



Abstract
Haloalkane dehalogenases catalyse environmentally important dehalogenation reactions. These microbial enzymes represent objects of interest for protein engineering studies, attempting to improve their catalytic efficiency or broaden their substrate specificity towards environmental pollutants. This paper presents the results of a comparative study of haloalkane dehalogenases originating from different organisms. Protein sequences and the models of tertiary structures of haloalkane dehalogenases were compared to investigate the protein fold, reaction mechanism and substrate specificity of these enzymes. Haloalkane dehalogenases contain the structural motifs of α/β-hydrolases and epoxidases within their sequences. They contain a catalytic triad with two different topological arrangements. The presence of a structurally conserved oxyanion hole suggests the two-step reaction mechanism previously described for haloalkane dehalogenase from Xanthobacter autotrophicus GJ10. The differences in substrate specificity of haloalkane dehalogenases originating from different species might be related to the size and geometry of an active site and its entrance and the efficiency of the transition state and halide ion stabilization by active site residues. Structurally conserved motifs identified within the sequences can be used for the design of specific primers for the experimental screening of haloalkane dehalogenases. Those amino acids which were predicted to be functionally important represent possible targets for future site-directed mutagenesis experiments.
Introduction
The activities and substrate specificities of enzymes involved in the catalysis of biodegradation reactions can determine the biodegradability of organic substances in the environment. Recalcitrance to biodegradation can be caused by biochemical blocks in catabolic pathways (Timmis et al., 1994; Janssen et al., 1999), when degrading organisms miss one or more essential enzymes for the consecutive conversion of the parent compound to metabolic products. Such cases are expected to occur mainly in the catabolic pathways involved in the degradation of `true' xenobiotic compounds (Providenti et al., 1993; Singleton, 1994) which were unknown to nature before humans started their production. It is possible that microorganisms did not have a sufficient amount of time to evolve the enzymes with specificities and activities required for the catalysis of these synthetic compounds and their metabolic intermediates.
The study of the substrate specificities of degrading enzymes might bring new information about the evolutionary adaptation. Comparative methods are often used to infer evolutionary changes that occurred in the past (Lenski et al., 1997). Understanding the molecular mechanisms of substrate specificities is also important for the improvement of the current enzymes by protein engineering (Janssen and Schanstra, 1994). Recent advances in the structural analysis of biomolecules, both experimental and theoretical, present new possibilities for understanding enzyme specificities at a molecular level.
Haloalkane dehalogenases are bacterial enzymes cleaving the carbon–halogen bond of halogenated aliphatic compounds by a hydrolytic mechanism. The water molecule serves as the only co-substrate during the catalysis and there is no evidence indicating the involvement of co-factors or metal ions in the catalytic mechanism (Janssen et al., 1994). Haloalkane dehalogenases have been isolated from a number of bacterial strains (for reviews see Hardman, 1991; Fetzner and Lingens, 1994). The classification of the various dehalogenases according to their substrate specificities, reaction kinetics, molecular mechanisms and DNA or amino acid sequence information was proposed by Slater et al. (1995). The quantitative classification of haloalkane dehalogenases using statistical analysis of activity data (Damborský et al., 1997a; Nagata et al., 1997) indicated the presence of at least three unique specificity classes within this family of enzymes: (i) haloalkane dehalogenase of Xanthobacter autotrophicus GJ10 (Janssen et al., 1988), Xanthobacter autotrophicus GJ11 (Van den Wijngaard et al., 1992), Ancylobacter aquaticus AD20 (Van den Wijngaard et al., 1992), Ancylobacter aquaticus AD25 (Van den Wijngaard et al., 1992) and Pseudomonas sp. E4M (Vienravi, 1993); (ii) haloalkane dehalogenase of Rhodococcus sp. HA1 (Scholtz et al., 1987), Rhodococcus sp. m15-3 (Yokota et al., 1987), Acinetobacter sp. GJ70 (Janssen et al., 1988), Rhodococcus erythropolis Y2 (Sallis et al., 1990), Rhodococcus sp. CP9 (Nyandoroh, 1996), Rhodococcus rhodochrous NCIMB 13064 (Kulakova et al., 1997), Pseudomonas pavonaceae 170 (Poelarends et al., 1998) and Mycobacterium sp. GP1 (Poelarends et al., 1999); and (iii) haloalkane dehalogenase of Sphingomonas paucimobilis UT26 (Nagata et al., 1997).
This paper describes a comparison of haloalkane dehalogenases from three different specificity classes: the dehalogenase of Xanthobacter autotrophicus GJ10, the dehalogenase of Rhodococcus rhodochrous NCIMB 13064 and the dehalogenase of Sphingomonas paucimobilis UT26, at the level of primary sequences and tertiary structures. Computer modelling was used for the construction of three-dimensional models of the proteins for which only the primary sequence is known at the moment, and 1D and 3D comparisons of the proteins from different specificity classes were used to gain new information about the molecular mechanisms of substrate specificity and to propose functionally important amino acids as potential targets for site-directed mutagenesis experiments.
Methods
Primary sequences
Protein sequences of the dehalogenases were obtained from genetic databases using the following Genbank-EMBL accession codes: D14594, the haloalkane dehalogenase from Sp.paucimobilis UT2 (LinB) (Nagata et al., 1993); Z77724, Z78020, Z77163, the putative haloalkane dehalogenases from Mycobacterium tuberculosis H37Rv (Rv2579, Rv1833c, Rv2296) (Philipp et al., 1996); AJ243259, the putative haloalkane dehalogenase from Mycobacterium bovis MU11 (Iso-Rv2579) (Jesenská,A., Rychlík,I., Pavlik,I., Damborský,J., manuscript in preparation); L49435, the haloalkane dehalogenase from R.rhodochrous NCIMB 13064 (DhaA) (Kulakova et al., 1997); AJ012627, the haloalkane dehalogenase from Mycobacterium sp. GP1 (DhaAf) (Poelarends et al., 1999); M26950, the haloalkane dehalogenase from X.autotrophicus GJ10 (DhlA) (Janssen et al., 1988) and D90422, the haloacetate dehalogenase from Moraxella sp. B (DehH1) (Kawasaki et al., 1992). The haloalkane dehalogenase from R.rhodochrous NCIMB 1304 has an identical sequence with the haloalkane dehalogenase from P.pavonaceae 170 (Poelarends et al., 1998) and R.erythropolis Y2 (Y.Nagata, personal communication).
Solvent accessibility and secondary elements
Solvent accessibility and secondary structure were predicted using the modelling servers PredAcc 1.0 (Mucchielli et al., 1999) and JPred (Cuff et al., 1998), respectively. PredAcc employs a logistic function to predict the solvent accessibility of the amino acid residues of a protein from its sequence. An accessibility threshold of 25% and a gamma risk value of 10% were used in the calculations. Jpred is an Internet web server that takes either a protein sequence or a multiple alignment of protein sequences and predicts secondary structure. It works by combining a number of high-quality prediction methods to form a consensus. Jpred employs DSC (King and Sternberg, 1996), PHD (Rost and Sander, 1993), PREDATOR (Frishman and Argos, 1996) and NNSSP (Salamov and Solovyev, 1995) for consensus prediction.
Multiple alignment
The initial alignment of protein sequences was conducted using multiple alignment algorithms CLUSTALW 1.7 (Thompson et al., 1994) and MSA 2.1 (Lipman et al., 1989). The initial alignment was further modified manually in the program Cameleon 3.14a (Oxford Molecular, UK). Manual re-alignment was done in an iterative way: a 3D model was constructed from each new alignment, incorrectly modelled regions were identified using validation methods described below and re-aligned (alignment → 3D models → evaluation of models → new alignment). This procedure was repeated until no further improvements in the quality of the 3D models could be obtained.
Structurally conserved motifs (fingerprints)
Conserved motifs within the sequences of haloalkane dehalogenase were identified using an Internet search of the Protein Motif Fingerprint Database (PRINTS) (Attwood et al., 1998). This database is a compendium of protein fingerprints. A fingerprint is a group of conserved motifs used to characterize a protein family.
Homology models
The models of haloalkane dehalogenases DhaA and LinB were constructed using the homology modelling approach as implemented in the program Modeller 3.0 (Sali and Blundell, 1993). This program uses the method of the satisfaction of spatial restraints for model building. The crystal structure of haloalkane dehalogenase from X.autotrophicus GJ10 (Verschueren et al., 1993a) obtained from the Brookhaven Protein Database served as a template (accession code 1ede). The sequences of DhaA and LinB were used as target sequences. The alignment of the template and the target sequences was derived from the multiple sequence alignment of eight dehalogenases. The constructed models were refined by the molecular dynamic `refine3' option of the program Modeller. The protein structures were visualized and manipulated in the modelling package InsightII 95.0 (MSI/Biosym, USA).
Validation of the models
The correctness of the models was tested by analysing (1) their stereochemical accuracy, (2) their packing quality and (3) their folding reliability. The stereochemical quality was checked by PROCHECK 3.0 (Laskowski et al., 1993). The packing quality was accessed by `bump checks' and by visual inspection of the distribution of the hydrophobic and hydrophilic residues within the protein. The folding reliability was accessed by calculation of the 3D–1D profile (Luthy et al., 1992) and the total energy of the amino acid profile using PROSAII 3.0 software (Sippl, 1993).
Size of active site cavities
The volumes of the active site cavities of the crystal structure and homology models were calculated by the program CAST 1.0 (Liang et al., 1998a). CAST uses the weighted Delaunay triangulation and the alpha complex for shape measurements (Liang et al., 1998b). A probe sphere radius of 1.4 Å and the parameters based on the molecular surface (Connolly, 1983) were employed in the calculations.
Results
Multiple alignment of haloalkane dehalogenase sequences
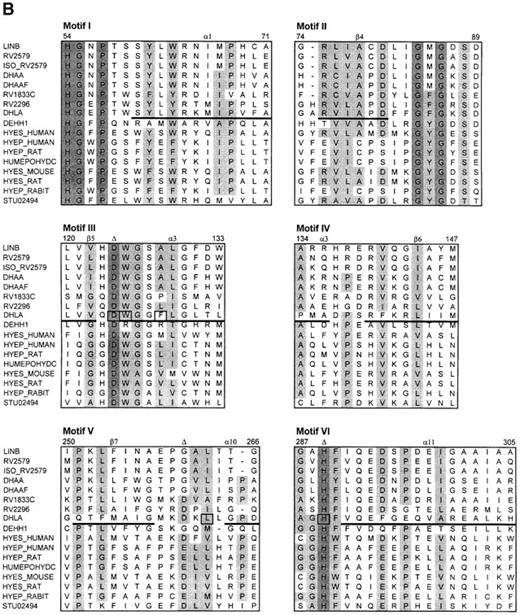
The starting alignments of four haloalkane dehalogenases (LinB, DhaA, DhaAf and DhlA), four putative haloalkane dehalogenases (Rv2579, Iso-Rv2579, Rv1833c, Rv2296) and haloalocetate dehalogenase (DehH1) were derived automatically using the CLUSTALW and MSA algorithms. Multiple alignment was preferred over the pairwise alignment to minimize misalignments in the regions of low sequential identity. The initial alignments were combined and further improved manually. An effort was made to avoid the gaps in the regions of secondary elements and to align the amino acid residues according to their solvent accessibility. The final alignment is shown in Figure 1. The highly conserved regions correspond mainly to the strands in DhlA (β2–β7). These β-strands form the core of this protein and they are apparently also well preserved in all proteins included in the alignment. The less conserved region corresponds to the cap domain of DhlA (helices α4–α8). It has been proposed that cap domain participates in the adaptation of the enzyme DhlA to new substrates (Pries et al., 1994a). The similarity among the sequences in this region drops below the level which makes it possible to derive the optimal alignment with high certainty. Consequently, the modelling of these regions by homology will also be uncertain. The least conserved region is in the surrounding vicinity of helix α4. A number of gaps had to be inserted in this region in all sequences. The prediction of secondary elements indicates the shift in the position of helices α4 and α5 among different dehalogenases. Direct repeats are present in this region of the dhlA gene. The mutant protein carrying a 10 amino acid deletion (Phe164–Ala174) in this region has been obtained by Pries et al. (1994a) during an in vivo adaptation experiment.
Fingerprints within haloalkane dehalogenase sequences
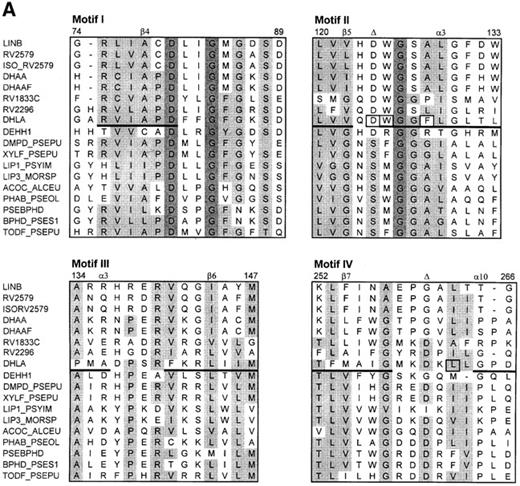
The search for the structurally conserved regions (fingerprints) in the Protein Motif Fingerprint Database (Attwood et al., 1998) using the dehalogenase sequences as templates revealed two sets of conserved motifs. The first fingerprint comprises four motifs (Figure 2A) and provides the signature for members of the α/β-hydrolase family (Ollis et al., 1992). This fingerprint is typical for hydrolases, dehalogenases, lipases and the depolymerase and also partially matches the sequence of magnesium chelatase, chloroperoxidase, atropine acylhydrolase and haloacetate dehalogenase. Motif II contains a catalytic nucleophile (Asp124 in DhlA), while catalytic acid (Asp260 in DhlA) is found in motif IV. The second fingerprint contains six motifs which resemble the epoxide hydrolase signature (Figure 2B). Motif I contains conserved residues that are involved in forming an oxyanion hole (His54, Gly55 and Pro56). Motifs II–V correspond to the motifs I–IV of the α/β-hydrolase fingerprint, while motif VI contains a catalytic base (His289 in DhlA). The regions of the dehalogenase sequences which correspond to the α/β-hydrolase and epoxide hydrolase fingerprints presumably play an important structural role. This is supported by the fact that no gaps are present within these regions in the alignment of haloalkane dehalogenases (Figure 1). On the other hand, some substitutions are also apparent in the motif regions (e.g. positions 134, 141, 251–256 and 294, numbered according to DhlA) indicating the amino acid residues which have a special structural role or the residues which determine the substrate specificity. The strand β7 of DhlA probably has different evolutionary origin compared to all other haloalkane dehalogenases.
Homology models of haloalkane dehalogenases
Three-dimensional models of LinB and DhaA were constructed by homology modelling using the alignment presented in Figure 1. The crystallographic structure of DhlA served as a template. Ten models were generated and evaluated for each protein. The best structure for each protein was selected and used for further analysis. The distribution of ϕ and Ψ torsion angles in the template structure and homology models is shown in Figure 3. Both modelled structures show favourable stereochemistry. The calculation of 3D–1D and PROSAII profiles (not shown) revealed a somewhat better quality for the DhaA structure, which may be the result of the slightly higher sequential identity of DhlA and DhaA (30%) over the identity of DhlA and LinB (28%). The disproportional distribution of conserved residues along the sequences results in the different reliability of the modelled structures. The highest reliability of the LinB and DhaA structures is achieved in the conserved regions indicated by the boxes in Figure 1. The position of these conserved regions within the tertiary structures of the modelled proteins is shown in Figure 4. Differences in the reliability of the predictions was taken into account in the structural comparisons of haloalkane dehalogenases presented below.
The catalytic triad of haloalkane dehalogenases
The catalytic residues of haloalkane dehalogenase DhlA are arranged in a so-called catalytic triad. This triad consists of nucleophile, base and acid. The nucleophile (Asp124) initiates the dehalogenation reaction by nucleophilic attack on the halogen-bound carbon atom of the substrate leading to the formation of a covalent alkyl-enzyme intermediate and a halide ion. The base (His289) activates the water molecule which subsequently hydrolyses the alkyl-enzyme intermediate. The catalytic acid (Asp260) assists the histidine in its function by stabilizing the charge developed on the imidazole ring and by keeping it in its proper orientation. Two out of the three catalytic residues of DhlA (nucleophile and base) are conserved in all dehalogenase sequences (Figure 1). The third member of the triad, the catalytic acid, is conserved only in the Rv1833c and Rv2296 sequences. The catalytic acid in LinB, Rv2579, Iso-Rv2579, DhaA, DhaAf and DehH1 presumably migrated to the position equivalent to Asn148 in DhlA. The catalytic acid is located after the strand β6 instead of strand β7 and corresponds to the location of the catalytic acid in human pancreatic lipase (Schrag et al., 1992). Experimental evidence for the feasibility of the migration of the catalytic acid within the protein structure has been provided by Krooshof et al. (1997), who constructed the D260N + N148D double mutant of DhlA. The position of the catalytic acid after the strand β6 in LinB dehalogenase has been experimentally confirmed by Hynková et al. (1999). The arrangement of the catalytic triads in the proteins discussed above is shown in Figure 5.
The oxyanion hole of haloalkane dehalogenases
The haloalkane dehalogenase DhlA, and also all members of the α/β-hydrolase family, have a small pocket near the amide nitrogen of the residue following the nucleophile. This pocket, called an oxyanion hole, stabilizes an alkyl-enzyme tetrahedral intermediate. In the DhlA, the partially positively charged NH1 of Glu56 and NH of Trp125 stabilize the partial negative charge on the O2 of Asp124 which is developed during the dehalogenation reaction (Verschueren et al., 1993b; Kutýet al., 1998). The regions around these stabilizing residues are highly conserved in all the proteins studied (Figure 1). The homology models of the LinB and DhlA proteins contain an oxyanion hole pocket and the nucleophile-stabilizing residues are in the correct orientation to facilitate the stabilization of an akyl-enzyme intermediate (Figure 6). Rink et al. (1997) recently proposed that Phe85 in DhlA stabilizes the oxyanion hole by interacting with His54 which stands perpendicular to the aromatic ring. This stabilization does not occur in the homology models where Met occurs in the position equivalent to the Phe85 of DhlA.
Transition state and halide stabilizing residues of haloalkane dehalogenases
The stabilization of the transition state (TS) occurring during the formation of the covalent intermediate and also the stabilization of the halide product by the active site residues of DhlA are among the most important catalytic phenomena of the haloalkane dehalogenase. Crystallographic and fluorescence studies suggested that two tryptophan residues, Trp125 and Trp175, could be involved in this stabilization (Verschueren et al., 1993b,c). Site-directed mutagenesis experiments confirmed the essential role of tryptophan residues for the enzyme activity (Kennes et al., 1995). Quantum-chemistry calculations showed the side-chain movements of tryptophan residues and the redistribution of electrons on the stabilizing atoms upon the formation of the transition state and the halide release (Damborský et al., 1997b, 1998). A third residue (Phe172) which stabilizes the TS and halide product in a similar way to the two tryptophans has been identified. Homology models suggest that all three stabilizing residues of DhlA could possibly be present in the active sites of LinB and DhaA. Trp125 is absolutely conserved among different haloalkane dehalogenases while Trp175 is systematically substituted by Phe. The residues Phe172 and Trp175 lie outside the regions conserved among different dehalogenases. Putative halide-stabilizing residues in LinB and DhaA predicted from the homology models therefore need experimental validation.
Discussion
A number of haloalkane dehalogenases from different bacteria have been purified and biochemically characterized to date (Scholtz et al., 1987; Yokota et al., 1987; Janssen et al., 1988; Sallis et al., 1990; Van den Wijngaard et al., 1992; Vienravi, 1993; Nyandoroh, 1996; Kulakova et al., 1997; Nagata et al., 1997; Poelarends et al., 1998, 1999). The first experimental structure of haloalkane dehalogenase (from X.autotrophicus GJ10) was determined by protein crystallography in 1991 (Franken et al., 1991). Three different genes encoding haloalkane dehalogenases have been cloned and sequenced (Janssen et al., 1988; Nagata et al., 1993; Kulakova et al., 1997). The N-terminal protein sequences of haloalkane dehalogenases and putative haloalkane dehalogenases (from M.tuberculosis and M.bovis) which are currently available are listed in Table I. Specificity classes assigned to the enzymes using principal component analysis of the activity data (Damborský et al., 1997a) are provided in the last column of this table. There is good correspondence between the N-terminal sequence composition and the substrate specificities. To understand further the specificity of different haloalkane dehalogenases, we have compared the available haloalkane dehalogenase-like sequences and the three-dimensional models derived from these sequences.
The protein fold of haloalkane dehalogenases
Haloalkane dehalogenases structurally belong to the α/β-hydrolase fold enzymes (Ollis et al., 1992). Without exception, they all contain a nucleophile elbow (Damborský, 1998) which is the most conserved structure within the α/β-hydrolase fold. The other highly conserved regions in all haloalkane dehalogenases are eight central β-strands creating the hydrophobic core (Figures 1 and 4). These strands form the catalytic domain carrying the catalytic triad Asp–His–Asp(Glu). Two different topological arrangements of the catalytic triad can be found within the haloalkane dehalogenase family. While the nucleophile is always located after the strand β5 and the base is always after the strand β8, the catalytic acid is either after the strand β6 (in LinB, Rv2579, Iso-Rv2579, DhaA, DhaAf and DehH1) or after the strand β7 (in DhlA, Rv1833c and Rv2296). The former location of the catalytic acid corresponds to the position of catalytic acid in human pancreatic lipase (Schrag et al., 1992), while the latter location corresponds to all other α/β-hydrolases (Ollis et al., 1992). Krooshof et al. 1997) proposed that the catalytic acid shifted from strand β6 to strand β7 in haloalkane dehalogenases and that this might be an important event in the adaptation to 1,2-dichloroethane. The M.tuberculosis enzymes Rv1833c and Rv2296 may present evolutionary intermediates of this event and their biochemical characterization might provide important information about the molecular adaptation of haloalkane dehalogenases. Dehalogenation experiments using the intact cells of M.tuberculosis H37Rv suggest that this strain is able to dehalogenate 1-chlorobutane, 1-chlorodecane, 1-bromobutane and 1,2-dibromoethane, but not 2-chloropropane and 1,2-dichloroethane (Jesenska et al., 1999).
The classification of haloalkane dehalogenases as belonging to the α/β-hydrolase fold enzymes is further supported by the presence of the α/β-hydrolases fingerprint in their sequences (Figure 2). Haloalkane dehalogenases also share common fingerprints with epoxidase hydrolases. The evolutionary relationship between haloalkane dehalogenases and epoxide hydrolases has been established previously (Rink et al., 1997; Archelas and Furstoss, 1998). The alignment of haloalkane dehalogenases with structurally conserved regions of α/β-hydrolases and epoxide hydrolases can be used for designing primers for the selective screening of haloalkane dehalogenases in bacterial genomes using a polymerase chain reaction technique.
The reaction mechanism of haloalkane dehalogenases
The reaction mechanism of haloalkane dehalogenase DhlA has been proposed from X-ray crystallography (Verschueren et al., 1993a,b) and site-directed mutagenesis experiments (Pries et al., 1994b, 1995a,b). Catalysis proceeds by the nucleophilic attack of the carboxylate oxygen of Asp124 on the carbon atom of the substrate, yielding a covalent alkyl-enzyme intermediate. The tetrahedral intermediate is stabilized by the amide nitrogens of Glu56 and Trp125 present in the oxyanion pocket. The negative charge which develops on the halogen atom is stabilized by the Trp125, Phe172 and Trp175 side chains. The alkyl-enzyme intermediate is subsequently hydrolysed by the water molecule activated by His289. Catalytic acid Asp260 stabilizes the charge developed on the imidazole ring of His289.
A number of residues involved in the reaction mechanism of DhlA are also conserved in other haloalkane dehalogenases (Table II), suggesting a very similar or identical reaction mechanism. All dehalogenases in the study contain a catalytic triad. Nucleophilic aspartate is positioned on the extremely sharp γ-like turn called the nucleophile elbow. The regions around the two tetrahedral-intermediate-stabilizing residues, i.e. the loop HGE(N)PT and the nucleophile elbow, are highly conserved and create the oxyanion hole (Figure 6). These structural observations clearly support a two-step reaction mechanism. The single halide-stabilizing residue (equivalent to Trp125 of DhlA) is fully conserved in all haloalkane dehalogenases. Two later halide-stabilizing residues (Phe172 and Trp175 of DhlA) occur in the structurally non-conserved regions. We attempted to predict the position and structural arrangement of the equivalent residues in the LinB and DhaA enzymes (Table II), but these predictions need further experimental validation. A study of the mutagenesis of the proposed halide-binding residues in LinB is currently in progress.
Substrate specificity of haloalkane dehalogenases
Structure–specificity relationships are very important in attempts to design improved environmental biocatalysts by means of protein engineering. Unfortunately, they are also among the most difficult to uncover by theoretical modelling. Structural dissimilarity rather than similarity has to be accessed, but unfortunately the modelling of non-conserved regions is often uncertain. The substrate specificity of an enzyme can be the function of several structural features and three of them will be discussed for the haloalkane dehalogenases: (1) the size and geometry of an active site, (2) the size and geometry of the entrance to the active site and (3) the efficiency of TS and halide stabilization by active site residues.
The active site of DhlA is a small cavity buried inside the protein. The volume of the cavity calculated by program CAST is 112 Å3. The walls of the active site of DhlA are lined by large hydrophobic residues (Table II). The size, shape and hydrophobic character of the active site are optimized for good binding and stabilization of the natural substrate 1,2-dichloroethane (DCE). The longer compounds (>C6) are poor substrates for DhlA. LinB and DhaA can dehalogenate compounds with chain lengths of up to 10 carbon atoms (with the optimum activity for compounds with seven or eight carbon atoms), but they cannot catalyse the dehalogenation of DCE. The volumes of the LinB and DhaA active sites are about 2.5 and, 2 times larger, respectively, than the active site of DhlA according to CAST calculations conducted with homology models. The larger volumes of the LinB and DhaA active sites may be due to the involvement of smaller amino acids (Table II), indels in the region around helix α4 and a different arrangement of the helices α4 and α5 (Figure 1). For example, small Ala is present in LinB and DhaA in the position equivalent to Phe128 of DhlA. This residue lies in a well conserved region within the whole α/β-hydrolase and epoxidase family and DhlA is the only enzyme carrying a large aromatic residue in this position (Figure 2). Ala→Phe substitution is possibly the result of the molecular adaptation of DhlA towards the efficient binding and catalysis of DCE. There are further positions, e.g. 134, 141, 146, 226, 251–256 and 294 (numbered according to DhlA) where the dehalogenases from different specificity classes show amino acid replacements. These replacements may relate to differences not only in specificity, but also in function. A representative of the latter type of replacement might be Phe294 substituting the polar residue (Asp, Glu, respectively) found in all other haloalkane dehalogenases and epoxide hydrolases (Figure 2B). Asp→Phe substitution prevents the formation of a hydrogen bond or even a salt bridge between the main and cap domain of DhlA and in this way it may facilitate the conformation change taking place in the cap domain during the halide release. The residues showing replacements in conserved regions represent suitable targets for site-directed mutagenesis experiments.
Verschueren et al. (1993a) proposed that the substrate molecule enters the active site of DhlA through the narrow tunnel connecting the buried cavity with the solvent. Binding and catalysis of DCE outside the bulk solvent are crucial for the efficient catalysis of the dehalogenation reaction. The export of the halide ion is believed to proceed by means of the conformational change occurring in the cap domain of the enzyme (Schanstra and Janssen, 1996). The exact anatomies of the active site entrances of LinB and DhaA are difficult to predict owing to low sequence similarity with the template structure and shift in the position of helices α4 and α5. Current models suggest that the active sites of LinB and DhaA are less buried and closer to the protein suface. Large substrates may have good access to the active site and the products of the catalysis should be able to leave the enzyme without conformational change. Information obtained from the homology models is limited, however, and experimental determination of the structure of the substrate-binding domain of LinB or DhaA is required to obtain more reliable data. It is possible that the dehalogenases from the different specificity classes acquired the substrate-binding domains from different origins. The substrate-binding domain in DhlA (called the cap domain) shares structural homology with uteroglobin (Russell and Sternberg, 1997), while LinB and DhaA share structural homology with renilla-luciferin 2-monooxygenase (Lorenz et al., 1991).
Catalytic efficiency can determine the specificity of particular enzymes in the sense that some substrates cannot be converted by an enzyme owing to poor kinetics or unfavourable thermodynamics. Electrostatic stabilization of TS and the halide ion by Trp125, Phe172 and Trp175 during conversion DCE by DhlA has been discussed above. Substitution of Trp175 in LinB and DhaA by a similar, but still chemically and sterically different residue (Phe) might be an additional structural feature responsible for the different substrate specificity of these enzymes. Quantum-chemical calculations show that Phe stabilizes the charge which develops on the halide atom less efficiently than Trp, resulting in a higher activation energy barrier and inefficient catalysis of the carbon–halogen bond cleavage of the DCE molecule (Damborský et al., 1997b; Damborský, 1998). Consequently, LinB and DhaA do not dehalogenate DCE and they may also not need to employ conformational change during the release of the less stabilized halide ion from the enzyme active site.
Note added in proof
The homology model of LinB was validated against the crystal structure solved in our laboratory at 1.6 Å resolution during the review process of this paper (Marek,M., Vévodová,J., Smatanová,I., Svensson,L.A., Newman,J., Nagata,Y., Damborský,J., Takagi,M., in preparation). Investigation of the experimental structure confirms the α/β-hydrolase fold of LinB and the proposed reaction mechanism. The X-ray structure supports the prediction of the catalytic triad (Asp108, His272 and Glu132) and the oxyanion hole (Asn38 and Trp109). All but one active site residues listed in Table II are present in the active site of LinB (except Phe154). Incorrect location of Phe154 is due to the misalignment in the sequentially non-conserved region around helices α4 and α5. Proper alignment of this region is provided in Figure 7. Additional residues forming the active site of LinB are Asn38, Ile134, Phe143, Pro144, Gln146, Asp147, Phe169, Val173, Leu177, Ala247 and Phe273. The active site of LinB (276 Å3 by CAST) is larger than the active site of DhlA, which is in agreement with the prediction. The X-ray structure of LinB confirms the shift in position of the helices α4 and α5 (with respect to DhlA) deduced from the secondary element prediction. Arrangement of these helices in LinB appears to be one of the key features determining its high affinity towards long-chain hydrocarbons.
A recently published kinetic analysis of DhaA dehalogenase revealed that this enzyme shows different halide binding from DhlA due to a missing tryptophan equivalent to the Trp175 of DhlA (Schindler et al., 1999). This observation is in agreement with our proposal of less efficient TS and halide ion stabilization by LinB and DhaA dehalogenases due to substitution of halide-stabilizing Trp by Phe.
N-terminal protein sequences of haloalkane dehalogenases and putative haloalkane dehalogenases
| Organism | Strain | Gene | N-terminal protein sequencea | Classb | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aProtein sequences determined directly by protein sequencing or deduced from a DNA sequence. | |||||||||||||||||||||||||
| bClassification according to substrate specificity (Damborský et al., 1997a). | |||||||||||||||||||||||||
| cPutative haloalkane dehalogenases. | |||||||||||||||||||||||||
| Xanthobacter autotrophicus | GJ10 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | I | |
| Xanthobacter autotrophicus | GJ11 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Ancylobacter aquaticus | AD20 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Ancylobacter aquaticus | AD25 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Pseudomonas sp | E4M | – | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | I | |
| Rhodococcus rhodochrous | NCIMB 13064 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Rhodococcus erythropolis | Y2 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Pseudomonas pavonaceae | 170 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | – | |
| Mycobacterium sp. | GP1 | dhaAf | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | – | |
| Rhodococcus sp. | HA1 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | ? | V | E | V | L | G | ? | ? | II | |
| Rhodococcus sp. | m15-3 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Rhodococcus sp. | CP9 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | ? | L | G | E | R | II/III | |
| Sphingomonas paucimobilis | UT26 | linB | M | S | L | G | A | K | P | F | G | E | K | K | F | I | E | I | K | G | R | R | M | III | |
| Mycobacterium tuberculosisc | H37Rv | rv2579 | M | T | A | F | G | V | E | P | Y | G | Q | P | K | Y | L | E | I | A | G | K | R | – | |
| Mycobacterium bovisc | MU11 | iso-rv2579 | M | T | A | F | G | V | E | P | Y | G | Q | P | K | Y | L | E | I | A | G | K | R | – | |
| Mycobacterium tuberculosisc | H37Rv | rv1833c | M | S | I | D | F | T | P | D | P | Q | L | Y | P | F | E | S | R | W | F | D | S | – | |
| Mycobacterium tuberculosisc | H37Rv | rv2296 | M | D | V | L | R | T | P | D | S | R | F | E | H | L | V | G | Y | P | F | A | P | – | |
| Organism | Strain | Gene | N-terminal protein sequencea | Classb | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aProtein sequences determined directly by protein sequencing or deduced from a DNA sequence. | |||||||||||||||||||||||||
| bClassification according to substrate specificity (Damborský et al., 1997a). | |||||||||||||||||||||||||
| cPutative haloalkane dehalogenases. | |||||||||||||||||||||||||
| Xanthobacter autotrophicus | GJ10 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | I | |
| Xanthobacter autotrophicus | GJ11 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Ancylobacter aquaticus | AD20 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Ancylobacter aquaticus | AD25 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Pseudomonas sp | E4M | – | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | I | |
| Rhodococcus rhodochrous | NCIMB 13064 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Rhodococcus erythropolis | Y2 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Pseudomonas pavonaceae | 170 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | – | |
| Mycobacterium sp. | GP1 | dhaAf | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | – | |
| Rhodococcus sp. | HA1 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | ? | V | E | V | L | G | ? | ? | II | |
| Rhodococcus sp. | m15-3 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Rhodococcus sp. | CP9 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | ? | L | G | E | R | II/III | |
| Sphingomonas paucimobilis | UT26 | linB | M | S | L | G | A | K | P | F | G | E | K | K | F | I | E | I | K | G | R | R | M | III | |
| Mycobacterium tuberculosisc | H37Rv | rv2579 | M | T | A | F | G | V | E | P | Y | G | Q | P | K | Y | L | E | I | A | G | K | R | – | |
| Mycobacterium bovisc | MU11 | iso-rv2579 | M | T | A | F | G | V | E | P | Y | G | Q | P | K | Y | L | E | I | A | G | K | R | – | |
| Mycobacterium tuberculosisc | H37Rv | rv1833c | M | S | I | D | F | T | P | D | P | Q | L | Y | P | F | E | S | R | W | F | D | S | – | |
| Mycobacterium tuberculosisc | H37Rv | rv2296 | M | D | V | L | R | T | P | D | S | R | F | E | H | L | V | G | Y | P | F | A | P | – | |
N-terminal protein sequences of haloalkane dehalogenases and putative haloalkane dehalogenases
| Organism | Strain | Gene | N-terminal protein sequencea | Classb | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aProtein sequences determined directly by protein sequencing or deduced from a DNA sequence. | |||||||||||||||||||||||||
| bClassification according to substrate specificity (Damborský et al., 1997a). | |||||||||||||||||||||||||
| cPutative haloalkane dehalogenases. | |||||||||||||||||||||||||
| Xanthobacter autotrophicus | GJ10 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | I | |
| Xanthobacter autotrophicus | GJ11 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Ancylobacter aquaticus | AD20 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Ancylobacter aquaticus | AD25 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Pseudomonas sp | E4M | – | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | I | |
| Rhodococcus rhodochrous | NCIMB 13064 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Rhodococcus erythropolis | Y2 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Pseudomonas pavonaceae | 170 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | – | |
| Mycobacterium sp. | GP1 | dhaAf | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | – | |
| Rhodococcus sp. | HA1 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | ? | V | E | V | L | G | ? | ? | II | |
| Rhodococcus sp. | m15-3 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Rhodococcus sp. | CP9 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | ? | L | G | E | R | II/III | |
| Sphingomonas paucimobilis | UT26 | linB | M | S | L | G | A | K | P | F | G | E | K | K | F | I | E | I | K | G | R | R | M | III | |
| Mycobacterium tuberculosisc | H37Rv | rv2579 | M | T | A | F | G | V | E | P | Y | G | Q | P | K | Y | L | E | I | A | G | K | R | – | |
| Mycobacterium bovisc | MU11 | iso-rv2579 | M | T | A | F | G | V | E | P | Y | G | Q | P | K | Y | L | E | I | A | G | K | R | – | |
| Mycobacterium tuberculosisc | H37Rv | rv1833c | M | S | I | D | F | T | P | D | P | Q | L | Y | P | F | E | S | R | W | F | D | S | – | |
| Mycobacterium tuberculosisc | H37Rv | rv2296 | M | D | V | L | R | T | P | D | S | R | F | E | H | L | V | G | Y | P | F | A | P | – | |
| Organism | Strain | Gene | N-terminal protein sequencea | Classb | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aProtein sequences determined directly by protein sequencing or deduced from a DNA sequence. | |||||||||||||||||||||||||
| bClassification according to substrate specificity (Damborský et al., 1997a). | |||||||||||||||||||||||||
| cPutative haloalkane dehalogenases. | |||||||||||||||||||||||||
| Xanthobacter autotrophicus | GJ10 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | I | |
| Xanthobacter autotrophicus | GJ11 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Ancylobacter aquaticus | AD20 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Ancylobacter aquaticus | AD25 | dhlA | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | – | |
| Pseudomonas sp | E4M | – | M | I | N | A | I | R | T | P | D | Q | R | F | S | N | L | D | Q | Y | P | F | S | I | |
| Rhodococcus rhodochrous | NCIMB 13064 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Rhodococcus erythropolis | Y2 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Pseudomonas pavonaceae | 170 | dhaA | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | – | |
| Mycobacterium sp. | GP1 | dhaAf | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | – | |
| Rhodococcus sp. | HA1 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | ? | V | E | V | L | G | ? | ? | II | |
| Rhodococcus sp. | m15-3 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | V | L | G | E | R | II | |
| Rhodococcus sp. | CP9 | – | M | S | E | I | G | T | G | F | P | F | D | P | H | Y | V | E | ? | L | G | E | R | II/III | |
| Sphingomonas paucimobilis | UT26 | linB | M | S | L | G | A | K | P | F | G | E | K | K | F | I | E | I | K | G | R | R | M | III | |
| Mycobacterium tuberculosisc | H37Rv | rv2579 | M | T | A | F | G | V | E | P | Y | G | Q | P | K | Y | L | E | I | A | G | K | R | – | |
| Mycobacterium bovisc | MU11 | iso-rv2579 | M | T | A | F | G | V | E | P | Y | G | Q | P | K | Y | L | E | I | A | G | K | R | – | |
| Mycobacterium tuberculosisc | H37Rv | rv1833c | M | S | I | D | F | T | P | D | P | Q | L | Y | P | F | E | S | R | W | F | D | S | – | |
| Mycobacterium tuberculosisc | H37Rv | rv2296 | M | D | V | L | R | T | P | D | S | R | F | E | H | L | V | G | Y | P | F | A | P | – | |
Functionally important residues in halokane dehalogenases and putative haloalkane dehalogenases deduced from the homology models
| Enzyme | Catalytic triad | Oxyanion hold | Putative active sites residues | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aFor complete list of active sites see Note added in proof. | ||||||||||||||||
| bFunctionally important residues were deduced from the X-ray structure (Verschueren et al., 1993a). | ||||||||||||||||
| LinBa | Asp108 | His272 | Glu132 | Asn38 | Trp109 | Asp108 | Trp109 | – | – | Phe151 | Phe154 | Trp207 | Pro208 | Ile211 | Leu248 | His272 |
| Rv2579 | Asp109 | His273 | Glu133 | Asn39 | Trp110 | Asp109 | Trp110 | – | – | Phe152 | Phe155 | Trp208 | Pro209 | Leu212 | Ile249 | His273 |
| Iso-Rv2579 | Asp109 | His273 | Glu133 | Asn39 | Trp110 | Asn109 | Trp110 | – | – | Phe152 | Phe155 | Trp208 | Pro209 | Leu212 | Ile249 | His273 |
| DhaA | Asp106 | His272 | Glu130 | Asn41 | Trp107 | Asp106 | Trp107 | – | – | Phe149 | Phe152 | Phe205 | Pro206 | Leu209 | Leu246 | His272 |
| DhaAf | Asp106 | His272 | Glu130 | Asn41 | Trp107 | Asp106 | Trp107 | – | – | Phe149 | Phe152 | Phe205 | Pro206 | Leu209 | Leu246 | His272 |
| Rv1833c | Asp109 | His267 | Asp238 | Asn43 | Trp110 | Asp109 | Trp110 | – | – | Phe147 | Val150 | Met203 | Pro204 | Ile207 | Ala240 | His267 |
| Rv2296 | Asp123 | His279 | Asp250 | Glu55 | Trp124 | Asp123 | Trp124 | Leu127 | – | Phe167 | Tyr170 | Phe212 | Pro213 | Val216 | Ile252 | His279 |
| DhlAb | Asp124 | His289 | Asp260 | Glu56 | Trp125 | Asp124 | Trp125 | Phe128 | Phe164 | Phe172 | Trp175 | Phe222 | Pro223 | Val226 | Leu262 | His289 |
| DehH1 | Asp105 | His272 | Asp129 | Phe35 | Arg106 | Asp105 | – | – | – | Tyr148 | Trp151 | Tyr213 | – | Ala216 | Met246 | His272 |
| Enzyme | Catalytic triad | Oxyanion hold | Putative active sites residues | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aFor complete list of active sites see Note added in proof. | ||||||||||||||||
| bFunctionally important residues were deduced from the X-ray structure (Verschueren et al., 1993a). | ||||||||||||||||
| LinBa | Asp108 | His272 | Glu132 | Asn38 | Trp109 | Asp108 | Trp109 | – | – | Phe151 | Phe154 | Trp207 | Pro208 | Ile211 | Leu248 | His272 |
| Rv2579 | Asp109 | His273 | Glu133 | Asn39 | Trp110 | Asp109 | Trp110 | – | – | Phe152 | Phe155 | Trp208 | Pro209 | Leu212 | Ile249 | His273 |
| Iso-Rv2579 | Asp109 | His273 | Glu133 | Asn39 | Trp110 | Asn109 | Trp110 | – | – | Phe152 | Phe155 | Trp208 | Pro209 | Leu212 | Ile249 | His273 |
| DhaA | Asp106 | His272 | Glu130 | Asn41 | Trp107 | Asp106 | Trp107 | – | – | Phe149 | Phe152 | Phe205 | Pro206 | Leu209 | Leu246 | His272 |
| DhaAf | Asp106 | His272 | Glu130 | Asn41 | Trp107 | Asp106 | Trp107 | – | – | Phe149 | Phe152 | Phe205 | Pro206 | Leu209 | Leu246 | His272 |
| Rv1833c | Asp109 | His267 | Asp238 | Asn43 | Trp110 | Asp109 | Trp110 | – | – | Phe147 | Val150 | Met203 | Pro204 | Ile207 | Ala240 | His267 |
| Rv2296 | Asp123 | His279 | Asp250 | Glu55 | Trp124 | Asp123 | Trp124 | Leu127 | – | Phe167 | Tyr170 | Phe212 | Pro213 | Val216 | Ile252 | His279 |
| DhlAb | Asp124 | His289 | Asp260 | Glu56 | Trp125 | Asp124 | Trp125 | Phe128 | Phe164 | Phe172 | Trp175 | Phe222 | Pro223 | Val226 | Leu262 | His289 |
| DehH1 | Asp105 | His272 | Asp129 | Phe35 | Arg106 | Asp105 | – | – | – | Tyr148 | Trp151 | Tyr213 | – | Ala216 | Met246 | His272 |
Functionally important residues in halokane dehalogenases and putative haloalkane dehalogenases deduced from the homology models
| Enzyme | Catalytic triad | Oxyanion hold | Putative active sites residues | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aFor complete list of active sites see Note added in proof. | ||||||||||||||||
| bFunctionally important residues were deduced from the X-ray structure (Verschueren et al., 1993a). | ||||||||||||||||
| LinBa | Asp108 | His272 | Glu132 | Asn38 | Trp109 | Asp108 | Trp109 | – | – | Phe151 | Phe154 | Trp207 | Pro208 | Ile211 | Leu248 | His272 |
| Rv2579 | Asp109 | His273 | Glu133 | Asn39 | Trp110 | Asp109 | Trp110 | – | – | Phe152 | Phe155 | Trp208 | Pro209 | Leu212 | Ile249 | His273 |
| Iso-Rv2579 | Asp109 | His273 | Glu133 | Asn39 | Trp110 | Asn109 | Trp110 | – | – | Phe152 | Phe155 | Trp208 | Pro209 | Leu212 | Ile249 | His273 |
| DhaA | Asp106 | His272 | Glu130 | Asn41 | Trp107 | Asp106 | Trp107 | – | – | Phe149 | Phe152 | Phe205 | Pro206 | Leu209 | Leu246 | His272 |
| DhaAf | Asp106 | His272 | Glu130 | Asn41 | Trp107 | Asp106 | Trp107 | – | – | Phe149 | Phe152 | Phe205 | Pro206 | Leu209 | Leu246 | His272 |
| Rv1833c | Asp109 | His267 | Asp238 | Asn43 | Trp110 | Asp109 | Trp110 | – | – | Phe147 | Val150 | Met203 | Pro204 | Ile207 | Ala240 | His267 |
| Rv2296 | Asp123 | His279 | Asp250 | Glu55 | Trp124 | Asp123 | Trp124 | Leu127 | – | Phe167 | Tyr170 | Phe212 | Pro213 | Val216 | Ile252 | His279 |
| DhlAb | Asp124 | His289 | Asp260 | Glu56 | Trp125 | Asp124 | Trp125 | Phe128 | Phe164 | Phe172 | Trp175 | Phe222 | Pro223 | Val226 | Leu262 | His289 |
| DehH1 | Asp105 | His272 | Asp129 | Phe35 | Arg106 | Asp105 | – | – | – | Tyr148 | Trp151 | Tyr213 | – | Ala216 | Met246 | His272 |
| Enzyme | Catalytic triad | Oxyanion hold | Putative active sites residues | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aFor complete list of active sites see Note added in proof. | ||||||||||||||||
| bFunctionally important residues were deduced from the X-ray structure (Verschueren et al., 1993a). | ||||||||||||||||
| LinBa | Asp108 | His272 | Glu132 | Asn38 | Trp109 | Asp108 | Trp109 | – | – | Phe151 | Phe154 | Trp207 | Pro208 | Ile211 | Leu248 | His272 |
| Rv2579 | Asp109 | His273 | Glu133 | Asn39 | Trp110 | Asp109 | Trp110 | – | – | Phe152 | Phe155 | Trp208 | Pro209 | Leu212 | Ile249 | His273 |
| Iso-Rv2579 | Asp109 | His273 | Glu133 | Asn39 | Trp110 | Asn109 | Trp110 | – | – | Phe152 | Phe155 | Trp208 | Pro209 | Leu212 | Ile249 | His273 |
| DhaA | Asp106 | His272 | Glu130 | Asn41 | Trp107 | Asp106 | Trp107 | – | – | Phe149 | Phe152 | Phe205 | Pro206 | Leu209 | Leu246 | His272 |
| DhaAf | Asp106 | His272 | Glu130 | Asn41 | Trp107 | Asp106 | Trp107 | – | – | Phe149 | Phe152 | Phe205 | Pro206 | Leu209 | Leu246 | His272 |
| Rv1833c | Asp109 | His267 | Asp238 | Asn43 | Trp110 | Asp109 | Trp110 | – | – | Phe147 | Val150 | Met203 | Pro204 | Ile207 | Ala240 | His267 |
| Rv2296 | Asp123 | His279 | Asp250 | Glu55 | Trp124 | Asp123 | Trp124 | Leu127 | – | Phe167 | Tyr170 | Phe212 | Pro213 | Val216 | Ile252 | His279 |
| DhlAb | Asp124 | His289 | Asp260 | Glu56 | Trp125 | Asp124 | Trp125 | Phe128 | Phe164 | Phe172 | Trp175 | Phe222 | Pro223 | Val226 | Leu262 | His289 |
| DehH1 | Asp105 | His272 | Asp129 | Phe35 | Arg106 | Asp105 | – | – | – | Tyr148 | Trp151 | Tyr213 | – | Ala216 | Met246 | His272 |
The protein sequence alignment of haloalkane dehalogenase from Sp.paucimobilis UT26 (LinB), putative haloalkane dehalogenases from M.tuberculosis H37Rv (Rv2579, Rv1833c, Rv2296) and M.bovis MU11 (Iso-Rv2579), haloalkane dehalogenase from R.rhodochrous NCIMB 13064, R.erythropolis Y2 and Ps.pavonaceae 170 (DhaA), haloalkane dehalogenase from Mycobacterium sp. GP1 (DhaAf), haloalkane dehalogenase from X.autotrophicus GJ10 (DhlA) and haloacetate dehalogenase from Moraxella sp. B (DehH1). Conserved residues are shaded. Conserved regions are boxed. The positions of the secondary elements (in DhlA deduced from the 3D structure and in other dehalogenases predicted from the sequence) are indicated by the lines under the sequences. The secondary structure elements are numbered according to DhlA. The amino acids present in the active site of DhlA are squared. The triangles indicate the position of the catalytic residues. The arrow indicates the position of the direct repeat in DhlA.
The alignment of dehalogenase sequences with the structurally conserved motifs of the α/β-hydrolase fold (A) and the epoxide hydrolase fold (B). Conserved residues are shaded. The positions of the secondary elements of X.autotrophicus dehalogenase known from crystallographic analysis are indicated by the lines under the DhlA sequence. The amino acids positioned in the active site of DhlA are squared. The triangles indicate the position of the catalytic residues. The numbers correspond to the sequence of DhlA. The sequence codes are as follows: LINB, haloalkane dehalogenase from Sp.paucimobilis; RV2579, RV1833C, RV2296; putative haloalkane dehalogenases from M.tuberculosis; ISO_RV2579, putative haloalkane dehalogenases from M.bovis; DHAA, haloalkane dehalogenase from R.rhodochrous, R.erythropolis and Ps.pavonaceae; DHAAF, haloalkane dehalogenase from Mycobacteroum sp.; DHLA, haloalkane dehalogenase from Xanthobacter autotrophicus; DEHH1, haloacetate dehalogenase from Moraxella sp.; DMPD_PSEPU, XYLF_PSEPU, 2-hydroxymuconic semialdehyde hydrolases from Pseudomonas putida; LIP1_PSYIM, lipase 1 precursor (triacylglycerol lipase) from Psychrobacter immobilis; LIP3_MORSP, lipase 3 precursor (triacylglycerol lipase) from Moraxella sp.; ACOC_ALCEU, putative dihydrolipoamide acetyltransferase from Alcaligenes eutrophus; PHAB_PSEOL, poly(3-hydroxyalkanoate) depolymerase from Pseudomonas oleovorans; PSEBPHD, 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoic acid hydrolase from Comamonas testosteroni; BPHD_PSES1, 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate hydrolase from Pseudomonas sp.; TODF_PSEPU, 2-hydroxy-6-oxo-2,4-heptadienoate hydrolase from Pseudomonas putida; HYES_HUMAN, soluble epoxide hydrolase from Homo sapiens; HYEP_HUMAN, microsomal epoxide hydrolase from Homo sapiens; HYEP_RAT, microsomal epoxide hydrolase from rat; HUMEPOHYDC, epoxide hydrolase from Homo sapiens; HYES_MOUSE, soluble epoxide hydrolase from mouse; HYES_RAT, soluble epoxide hydrolase from rat; HYEP_RABIT, microsomal epoxide hydrolase from rabbit; STU02494, epoxide hydrolase from Solanum tuberosum.
Ramachandran plot of the crystal structure of DhlA, the homology model of LinB and the homology model of DhaA. The shading represents the different regions of the plot: the darker the region the more favoured is the corresponding ϕ, Ψ combination. Amino acid residues lying in less favoured regions are labelled.
Stereoview of the tertiary structure of DhlA, the homology model of LinB and the homology model of DhaA. Only Cα atoms are shown for clarity. The highly reliable regions of the homology models are in black and less reliable regions are in grey.
The arrangement of the catalytic triad in the crystal structure of DhlA (A), the homology model of LinB (B), the homology model of DhaA (C), the theoretical model of the D260N + N148D double mutant of DhlA published by Krooshof et al. (1997) (D) and the crystal structure of human pancreatic lipase (E). The arrow indicates the orientation of the catalytic acid residue within each triad.
The arrangement of the nucleophile-stabilizing residues of the oxyanion hole in the crystal structure of DhlA, the homology model of LinB and the homology model of DhaA. The partially positively charged atoms Trp-NH and Glu(Asn)-NH1 stabilize the partially negatively charged atom Asp-O2.
Structurally derived alignment of haloalkane dehalogenases in the region around helices α4 and α5. Labeling of haloalkane dehalogenases is the same as in Figure 1. Solvent-accessible residues are blank and buried residues are shaded. The positions of the secondary elements in LinB and DhlA deduced from the 3D structures are indicated by the lines under the sequences. The amino acids present in the active sites of LinB and DhlA are squared.
To whom correspondence should be addressed
The authors thank the operators of the Czech Academic Supercomputing Centres in Brno and Prague for providing excellent service. The Grant Agency of the Czech Republic and the Ministry of Education of the Czech Republic are acknowledged for financial support through grants 203/97/P149 and ME276/1998. The Laboratory of Biomolecular Structure and Dynamics was established by the Ministry of Education of the Czech Republic under contract VS96095. Laurence Benjamin (Masaryk University, Brno) is gratefully acknowledged for help with the linguistic revision of the manuscript.
References
Attwood,T.K., Beck,M.E., Flower,D.R., Scordis,P. and Selley,J. (
Damborský,J., Nyandoroh,M.G., Němec,M., Holoubek,I., Bull,A.T. and Hardman,D.J. (
Janssen,D.B., Gerritse,J., Brackman,J., Kalk,C., Jager,D. and Witholt,B. (
Janssen,D.B., Damborský,J., Rink,R. and Krooshof,G.H. (1999) In Alberghina,L. (ed.), Protein Engineering in Industrial Biotechnology. Harwood Academic Publishers, in press.
Kennes,C., Pries,F., Krooshof,G.H., Bokma,E., Kingma,J. and Janssen,D.B. (
Krooshof,G.H., Kwant,E.M., Damborský,J., Koča,J. and Janssen,D.B. (
Laskowski,R.A., McArthur,M.W., Moss,D.S. and Thornton,J.M. (
Lenski,R.E., Velicer,G.J. and Korona,R. (1997) In Horikoshi,K., Fukuda,M. and Kudo,T. (eds.), Microbial Diversity and Genetics of Biodegradation. Japan Scientific Societies Press, Tokyo, pp. 83–95.
Liang,J., Edelsbrunner,H., Fu,P., Sudhakar,P.V. and Subramaniam,S. (
Lorenz,W.W., McCann,R.O., Longiaru,M. and Cormier,M.J. (
Nagata,Y., Nariya,T., Ohtomo,R., Fukuda,M., Yano,K. and Takagi,M. (
Nagata,Y., Miyauchi,K., Damborský,J., Manová,K., Ansorgová,A. and Takagi,M. (
Poelarends,G.J., Wilkens,M., Larkin,J., van Elsas,J.D. and Janssen,D.B. (
Poelarends,G.J., van Hylckama Vlieg,J.E.T., Marchesi,J.R., Freitas Dos Santos,L.M. and Janssen,D.B. (
Pries,F., Kingma,J., Pentega,M., Van Pouderoyen,G., Jeronimus-Stratingh,C.M., Bruins,A.P. and Janssen, D.B. (
Pries,F., Van den Wijngaard,A.J., Bos,R., Pentenga,M. and Janssen,D.B. (
Pries,F., Kingma,J., Krooshof,G.H., Jeronimus-Stratingh,C.M., Bruins,A.P. and Janssen,D.B. (
Schindler,J.F., Naranjo,P.A., Honaberger,D.A., Chang,C.H., Brainard,J.R., Vanderberg,L.A. and Unkefer,C.J. (
Van den Wijngaard,A.J., Kamp,K.W.H.J., Ploeg,J., Pries,F., Kazemier,B. and Janssen,D. (
Verschueren,K.H.G., Franken,S.M., Rozeboom,H.J., Kalk,K.H. and Dijkstra,B.W. (
Verschueren,K.H.G., Kingma,J., Rozeboom,H.J., Kalk,K.H., Janssen,D.B. and Dijkstra,B.W. (
Verschueren,K.H.G., Seljee,F., Rozeboom,H.J., Kalk,K.H. and Dijkstra,B.W. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}