth plate is unclear at present. Transmission of external signals from the cell surface to the internal cellular environment occurs via tightly controlled complex transduction pathways. Alterations in these highly regulated signalling cascades in chondrocytes may play a fundamental role in the functional abnormalities of growth plate cartilage that underlie cytokine-induced growth retardation.
Sox9 gene expression
Sox9 is a master regulatory factor for chondrocyte differentiation and cartilage formation [85]. In mouse chimeric embryos, Sox9-null cells are unable to express the genes for chondrocyte-specific markers, such as collagen types II, IX and XI and aggrecan [85]. Both IL-1β and TNF-α markedly down-regulate the expression of Sox9 in both costal chondrocytes and a chondrocytic cell line [86]. These effects appear to be mediated by the NF-κB pathway, although the precise mechanism remains to be elucidated [86].
TNF-α and apoptosis
TNF-α has two cell surface receptors, TNFR1 and TNFR2. Binding of TNF-α to TNFR1 initiates an apoptotic response, which is simultaneously activated by TNF-α through the Fas-associated death domain (FADD), a protein that triggers the pro-apoptotic caspases and cell death. TNF-α has been reported to induce apoptosis in chondrogenic cells [75] and fetal rat metatarsal cultures [77]. TNF-α-induced apoptosis may be a major contributor to the growth abnormalities observed in children suffering from inflammatory diseases.
Sex steroids
Proinflammatory cytokines can interfere with steroidogenesis at the level of the testes and ovaries, which may in turn modulate the effects of sex steroids on the growth plate. TNF-α, IL-1β and IL-6 inhibit the steroidogenesis of Leydig cells [87], which synthesize and secrete the male sex steroid hormones. TNF-α and IL-1β also inhibit gonadotrophin-stimulated steroidogenesis of undifferentiated ovarian cells [88]. TNF-α has also been shown to decrease androgen receptor protein and mRNA levels in a prostate cancer line, and inhibits the ability of dihydrotestosterone to induce cell proliferation and activate the prostate-specific antigen gene promoter [89]. In contrast, IL-6 up-regulates androgen receptor expression and activates androgen receptor-mediated gene expression in this cell line [90, 91].
Oestrogen also functions as a specific inhibitor of inflammatory pathways. It is not known if oestrogen modulates the direct effects of proinflammatory cytokines on growth plate chondrocytes. However, it is likely that the mechanisms elucidated in other tissues may also be present in the epiphyseal growth plate. Raloxifene, a selective oestrogen receptor modulator, inhibits both IL-6 and TNF-α expression and activity in osteoblasts and osteoclasts in vitro [92]. Oestrogen inhibits IL-1β induction of IL-6 expression in both rodent and human bone marrow-derived cell lines [93]. This decrease in IL-6 expression occurs through oestrogen receptor α (ERα) inhibition of both NF-κB and NF-IL6 activation of the IL-6 promoter [94, 95]. The carboxyl-terminal activation function-2 (AF2) domain of ERα is necessary for this inhibition [96]. Similarly, ERα can inhibit TNF-α induced gene expression [97] in an AF2 domain-dependent manner [98]. ERα also inhibits IL-1β induction of gene expression in the mouse liver, through a coactivator-based mechanism [99].
IGF-1 signalling
The IGF-1 signalling pathway is unequivocally a major autocrine/paracrine regulator of bone growth [100]. Proinflammatory cytokines may modulate the IGF-1 signalling cascade at one or more junctures: IGF-1 receptor binding; insulin receptor substrate phosphorylation; the p44/42 mitogen-activated protein kinase (MAPK) signalling pathway; the phosphatidylinositol 3-kinase (PI-3K) signalling pathway; and Akt phosphorylation (Fig. 2). The effects of proinflammatory cytokines on the major IGF-1 signalling pathways in growth plate chondrocytes have yet to be described in the literature. However, the effects of cytokines on IGF-1 signalling have been investigated in a number of other cell types, which are reviewed in the following text.
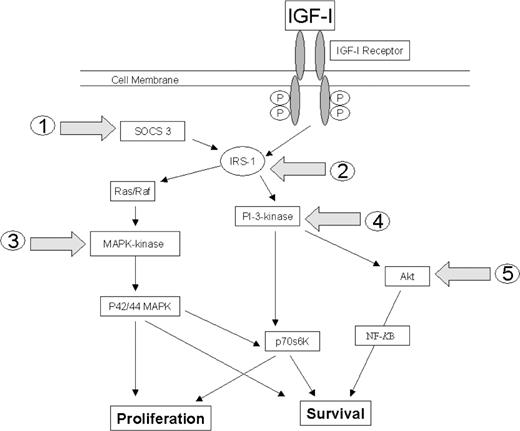
Potential junctures at which proinflammatory cytokines may interrupt IGF-1 signalling: (1) increased SOCS 3 expression; (2) insulin receptor substrate phosphorylation (IRS); (3) MAPK-kinase phosphorylation within the p44/42 mitogen-activated protein kinase (MAPK) signalling pathway; (4) the phosphatidylinositol 3-kinase (PI-3K) signalling pathway; and (5) Akt phosphorylation.
The cellular actions of IGF-1 are mediated by a receptor tyrosine kinase (IGF-1R), which is expressed in growth plate chondrocytes. However IL-1β and TNF-α do not down-regulate IGF-1 receptor expression [101] or affect IGF-1 receptor affinity [102] in articular cartilage. Furthermore, IL-1β and TNF-α do not impair the intrinsic tyrosine kinase activity of the IGF-1 receptor in either breast cancer cells [103] or myoblasts [104, 105]. Therefore, it is unlikely that proinflammatory cytokines alter the IGF-1 signalling cascade at the level of the IGF-1 receptor in growth plate chondrocytes.
Binding of IGF-1 to its receptor utilizes a family of soluble receptors, known as insulin receptor substrates (IRSs), to initiate a series of autophosphorylation events. The mammalian IRS family contains at least four members, but only IRS-1 is expressed in epiphyseal cartilage [106]. TNF-α and IL-1β have been shown to induce IGF-1 resistance by inhibiting IRS-1 phosphorylation in both myoblasts [104, 105] and breast cancer epithelial cells [103]. Proinflammatory cytokines may therefore also induce IGF-1 resistance by inhibiting IRS-1 phosphorylation in growth plate chondrocytes.
TNF-α and IL-1β receptor activation has been shown to elevate ceramide, a sphingosine-based lipid second messenger, through de novo pathways in breast carcinoma, fibrosarcoma and hepatic cells [107, 108] and sphingomyelinase pathways in leukaemic T-cell and pre-B cell lines and breast carcinoma, fibrosarcoma and thymoma cells [107, 109, 110]. Ceramide also inhibits IGF-1-induced tyrosine phosphorylation of IRS-1 in myoblast and hepatic cells [105, 111] and may also be a key intermediate by which proinflammatory cytokines impair IGF-1 action in growth plate chondrocytes.
Many proinflammatory cytokines, including TNF-α and IL-1β, stimulate the tissue-specific expression of suppressor of cytokine signalling proteins (SOCS) [112]. SOCS are a group of signalling proteins characterized by their ability to down-regulate cytokine signalling and are critical in modulating GH signalling [113, 114]. Cytokine binding to its receptor activates the JAK-STAT signalling pathway, leading to induction of SOCS mRNA and protein [115]. Studies in adipocytes have shown that TNF-α and IL-1B increase SOCS 3 expression, and this directly inhibits IRS-1 phosphorylation by IRS-1 protein degradation [112].
Autophosphorylation of IRS-1 results in the activation of two distinct signalling pathways, PI-3K and p42/p44 MAPK, leading to proliferative and anti-apoptotic effects. Therefore cytokine-induced inhibition of IRS-1 phosphorylation in growth plate chondrocytes would be likely to reduce the downstream activity of both pathways. However, proinflammatory cytokines may also act directly on the PI-3K and/or p44/p42 MAPK pathways of growth plate chondrocytes. The MAPK pathway is strongly dependent on tyrosine phosphorylation steps and involves p21ras, c-Raf-1 and MAPK-kinase (MKK), which is a direct upstream activator of p44/p42 MAPK. Phosphorylated MAPK translocates to the nucleus, where it participates in the phosphorylation of retinoblastoma pocket protein, resulting in the release of transcription factors and the activation of genes necessary for cell cycle progression and DNA replication [116]. TNF-α has been shown to inhibit MKK phosphorylation in neuronal cells [117]. Proinflammatory cytokines may therefore also act directly on the p44/p42 MAPK pathway of growth plate chondrocytes.
TNF-α has also been shown to prevent the intranuclear translocation of PI3K in an osteoblast cell line [118]. Activation of PI-3K activity results in a series of intracellular downstream events, generating phosphorylated phosphatidylinositol (PI) intermediates (such as PI 3,4,5-triphosphate, PIP3) in the cytosolic leaflet of the plasma membrane. PIP3 intermediates recruit other downstream signalling molecules, such as Akt, to the plasma membrane, and the subsequent phosphorylation of its several downstream effectors (e.g. NFκB, caspase-9) mediates the effects of Akt on cell growth, proliferation and protection from pro-apoptotic stimuli [119]. Proinflammatory cytokines may inhibit Akt phosphorylation in growth pate chondrocytes through either PI-3K-dependent and/or PI-3K-independent mechanisms. TNF-α has been shown to inhibit the phosphorylation and activation of Akt in neuronal cells [117]. Additionally, studies in erythroleukaemic, adrenal phaeochromocytoma and glioblastoma cell lines have suggested that ceramide induces growth arrest via dephosphorylation of Akt [120–122]. In neuronal cells, ceramide has been shown to inhibit Akt indirectly [123], leading to the proposal of multiple indirect mechanisms for ceramide regulation of Akt. Conflicting results have suggested PI-3K-dependent regulation of Akt by ceramide in fibroblasts [124] as well as PI-3K-independent regulation of Akt by ceramide in kidney cells, fibroblasts and adipocytes [125, 126]. Two molecular PI-3K-independent mechanisms by which ceramide inhibits Akt activation by insulin have been elucidated in pre-adipocytes [127]. Ceramide specifically blocks the translocation of Akt to the plasma membrane, while simultaneously promoting the dephosphorylation of Akt by protein phosphatase 2A.
Growth hormone (GH) signalling
Increased levels of inflammatory cytokines may inhibit the effects of GH on the growth plate. It has been suggested that cytokines may act on GH receptor signalling [128], although very little research has been undertaken in this area. Members of the SOCS family of proteins make important contributions to the negative regulation of the growth-promoting actions of GH. Mice lacking SOCS2 expression display gigantism accompanied by evidence of deregulated GH signalling [129]. Additionally, IL-6 has been proposed to inhibit liver GH signalling by inducing SOCS 3 [130], a phenomenon that may explain GH resistance in inflammatory disease.
Concluding remarks
The development of abnormal growth patterns in children with inflammatory diseases such as JIA may be modulated by proinflammatory cytokines through both systemic effects and effects acting locally at the level of the growth plate. An improved understanding of the crucial cellular events that may be affected in growth plate development will allow us to be in a stronger position to ameliorate disturbed growth in affected children.
We are grateful to Novo Nordisk UK Ltd. and the Biotechnology and Biological Sciences Research Council (BBSRC) for their generous support.
The authors have declared no conflicts of interest.
References
Grom AA, Murray KJ, Luyrink L et al. Patterns of expression of tumor necrosis factor alpha, tumor necrosis factor beta, and their receptors in synovia of patients with juvenile rheumatoid arthritis and juvenile spondylarthropathy.
Yetgin S, Ozen S, Saatci U et al. Evaluation of tumour necrosis factor alpha, interferon gamma and granulocyte-macrophage colony stimulating factor levels in juvenile chronic arthritis.
Rooney M, Varsani H, Martin K, Lombard PR, Dayer JM, Woo P. Tumour necrosis factor alpha and its soluble receptors in juvenile chronic arthritis.
Almawi WY, Beyhum HN, Rahme AA, Rieder MJ. Regulation of cytokine and cytokine receptor expression by glucocorticoids.
Corral LG, Muller GW, Moreira AL et al. Selection of novel analogs of thalidomide with enhanced tumor necrosis factor alpha inhibitory activity.
Kutukculer N, Caglayan S, Aydogdu F. Study of pro-inflammatory (TNF-alpha, IL-1 alpha, IL-6) and T-cell-derived (IL-2, IL-4) cytokines in plasma and synovial fluid of patients with juvenile chronic arthritis: correlations with clinical and laboratory parameters.
Wilkinson N, Jackson G, Gardner-Medwin J. Biologic therapies for juvenile arthritis.
Yilmaz M, Kendirli SG, Altintas D, Bingol G, Antmen B. Cytokine levels in serum of patients with juvenile rheumatoid arthritis.
Farquarson C. Bone growth. In: Scanes CG, ed.
Hunziker EB, Schnek RK. Physiological mechanisms adopted by chondrocytes in regulating longitudinal bone growth.
Breur GJ, Turgai J, Vanenkevort BA, Farnum CE, Wilsman NJ. Stereological and serial section analysis of chondrocytic enlargement in the proximal tibial growth-plate of the rat.
Buckwalter JA, Mower D, Ungar R, Schaeffer J, Ginsberg B. Morphometric analysis of chondrocyte hypertrophy.
Breur GJ, Vanenkevort BA, Farnum CE, Wilsman NJ. Linear relationship between the volume of hypertrophic chondrocytes and the rate of longitudinal bone-growth growth plates.
Bateman JF, Lamandé SR, Ramshaw JAM.
Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation.
Hunziker EB, Schenk RK, Cruzorive LM. Quantitation of chondrocyte performance in growth-plate cartilage during longitudinal bone-growth.
Nilsson O, Baron J. Fundamental limits on longitudinal bone growth: growth plate senescence and epiphyseal fusion.
Zezulak KM, Green H. The generation of insulin-like growth factor-1-sensitive cells by growth-hormone action.
Isaksson OGP, Jansson JO, Gause IAM. Growth-hormone stimulates longitudinal bone-growth directly.
Hunziker EB, Wagner J, Zapf J. Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo.
Reinecke M, Schmid AC, Heyberger-Meyer B, Hunziker EB, Zapf J. Effect of growth hormone and insulin-like growth factor I (IGF-1) on the expression of IGF-1 messenger ribonucleic acid and peptide in rat tibial growth plate and articular chondrocytes in vivo.
Smink JJ, Koster JG, Gresnigt MG, Rooman R, Koedam JA, Van Buul-Offers SC. IGF and IGF-binding protein expression in the growth plate of normal, dexamethasone-treated and human IGF-1I transgenic mice.
Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I.
Yakar S, Rosen CJ, Beamer WG et al. Circulating levels of IGF-1 directly regulate bone growth and density.
Lanske B, Karaplis AC, Lee K et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth.
Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation.
Vanderschueren D, Vandenput L, Boonen S, Lindberg MK, Bouillon R, Ohlsson C. Androgens and bone.
van der Eerden BCJ, Lowik CWGM, Wit JM, Karperien M. Expression of estrogen receptors and enzymes involved in sex steroid metabolism in the rat tibia during sexual maturation.
Blanchard O, Tsagris L, Rappaport R, Duval-Beaupere G, Corvol M. Age-dependent responsiveness of rabbit and human cartilage cells to sex steroids in vitro.
Krohn K, Haffner D, Hugel U, Himmele R, Klaus G, Mehls O, Schaefer F. 1,25(OH)2D3 and dihydrotestosterone interact to regulate proliferation and differentiation of epiphyseal chondrocytes.
Carani C, Qin K, Simoni M et al. Effect of testosterone and estradiol in a man with aromatase deficiency.
Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens.
Nasatzky E, Schwartz Z, Boyan BD, Soskolne WA, Ornoy A. Sex-dependent effects of 17-beta-estradiol on chondrocyte differentiation in culture.
Schwartz Z, Nasatzky E, Ornoy A, Brooks BP, Soskolne WA, Boyan BD. Gender-specific, maturation-dependent effects of testosterone on chondrocytes in culture.
Frank GR. Role of estrogen and androgen in pubertal skeletal physiology.
Mushtaq T, Ahmed SF. The impact of corticosteroids on growth and bone health.
Zak M, Müller J, Karup Pedersen F. Final height, armspan, subischial leg length and body proportions in juvenile chronic arthritis.
Simon D, Lucidarme N, Prieur A-M, Ruiz J-C, Czernichow P. Treatment of growth failure in juvenile chronic arthritis.
Ahmed SF, Tucker P, Mushtaq T, Wallace AM, Williams DM, Hughes IA. Short-term effects on linear growth and bone turnover in children randomized to receive prednisolone or dexamethasone.
Simon S, Whiffen J, Shapiro F. Leg-length discrepancies in monoarticular and pauciarticular juvenile rheumatoid arthritis.
Sherry DD, Stein LD, Reed AM, Schanberg LE, Kredich DW. Prevention of leg length discrepancy in young children with pauciarticular juvenile rheumatoid arthritis by treatment with intraarticular steroids.
Heuck C, Wolthers OD, Herlin T. Growth-suppressive effect of intra-articular glucocorticoids detected by knemometry.
Davies UM, Rooney M, Preece MA, Ansell BM, Woo P. Treatment of growth-retardation in juvenile chronic arthritis with recombinant human growth-hormone.
Touati G, Prieur AM, Ruiz JC, Noel M, Czernichow P. Beneficial effects of one-year growth hormone administration to children with juvenile chronic arthritis on chronic steroid therapy. I. Effects on growth velocity and body composition.
Bechtold S, Ripperger P, Muhlbayer D et al. GH therapy in juvenile chronic arthritis: results of a two-year controlled study on growth and bone.
Kuhns JG, Swaim LT. Disturbances in growth in chronic arthritis in children.
De Benedetti F, Alonzi T, Moretta A et al. Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I. A model for stunted growth in children with chronic inflammation.
De Benedetti F, Pignatti P, Vivarelli M et al. In vivo neutralization of human IL-6 (hIL-6) achieved by immunization of hIL-6-transgenic mice with a hIL-6 receptor antagonist.
De Benedetti F, Meazza C, Oliveri M et al. Effect of IL-6 on IGF binding protein-3: a study in IL-6 transgenic mice and in patients with systemic juvenile idiopathic arthritis.
Davies UM, Jones J, Reeve J et al. Juvenile rheumatoid arthritis—effects of disease activity and recombinant human growth hormone on insulin-like growth factor 1, insulin-like growth factor binding proteins 1 and 3, and osteocalcin.
Tsatsoulis A, Siamopoulou A, Petsoukis C, Challa A, Bairaktari E, Seferiadis K. Study of growth hormone secretion and action in growth-retarded children with juvenile chronic arthritis (JCA).
Li P, Schwarz EM. The TNF-alpha transgenic mouse model of inflammatory arthritis.
Siegel SA, Shealy DJ, Nakada MT et al. The mouse-human chimeric monoclonal-antibody ca2 neutralizes tnf in-vitro and protects transgenic mice from cachexia and tnf lethality in-vivo.
Fan J, Char D, Bagby GJ, Gelato MC, Lang CH. Regulation of insulin-like growth factor-1 (IGF-1) and IGF-binding proteins by tumour necrosis factor.
Barreca A, Ketelslegers JM, Arvigo M, Minuto F, Thissen JP. Decreased acid-labile subunit (ALS) levels by endotoxin in vivo and by interleukin-1 beta in vitro.
Delhanty PJD. Interleukin-1 beta suppresses growth hormone-induced acid-labile subunit mRNA levels and secretion in primary hepatocytes.
Jones JI, Dercole AJ, Camachohubner C, Clemmons DR. Phosphorylation of insulin-like growth-factor (IGF)-binding protein-1 in cell-culture and in vivo—effects on affinity for IGF-1.
Lang CH, Fan J, Cooney R, Vary TC. IL-1 receptor antagonist attenuates sepsis-induced alterations in the IGF system and protein synthesis.
Samstein B, Hoimes ML, Fan J, Frost RA, Gelato MC, Lang CH. IL-6 stimulation of insulin-like growth factor binding protein (IGFBP)-1 production.
Lang CH, Nystrom GJ, Frost RA. Regulation of IGF binding protein-1 in Hep G2 cells by cytokines and reactive oxygen species.
Frost RA, Nystrom GJ, Lang CH. Stimulation of insulin-like growth factor finding protein-1 synthesis by interleukin-1 beta: requirement of the mitogen-activated protein kinase pathway.
de Hooge ASK, van de Loo FAJ, Bennink MB et al. Growth plate damage, a feature of juvenile idiopathic arthritis, can be induced by adenoviral gene transfer of oncostatin M—a comparative study in gene-deficient mice.
Goldring MB, Birkhead J, Sandell LJ, Kimura T, Krane SM. Interleukin-1 suppresses expression of cartilage-specific type-II and type-IX collagens and increases type-I and type-III collagens in human chondrocytes.
Lefebvre V, Peetersjoris C, Vaes G. Modulation by interleukin-1 and tumor necrosis factor-alpha of production of collagenase, tissue inhibitor of metalloproteinases and collagen types in differentiated and dedifferentiated articular chondrocytes.
Lum ZP, Hakala BE, Mort JS, Recklies AD. Modulation of the catabolic effects of interleukin-1 beta on human articular chondrocytes by transforming growth factor-beta.
Bolton MC, Dudhia J, Bayliss MT. Quantification of aggrecan and link-protein mRNA in human articular cartilage of different ages by competitive reverse transcriptase-PCR.
Kato Y, Nakashima K, Iwamoto M et al. Effects of interleukin-1 on syntheses of alkaline-phosphatase, type-x collagen, and 1,25-dihydroxyvitamin-d(3) receptor, and matrix calcification in rabbit chondrocyte cultures.
Soder O, Madsen K. Stimulation of chondrocyte DNA-synthesis by interleukin-1.
Ikebe T, Hirata M, Koga T. Effects of human recombinant tumor necrosis factor-alpha and interleukin-1 on the synthesis of glycosaminoglycan and DNA in cultured rat costal chondrocytes.
Enomoto M, Pan HO, Kinoshita A, Yutani Y, Suzuki F, Takigawa M. Effects of tumor necrosis factor-alpha on proliferation and expression of differentiated phenotypes in rabbit costal chondrocytes in culture.
Aizawa T, Kon T, Einhorn TA, Gerstenfeld LC. Induction of apoptosis in chondrocytes by tumor necrosis factor-alpha.
Horiguchi M, Akiyama H, Ito H, Shigeno C, Nakamura T. Tumour necrosis factor-alpha up-regulates the expression of BMP-4 mRNA but inhibits chondrogenesis in mouse clonal chondrogenic EC cells, ATDC5.
van Bezooijen RL, van der Wee-Pals L, Papapoulos SE, Lowik CWGM. Interleukin 17 synergises with tumour necrosis factor alpha to induce cartilage destruction in vitro.
Martensson K, Chrysis D, Savendahl L. Interleukin-1 beta and TNF-alpha act in synergy to inhibit longitudinal growth in fetal rat metatarsal bones.
Lazarus DD, Moldawer LL, Lowry SF. Insulin-like growth factor-1 activity is inhibited by interleukin-1 alpha, tumor necrosis factor-alpha and interleukin-6. Lymphokine
Nietfeld JJ, Wilbrink B, Helle M et al. Interleukin-1-induced interleukin-6 is required for the inhibition of proteoglycan synthesis by interleukin-1 in human articular cartilage.
Malfait A-M, Verbruggen G, Veys EM, Lambert J, De Ridder L, Cornelissen M. Comparative and combined effects of interleukin 6, interleukin 1 beta, and tumor necrosis factor alpha on proteoglycan metabolism of human articular chondrocytes cultured in agarose.
Guerne PA, Desgeorges A, Jaspar JM et al. Effects of IL-6 and its soluble receptor on proteoglycan synthesis and NO release by human articular chondrocytes: comparison with IL-1. Modulation by dexamethasone.
Legendre F, Dudhia J, Pujol JP, Bogdanowicz P. JAK/STAT but not ERK1/ERK2 pathway mediates interleukin (IL)-6/soluble IL-6R down-regulation of Type II collagen, aggrecan core, and link protein transcription in articular chondrocytes. Association with a down-regulation of SOX9 expression.
Horan J, Dean DD, Kieswetter K, Schwartz Z, Boyan BD. Evidence that interleukin-1, but not interleukin-6, affects costochondral chondrocyte proliferation, differentiation, and matrix synthesis through an autocrine pathway.
Bi WM, Deng JM, Zhang ZP, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation.
Murakami S, Lefebvre V, de Crombrugghe B. Potent inhibition of the master chondrogenic factor Sox9 gene by interleukin-1 and tumor necrosis factor-alpha.
Terranova PF, Rice VM. Review: cytokine involvement in ovarian processes.
Mizokami A, Gotoh A, Yamada H, Keller ET, Matsumoto T. Tumor necrosis factor-alpha represses androgen sensitivity in the LNCaP prostate cancer cell line.
Lin DL, Whitney MC, Yao Z, Keller ET. Interleukin-6 induces androgen responsiveness in prostate cancer cells through up-regulation of androgen receptor expression.
Chen T, Wang LH, Farrar WL. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells.
Taranta A, Brama M, Teti A et al. The selective estrogen receptor modulator raloxifene regulates osteoclast and osteoblast activity in vitro.
Jilka RL, Hangoc G, Girasole G et al. Increased osteoclast development after estrogen loss: mediation by interleukin-6.
Pottratz ST, Bellido T, Mocharla H, Crabb D, Manolagas SC. 17 beta-estradiol inhibits expression of human interleukin-6 promoter-reporter constructs by a receptor-dependent mechanism.
Stein B, Yang MX. Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta.
Kurebayashi S, Miyashita Y, Hirose T, Kasayama S, Akira S, Kishimoto T. Characterization of mechanisms of interleukin-6 gene repression by estrogen receptor.
Cerillo G, Rees A, Manchanda N et al. The oestrogen receptor regulates NFkappaB and AP-1 activity in a cell-specific manner.
An J, Ribeiro RC, Webb P et al. Estradiol repression of tumor necrosis factor-alpha transcription requires estrogen receptor activation function-2 and is enhanced by coactivators.
Evans MJ, Lai K, Shaw LJ, Harnish DC, Chadwick CC. Estrogen receptor alpha inhibits IL-1beta induction of gene expression in the mouse liver.
Loveridge N, Farquharson C, Scheven BAA. Endogenous mediators of growth.
Blount S, Crawford A. Regulation of igf-1 receptors on rabbit articular chondrocytes by inflammatory mediators.
Matsumoto T, Tsukazaki T, Enomoto H, Iwasaki I, Yamashita S. Effects of interleukin-1-beta on insulin-like growth factor-I autocrine/paracrine axis in cultured rat articular chondrocytes.
Shen WH, Zhou JH, Broussard SR, Freund GG, Dantzer R, Kelley KW. Proinflammatory cytokines block growth of breast cancer cells by impairing signals from a growth factor receptor.
Broussard SR, McCusker RH, Novakofski JE et al. IL-1 beta impairs insulin-like growth factor I-induced differentiation and downstream activation signals of the insulin-like growth factor I receptor in myoblasts.
Strle K, Broussard SR, McCusker RH et al. Proinflammatory cytokine impairment of insulin-like growth factor I-induced protein synthesis in skeletal muscle myoblasts requires ceramide.
Hoshi K, Ogata N, Shimoaka T et al. Deficiency of insulin receptor substrate-1 impairs skeletal growth through early closure of epiphyseal cartilage.
Dbaibo GS, El-Assaad W, Krikorian A et al. Ceramide generation by two distinct pathways in tumor necrosis factor alpha-induced cell death.
Memon RA, Holleran WM, Moser AH et al. Endotoxin and cytokines increase hepatic sphingolipid biosynthesis and produce lipoproteins enriched in ceramides and sphingomyelin.
Wiegmann K, Schutze S, Machleidt T, Witte D, Kronke M. Functional dichotomy of neutral and acidic sphingomyelinases in tumor-necrosis-factor signaling.
Mathias S, Younes A, Kan CC, Orlow I, Joseph C, Kolesnick RN. Activation of the sphingomyelin signaling pathway in intact el4 cells and in a cell-free system by IL-1-beta.
Kanety H, Hemi P, Papa MZ, Karasik A. Sphingomyelinase and ceramide suppress insulin-induced tyrosine phosphorylation of the insulin receptor substrate-1.
Shi H, Tzameli I, Bjorbaek C, Flier JS. Suppressor of cytokine signaling 3 is a physiological regulator of adipocyte insulin signalling.
Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signalling.
Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signalling.
Pestell RG, Albanese C, Reutens AT, Segall JE, Lee RJ, Arnold A. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation.
Kenchappa P, Yadav A, Singh G, Nandana S, Banerjee K. Rescue of TNFalpha-inhibited neuronal cells by IGF-1 involves Akt and c-Jun N-terminal kinases.
Martelli AM, Borgatti P, Bortul R et al. Phosphatidylinositol 3-kinase translocates to the nucleus of osteoblast-like MC3T3-E1 cells in response to insulin-like growth factor I and platelet-derived growth factor but not to the proapoptotic cytokine tumor necrosis factor alpha.
Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts.
Schubert KM, Scheid MP, Duronio V. Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473.
Salinas M, Lopez-Valdaliso R, Martin D, Alvarez A, Cuadrado A. Inhibition of PKB/Akt1 by C2-ceramide involves activation of ceramide-activated protein phosphatase in PC12 cells.
Zinda MJ, Vlahos CJ, Lai MT. Ceramide induces the dephosphorylation and inhibition of constitutively activated Akt in PTEN negative U87MG cells.
Zhou HL, Summers SK, Birnbaum MJ, Pittman RN. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis.
Zundel W, Giaccia A. Inhibition of the anti-apoptotic PI(3)K/Akt/Bad pathway by stress.
Meier R, Thelen M, Brian A, Hemmings BA. Inactivation and dephosphorylation of protein kinase Bα (PKBα) promoted by hyperosmotic stress.
Chen D, Fucini RV, Olson AL, Hemmings BA, Pessin JE. Osmotic shock inhibits insulin signaling by maintaining akt protein kinase B in an inactive dephosphorylated state.
Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B.
von Laue S, Ross RJM. Inflammatory cytokines and acquired growth hormone resistance.
Horvat S, Medrano JF. Lack of Socs2 expression causes the high-growth phenotype in mice.
Author notes
1Bone Biology Group, Division of Gene Function and Development, Roslin Institute, Edinburgh and 2Bone and Endocrine Research Group, Royal Hospital for Sick Children, Glasgow, UK.

{kind=link}
{kind=link}
Comments