





Sir,
Hyponatraemia following traumatic brain injury (TBI) is a common complication, occurring in 13% of cases.1 The commonest cause of hyponatraemia is the syndrome of inappropriate antidiuretic hormone secretion (SIADH), which is responsible for over 90% of cases,1 whereas cerebral salt wasting, medications and injudicious use of intravenous fluids may also cause hyponatraemia following TBI. As glucocorticoid deficiency can present with hyponatraemia similar to that found in SIADH, it is essential to exclude adrenal insufficiency before making a diagnosis of SIADH.2 This may be of particular importance in the case of TBI, as recent data have indicated a high frequency of undiagnosed hypopituitarism among long-term survivors.3 In addition, 16% of acute head injury patients show biochemical evidence of adrenocorticotrophin (ACTH) deficiency.4 Acute hypopituitarism with ACTH deficiency may therefore be a potentially important, cause of hyponatraemia in patients with acute TBI, which is misdiagnosed as SIADH.
To illustrate this potential pitfall in diagnosing post-traumatic hyponatraemia, we report our recent experience of three cases where acute TBI was complicated by hyponatraemia of between 125–130 mmol/l (normal 135–145 mmol/l) with all the biochemical features of SIADH (clinical euvolaemia, inappropriately concentrated urine and a natriuresis).2 All three patients had significant head trauma. Patient A had a penetrating skull injury after falling from a ladder (Figure 1), patient B had diffuse axonal injury after a road traffic accident and patient C had intracerebral haematoma following a fall. At presentation, patients A and B were also hypotensive (blood pressures of 80/30 and 70/40 mmHg, respectively) and hypoglycaemic (plasma glucoses of 0.9 and 2.5 mmol/l, respectively) and required vasopressor support and continuous intravenous dextrose infusion. Patient C had normal blood pressure and plasma glucose. The diagnosis of post-traumatic hypopituitarism with ACTH deficiency was suspected in cases A and B because of the combination of hyponatraemia, hypoglycaemia and hypotension and in patient C because plasma sodium did not correct with fluid restriction. All three patients were found to have low basal serum cortisol concentrations: patient A, 110 nmol/l; patient B, 33 nmol/l; and patient C, 97 nmol/l (normal >200 nmol/l). All three patients had undetectable plasma ACTH levels. These findings are very suggestive of acute secondary adrenal (ACTH) deficiency, as the acute illness, particularly when associated with hypotension, activates the hypothalamic-pituitary-adrenal axis, resulting in high circulating cortisol concentrations.5 Patients B and C also showed a blunted cortisol response following glucagon stimulation. All three patients showed remarkable and rapid response to the administration of intravenous hydrocortisone, with normalization of plasma sodium and, blood pressure and plasma glucose (patients A and B). There was evidence of additional pituitary hormone abnormalities in all three cases: patients A and B had low growth hormone, insulin-like growth factor-1 and serum testosterone levels, and patient C had low serum testosterone concentration. Thyroid function and prolactin were normal in all three patients.
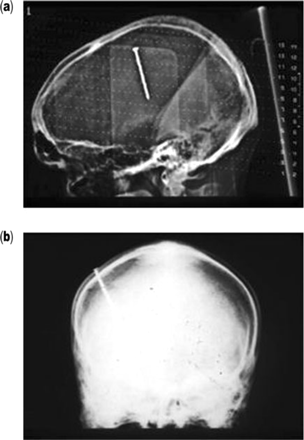
Tomographic (a) and frontal skull X-ray (b) views of patient A, showing a nail embedded through the skull into the brain. Note the translucency surrounding the nail in image a, which represents abscess formation.
These three case reports illustrate that hyponatraemia following traumatic brain injury may be a marker of acute hypopituitarism with secondary adrenal failure. Therefore, assessment of glucocorticoid reserves should be performed in all head injury patients who show an SIADH-like clinical picture. Moreover, as traumatic brain injury is increasingly recognised to be a high risk condition for the development of hypopituitarism,3,4 a persuasive case can be made for the assessment of pituitary function to become part of standard post-traumatic clinical care, in order to detect a potentially common but treatable cause of morbidity and even mortality following TBI.
References
Agha A, Thornton E, O’Kelly P, Tormey W, Phillips J, Thompson CJ. Posterior pituitary dysfunction following traumatic brain injury.
Agha A, Rogers B, Sherlock M, O’ Kelly P, Tormey W, Phillips J, Thompson CJ. Anterior Pituitary dysfunction in survivors of traumatic brain injury.
Agha A, Roger B, Mylotte D, Taleb F, Tormey W, Phillips J, Thompson CJ. Neuroendocrine dysfunction in the acute phase of traumatic brain injury.

{kind=link}