





Abstract
Influenza A, B, and C viruses are the etiological agents of influenza. Hemagglutinin (HA) is the major envelope glycoprotein of influenza A and B viruses, and hemagglutinin-esterase (HE) in influenza C viruses is a protein homologous to HA. Because influenza A virus pandemics in humans appear to occur when new subtypes of HA genes are introduced from aquatic birds that are known to be the natural reservoir of the viruses, an understanding of the origin and evolution of HA genes is of particular importance. We therefore conducted a phylogenetic analysis of HA and HE genes and showed that the influenza A and B virus HA genes diverged much earlier than the divergence between different subtypes of influenza A virus HA genes. The rate of amino acid substitution for A virus HAs from duck, a natural reservoir, was estimated to be 3.19 × 10−4 per site per year, which was slower than that for human and swine A virus HAs but similar to that for influenza B and C virus HAs (HEs). Using this substitution rate from the duck, we estimated that the divergences between different subtypes of A virus HA genes occurred from several thousand to several hundred years ago. In particular, the earliest divergence time was estimated to be about 2,000 years ago. Also, the A virus HA gene diverged from the B virus HA gene about 4,000 years ago and from the C virus HE gene about 8,000 years ago. These time estimates are much earlier than the previous ones.
Introduction
Influenza viruses are members of the viral family Orthomyxoviridae and have a segmented, single-stranded, and negative-sense RNA genome in an enveloped virion (Smith, Andrewes, and Laidlaw 1933 ). The genome encodes envelope glycoproteins, matrix proteins, nonstructural proteins, nucleoproteins, and polymerase proteins. According to the antigenic properties of matrix proteins or nucleoproteins, influenza viruses are classified into types A, B, and C. Influenza A viruses cause epidemics and pandemics of influenza in mammals and birds, and aquatic birds are known to be the natural reservoir of these viruses (Slemons et al. 1974 ; Webster et al. 1978 ; Hinshaw, Webster, and Turner 1980 ). Influenza B and C viruses are isolated mainly from humans and are less pathogenic than influenza A viruses. Phylogenetic analyses of nucleoproteins and polymerase proteins have indicated that influenza A and B viruses are more closely related to each other than to influenza C viruses (Gammelin et al. 1990 ; Krossoy et al. 1999 ; Cox et al. 2000 ).
Hemagglutinin (HA) is the major envelope glycoprotein of A and B viruses, and hemagglutinin-esterase (HE) in C viruses is a protein homologous to HA. HA (HE) is cleaved into the signal peptide (about 20 amino acids in influenza A viruses), protein HA1 (HE1) (about 320 amino acids), and protein HA2 (HE2) (about 220 amino acids) when mature proteins are produced (fig. 1 ). HA1 (HE1) is a receptor-binding protein and the major target of immune responses, whereas HA2 (HE2) is an anchor protein of the envelope and mediates fusion of the envelope and the cellular endosomal membrane. Influenza A virus HA genes are classified into 15 subtypes (H1–H15), according to their antigenic properties (WHO Memorandum 1980 ), whereas B and C virus HA (HE) genes are not classified into subtypes. Because influenza A virus pandemics in humans appear to occur when new subtypes of HA genes are introduced from aquatic birds, an understanding of the origin and evolution of HA genes is of particular importance.
From the phylogenetic analyses of A and B virus HA genes, Webster et al. (1992) suggested that the divergence between these two genes occurred later than the divergences between some subtypes of A virus HA genes. However, the reliability of their conclusion is unclear because they did not use any outgroup to root their phylogenetic tree. Saitou and Nei (1986) estimated the earliest divergence time between subtypes of A virus HA genes to be 200–300 years ago, using the rate of amino acid substitution for human A virus HAs. This estimate is also unreliable because the natural reservoir of these viruses is aquatic birds, and the evolutionary rate is known to be slower in birds than in humans (Saitou and Nei 1986 ; Bean et al. 1992 ; Schafer et al. 1993 ; Makarova et al. 1999 ; Reid et al. 1999 ; Suarez 2000 ).
The purpose of this paper is to study the evolutionary relationships of influenza A, B, and C virus HA (HE) genes. We are also interested in estimating the divergence times between these genes.
Materials and Methods
Phylogenetic Analyses
For constructing a phylogenetic tree for influenza A, B, and C virus HA (HE) genes, we used amino acid sequences because they are known to give more reliable results than nucleotide sequences when the sequence divergence is high (Nei and Kumar 2000, pp. 17–32 ). We also used only the HA2 (HE2) region of HA (HE) because in the multiple alignment for the entire region of A, B, and C virus HAs (HEs), the signal peptide and HA1 (HE1) protein regions generated many gaps and only the HA2 (HE2) region appeared reliable (fig. 1 ) (Nakada et al. 1984 ).
Amino acid sequences of influenza A, B, and C virus HA2s (HE2s) were collected from the international DNA databank (DDBJ release 43). After excluding sequences from laboratory-adapted viruses and identical sequences within species, we obtained 57, 34, 58, 10, 29, 2, 41, 1, 4, 2, 1, 1, 3, 1, and 2 amino acid sequences for the H1–H15 subtypes of A virus HA2s, respectively. We also obtained 15 sequences for B virus HA2s and 35 sequences for C virus HE2s. A total of 296 amino acid sequences were aligned by the computer program CLUSTAL W (Thompson, Higgins, and Gibson 1994 ). After removing all alignment gaps, 207 amino acid sites were used for estimating p, Poisson correction (PC), and gamma distances (Nei and Kumar 2000 ). The gamma shape parameter (a) was estimated to be 1.83 by Gu and Zhang's (1997) method. The phylogenetic tree was constructed by the neighbor-joining (NJ) method (Saitou and Nei 1987 ), and the reliability of each interior branch was tested by the bootstrap method with 1,000 resamplings (Felsenstein 1985 ; Kumar et al. 2001 ). The NJ trees were also constructed for 17 amino acid sequences which were randomly chosen from each subtype of A virus HA2s and from B virus HA2s and C virus HE2s (table 1 ).
Estimation of Divergence Times
For estimating the divergence times between subtypes of influenza A virus genes, we used only A virus sequences because B and C virus sequences were not necessary. We also used amino acid sequences for the entire region of HA because the alignment for A virus HAs appeared to be reliable (fig. 1 ) (Rohm et al. 1996 ), and longer sequences were expected to give more reliable estimates.
We obtained 50, 25, 24, 10, 21, 2, 25, 1, 4, 2, 1, 1, 3, 1, and 2 amino acid sequences for the H1–H15 subtypes of A virus HAs from the databank, respectively, and made a multiple alignment for a total of 172 sequences by CLUSTAL W. After removing all alignment gaps, 540 amino acid sites were used for estimating gamma distances with a = 1.20, which was obtained by Gu and Zhang's method. An NJ tree was constructed, and the branch lengths were recalculated by the ordinary least squares method (Rzhetsky and Nei 1993 ) to estimate the rate of amino acid substitution accurately (see subsequently).
When the years of isolation are available for viral sequences in a phylogenetic tree, the rate of amino acid substitution may be estimated by the regression coefficient of the numbers of amino acid substitutions from a common root on the years of isolation (Nei 1983 ; Suzuki, Wyndham, and Gojobori 2001 ). Using the phylogenetic tree for 172 sequences of influenza A virus HAs, we estimated the rate of amino acid substitution for duck A virus HAs because duck provided the largest number (28) of sequences among aquatic birds. For estimating the divergence times between subtypes of A virus HA genes, we constructed a linearized tree (Takezaki, Rzhetsky, and Nei 1995 ) for 28 amino acid sequences of duck A virus HAs using the gamma distance with a = 1.20. The standard errors (SEs) and 99% confidence intervals (CIs) of the rates and the divergence times were estimated by the bootstrap method, under the assumption that the topologies of the phylogenetic trees for 172 sequences of influenza A virus HAs and 28 sequences of duck A virus HAs were correct (Nei and Kumar 2000 ).
Results
Phylogenetic Relationships of Influenza A, B, and C Virus HA Genes
The NJ trees constructed by using p, PC, and gamma distances for 17 randomly chosen amino acid sequences of the HA2 (HE2) protein are shown in panels (a), (b), and (c) of figure 2 , respectively. All trees show the same topology and indicate that all influenza A virus HA genes diverged after they separated from B virus HA genes. The monophyly of A virus HA genes is supported by a bootstrap value of 100%, 99%, and 95% in trees (a), (b), and (c), respectively. This relationship was also supported by the NJ trees for 296 amino acid sequences of influenza A, B, and C virus HA2s (HE2s) with high bootstrap values (100%, 99%, and 86% for p, PC, and gamma distances, respectively) (data not shown).
Divergence Times Between Subtypes of A Virus HA Genes
For estimating the divergence times between subtypes of A virus HA genes, we first estimated the rate of amino acid substitution for duck A virus HAs because duck is one of the natural reservoirs of these viruses and provided the largest number of sequences among them. In the phylogenetic tree for 172 amino acid sequences of the entire region of A virus HAs, only the H1 and H2 subtypes included sufficient numbers of sequences for estimating the rate for duck A virus HAs and are shown in panels (a) and (b) of figure 3 , respectively. In this figure, avian sequences had generally shorter branch lengths than human and swine sequences in both subtypes, indicating that the rate for the former was slower than that for the latter. To estimate the rate of amino acid substitution, we conducted a regression analysis using duck sequences but failed to obtain the rate because it became negative in both H1 and H2 subtypes (data not shown). This happened probably because the evolutionary rate for duck sequences was too slow to give reliable estimates (Bean et al. 1992 ; Schafer et al. 1993 ; Suarez 2000 ).
To obtain a reliable rate for duck sequences, it was necessary to analyze duck sequences which were more distantly related from one another than those analyzed earlier in the article. For this purpose, we estimated the years of divergences between duck sequences and human and swine sequences in figure 3 , using the rates for the latter sequences and added these nodes to the regression analysis of duck sequences. The rates for human and swine sequences were easily estimated by the regression analysis using these sequences only because the rates were relatively high. In the H1 subtype, we first estimated the year of divergence at node M using the rate for human sequences (fig. 3a ). We used only human sequences which were isolated before 1977 because human A viruses circulating after 1977 are known to have originated from a laboratory-adapted virus (Kendal et al. 1978 ; Nakajima, Desselberger, and Palese 1978 ; Scholtissek, von Hoyningen, and Rott 1978 ; Palese and Young 1982 ; Hayashida et al. 1985 ). The rate of amino acid substitution for human sequences was estimated to be 1.20 × 10−3 per site per year (fig. 4a and table 2 ), and the year of divergence at node M was a.d. 1862. We also estimated the year of divergence at the same node using classical swine sequences. This gave a rate of 0.56 × 10−3 per site per year (fig. 4b ) and a date of 1836 at node M. Taking the average for human and classical swine sequences, we obtained 1849 as the final estimate of the year of divergence at node M (table 3 ). In addition, we estimated the year of divergence at node N using avian-like swine sequences. These sequences apparently evolved at a rate of 1.87 × 10−3 per site per year (fig. 4c ), and the year of divergence at node N was 1965. By adding nodes M and N to the regression analysis of duck sequences (fig. 4e ), the rate for these sequences was estimated to be 3.89 × 10−4 per site per year. We also estimated the rate for duck sequences in the H2 subtype. In this subtype, we estimated the year of divergence at node O using human sequences (fig. 3b ). The rate of amino acid substitution for these sequences was estimated to be 2.03 × 10−3 per site per year (fig. 4d and table 2 ) and the year of divergence at node O was 1946 (table 3 ). We added node O to the regression analysis of duck sequences (fig. 4 f ) and obtained a rate of 2.48 × 10−4 per site per year for these sequences. Taking the average for the H1 and H2 subtypes, we obtained 3.19 × 10−4 per site per year as the final estimate of the rate of amino acid substitution for duck influenza A virus HAs.
A linearized tree for 28 amino acid sequences of duck A virus HAs is shown in figure 5 . The topology of subtypes was the same as that shown in figure 1 , except for the branching pattern of the H11 subtype, which was supposed to make a cluster with the H12 subtype but made a cluster with the H1, H2, and H5 subtypes. The estimates of the divergence times between different subtypes of influenza A virus HA genes are listed in table 4 . Although the SE and 99% CI are large, all subtypes apparently diverged from several thousand to several hundred years ago. In particular, the earliest divergence (node X) is likely to have occurred about 2,000 years ago. We further estimated the divergence times between influenza A, B, and C virus HA (HE) genes by linearizing the phylogenetic tree in figure 2c. Assuming that the earliest divergence between subtypes of A virus HA genes occurred 1,971 years ago (table 4 ), A and B virus HA genes apparently diverged 3,832 years ago, and the separation of A and B virus HA genes from C virus HE genes occurred 7,919 years ago.
Discussion
The divergence between influenza A and B virus HA genes apparently occurred earlier than the divergences between different subtypes of A virus HA genes. This is different from the conclusion of Webster et al. (1992) that the divergence between A and B virus HA genes occurred later than the divergences between some subtypes of A virus HA genes. This difference is apparently caused by the fact that Webster et al. did not use an outgroup for their phylogenetic analysis. In the present study, the phylogenetic relationship was supported with a high bootstrap value, indicating that the relationship obtained is highly reliable.
The rate of amino acid substitution for duck A virus HAs (3.19 × 10−4 per site per year) was slower than that for human and swine A virus HAs ([0.56–2.03] × 10−3 per site per year) but similar to that for B virus HAs (5.3 × 10−4 per site per year [Air et al. 1990] ) and C virus HEs (2.3 × 10−4 per site per year [Muraki et al. 1996] ). These results suggest that the rate for HAs (HEs) is more or less constant in the natural reservoir but is accelerated in the newly infected host species. This is probably caused by variation in the strengths of immune responses and functional constraints on HAs (HEs) among different host species (Yamashita et al. 1988 ; Bean et al. 1992 ; Schafer et al. 1993 ; Scholtissek, Ludwig, and Fitch 1993 ; Makarova et al. 1999 ; Suzuki and Gojobori 1999 ).
The earliest divergence time between subtypes of influenza A virus HA genes was estimated to be about 2,000 years ago. Also, the divergence time between A and B virus HA genes was estimated to be about 4,000 years ago, whereas A and B virus HA genes and C virus HE genes diverged about 8,000 years ago. These estimates are substantially higher than those (200–300 years) by Saitou and Nei (1986) , who used human HA sequences. Because the evolutionary rate for human A virus HAs is known to be higher than that for aquatic birds, their estimates are considered to be underestimates. In fact, influenza pandemics in humans have been recorded as early as 412 B.C. (Kaplan and Webster 1977 ), suggesting that influenza A viruses existed more than 2,400 years ago. This observation is consistent with the estimates obtained in the present study.
We estimated the rates and the divergence times under the assumption that the molecular clock has held throughout the evolutionary history of HA (HE) genes. To examine whether this was really the case, we tested the linear relationship between the year of isolation and the number of amino acid substitutions in figure 4 and found that the linearity was not supported at the 1% significance level in both panels (a) and (f). However, the rate of amino acid substitution for human A virus HAs obtained from panel (a) (1.20 × 10−3 per site per year) was similar to that from previous studies (1.0 × 10−3 per site per year [Saitou and Nei 1986] ), and the rate for duck A virus HAs obtained from panel (f) (3.89 × 10−4 per site per year) was similar to that obtained from panel (e) (2.48 × 10−4 per site per year). These observations suggest that the rates obtained from panels (a) and (f) are approximately correct. Also, the molecular clock was not rejected at the 1% significance level for the phylogenetic tree in figure 5 by the likelihood-ratio test (Rambaut 2000 ; Yang 2000 ) but was rejected for the tree in figure 2c. The latter observation may reflect the fact that the biochemical functions are different between HAs and HEs and the natural reservoirs are not the same for influenza A, B, and C viruses. Therefore, some caution is necessary in estimating the divergence times between influenza A, B, and C virus HA (HE) genes. However, the rate of amino acid substitution for duck influenza A virus HAs was similar to that for B virus HAs and C virus HEs, as indicated previously. Also, in reality, no strict molecular clock is likely to hold for any protein but it is known that rough divergence times can be obtained even if the molecular clock is violated to some extent (Nei and Kumar 2000 , pp. 187–206; Nei, Xu, and Glazko 2001 ). Therefore, these estimates also appear to be appropriate as rough estimates.
In conclusion, influenza virus HA (HE) genes apparently evolved at a rate of amino acid substitution of 10−4 per site per year in the natural reservoir. These genes apparently diverged into influenza A, B, and C virus HA (HE) genes several thousand of years ago and subsequently into subtypes in influenza A viruses from several thousand to several hundred years ago.
Naruya Saitou, Reviewing Editor
Keywords: influenza virus hemagglutinin hemagglutinin-esterase rate of amino acid substitution divergence time
Address for correspondence and reprints: Yoshiyuki Suzuki, Institute of Molecular Evolutionary Genetics, The Pennsylvania State University, 328 Mueller Laboratory, University Park, Pennsylvania 16802. yis1@psu.edu
Table 1. Amino Acid Sequences of the HA or HE Genes used for a Phylogeneic Analysis of A Viruses (15 subtypes) and B and C Virusesa

Table 1. Amino Acid Sequences of the HA or HE Genes used for a Phylogeneic Analysis of A Viruses (15 subtypes) and B and C Virusesa
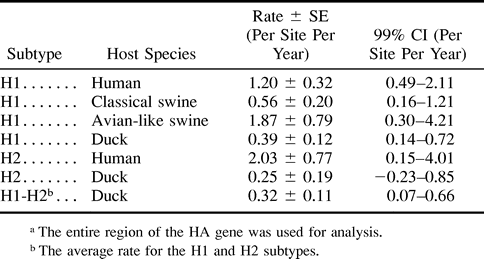
Table 3. Years of Divergences Between Influenza A Virus HA Genes in the H1 and H2 Subtypes

Table 3. Years of Divergences Between Influenza A Virus HA Genes in the H1 and H2 Subtypes
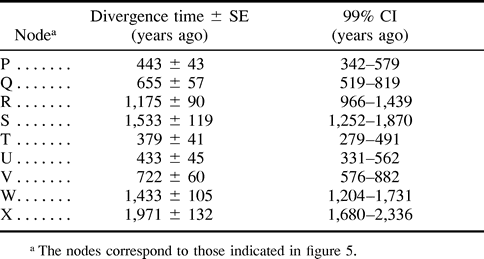
Fig. 1.—Multiple alignment for 17 randomly chosen amino acid sequences (see table 1 ) of the entire region of influenza A, B, and C virus HAs (HEs). Dashes indicate gaps. Stars indicate amino acid sites where the same amino acid exists for all sequences, and dots indicate amino acid sites where amino acids with similar properties exist for all sequences. HAs in A and B viruses (HEs in C viruses) are cleaved into the signal peptide and the proteins HA1 (HE1) and HA2 (HE2) when mature proteins are produced
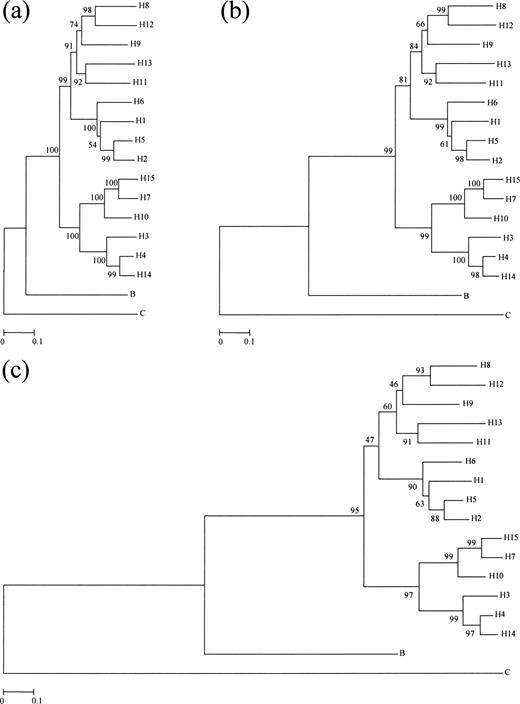
Fig. 2.—Phylogenetic trees for 17 randomly chosen amino acid sequences (see table 1 ) of the HA2 (HE2) region of influenza A, B, and C virus HAs (HEs). Trees (a), (b), and (c) were obtained by using p, PC, and gamma distances, respectively. The bootstrap value is indicated for each interior branch. The scale bars indicate the numbers of amino acid substitutions per site
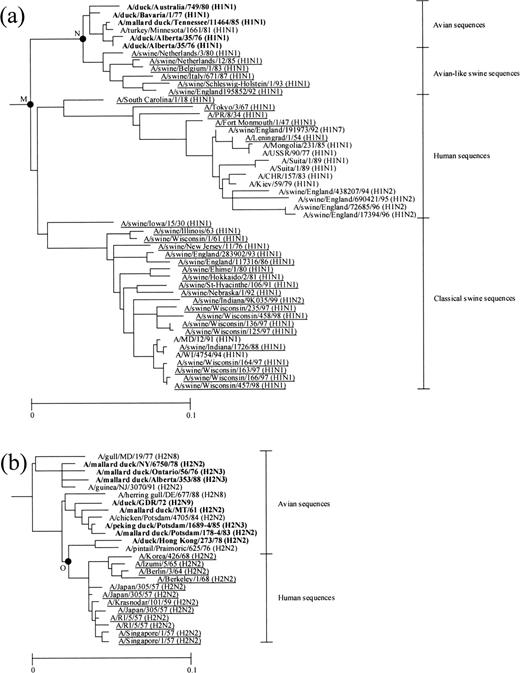
Fig. 3.—(a) H1 and (b) H2 subtype subtrees in the phylogenetic tree for 172 amino acid sequences of the entire region of influenza A virus HAs. For the nomenclature of influenza viruses, see the footnotes of table 1 . The nodes at which the year of divergence was estimated (table 3 ) are denoted by black circles and labeled with letters. The human and swine sequences used for estimating the rates and the years of divergences are underlined. The duck sequences used for estimating the rates are boldfaced. The scale bars indicate the numbers of amino acid substitutions per site
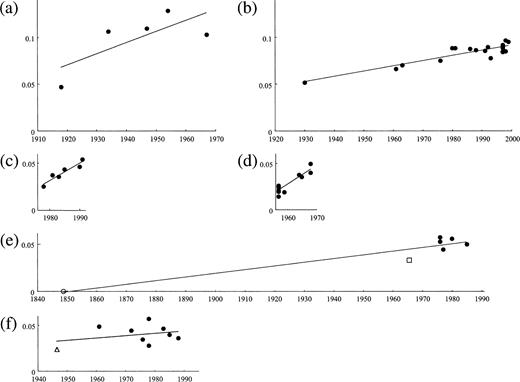
Fig. 4.—Regression analyses for estimating the rate of amino acid substitution for the entire region of (a) human, (b) classical swine, and (c) avian-like swine influenza A virus HAs in the H1 subtype and for (d) human sequences in the H2 subtype and for the (e) H1 and (f) H2 subtype sequences of duck HAs. In each panel, the abscissa indicates the year of isolation or divergence, and the ordinate indicates the number of amino acid substitutions from node (a) M, (b) M, (c) N, (d) O, or (e) M, or from (f) the earliest node of panel (b) in figure 3 . An open circle and an open square in panel (e) indicate nodes M and N in figure 3a, respectively, and an open triangle in panel (f) indicates node O in figure 3b
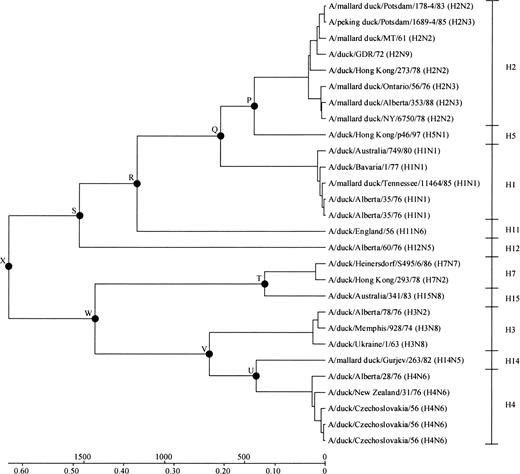
Fig. 5.—Linearized tree for 28 amino acid sequences of the entire region of duck influenza A virus HAs. For the nomenclature of influenza viruses, see the footnotes of table 1 . The nodes at which the divergence times were estimated (table 4 ) are denoted by black circles and labeled with letters. The scale bar indicates the number of years before present (above the line) and the number of amino acid substitutions per site (below the line)
The authors thank two anonymous reviewers for their valuable comments. This study was supported by grants from the National Institutes of Health to M.N. (GM20293). Y.S. is supported by the JSPS Research Fellowships for Young Scientists.
References
Air G. M., A. J. Gibbs, W. G. Laver, R. G. Webster,
Bean W. J., M. Schell, J. Katz, Y. Kawaoka, C. Naeve, O. Gorman, R. G. Webster,
Cox N. J.,, F. Fuller, N. Kaverin, H. D. Klenk, R. A. Lamb, B. W. J. Mahy, J. McCauley, K. Nakamura, P. Palese, R. Webster,
Felsenstein J.,
Gammelin M., A. Altmuller, U. Reinhardt, J. Mandler, V. R. Harley, P. J. Hudson, W. M. Fitch, C. Scholtissek,
Gu X., J. Zhang,
Hayashida H., H. Toh, R. Kikuno, T. Miyata,
Hinshaw V. S., R. G. Webster, B. Turner,
Kendal A. P., G. R. Noble, J. J. Skehel, W. R. Dowdle,
Krossoy B., I. Hordvik, F. Nilsen, A. Nylund, C. Endresen,
Kumar S., K. Tamura, I. B. Jakobsen, M. Nei,
Makarova N. V., N. V. Kaverin, S. Krauss, D. Senne, R. G. Webster,
Muraki Y., S. Hongo, K. Sugawara, F. Kitame, K. Nakamura,
Nakada S., R. S. Creager, M. Krystal, R. P. Aaronson, P. Palese,
Nakajima K., U. Desselberger, P. Palese,
Nei M.,
Nei M., S. Kumar,
Nei M., P. Xu, G. Glazko,
Rambaut A.,
Reid A. H., T. G. Fanning, J. V. Hultin, J. K. Taubenberger,
Rohm C., N. Zhou, J. Suss, J. Mackenzie, R. G. Webster,
Rzhetsky A., M. Nei,
Saitou N., M. Nei,
———.
Schafer J. R., Y. Kawaoka, W. J. Bean, J. Suss, D. Senne, R. G. Webster,
Scholtissek C., S. Ludwig, W. M. Fitch,
Scholtissek C. V., V. von Hoyningen, R. Rott,
Slemons R. D., D. C. Johnson, J. S. Osborn, F. Hayes,
Smith W., C. H. Andrewes, P. P. Laidlaw,
Suzuki Y., T. Gojobori,
Suzuki Y., A. Wyndham, T. Gojobori,
Takezaki N., A. Rzhetsky, M. Nei,
Thompson J. D., D. G. Higgins, T. J. Gibson,
Webster R. G., W. J. Bean, O. T. Gorman, T. M. Chambers, Y. Kawaoka,
Webster R. G., M. Yakhno, V. S. Hinshaw, W. J. Bean, K. G. Murti,
WHO Memorandum.
Yamashita M., M. Krystal, W. M. Fitch, P. Palese,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}