





Abstract
Background. Alport syndrome is a clinically and genetically heterogeneous nephropathy characterized by glomerular basement membrane lesions often associated with hearing loss and ocular anomalies. While the X-linked and the autosomal recessive forms are well known, the autosomal dominant form is not well acknowledged.
Methods. We have clinically investigated 38 patients with a diagnosis of autosomal dominant Alport syndrome belonging to eight different families. The analysis of the COL4A4 gene was performed by denaturing high performance liquid chromatography and automated DNA sequencing.
Results. In our cohort of patients, only 24.3% (9/37) reached end-stage renal disease, at the mean age of 51.2 years. Four patients had hearing loss (13.3%) and none ocular changes. Molecular analysis revealed eight novel private COL4A4 gene mutations: three frameshift, three missense and two splice-site mutations.
Conclusions. These data indicate autosomal dominant Alport syndrome as a disease with a low risk of ocular and hearing anomalies but with a significant risk to develop renal failure although at an older age than the X-linked form. We were unable to demonstrate a genotype–phenotype correlation. Altogether, these data make difficult the differential diagnosis with the benign familial haematuria due to heterozygous mutations of COL4A4 and COL4A3, especially in young patients, and with the X-linked form of Alport syndrome in families where only females are affected. A correct diagnosis and prognosis is based on a comprehensive clinical investigation in as many family members as possible associated with a broadly formal genetic analysis of the pedigree.
Introduction
Alport syndrome (ATS) is a progressive heterogeneous nephropathy characterized by the association of progressive haematuric nephritis with ultrastructural changes of the glomerular basement membrane (GBM), sensorineural deafness and variable ocular abnormalities [1–3]. ATS accounts for 1–2% of all patients who start renal replacement therapy in Europe, with an estimate frequency of about 1 in 5000 [4–6].
ATS is characterized by changes of type IV collagen α3, α4 and α5 network of the GBM, and three modes of inheritance are known. The X-linked inheritance, due to mutations in COL4A5 located in Xq22.3, is the most common mode of transmission (XLAS, OMIM 301050). In this form, 70% of affected males reach end-stage renal disease (ESRD) before 30 years (juvenile form), while only few cases (30%) progress towards ESRD after 30 years (rare adult form) [4,5,7]. Usually, females have only microhaematuria; however, a small percentage of females can develop renal failure. The autosomal recessive (ARAS, OMIM 203780) form of the disease is due to mutations in the COL4A3 and COL4A4 genes, located in 2q36–37, and is reported in 15% of families in European countries [8–10]. Autosomal recessive transmission due to COL4A3 and COL4A4 mutations is suggested by the presence of one of the following features: (i) severe early disease in both females and males; (ii) absence of severe signs in parents (they may be completely asymptomatic or may have isolated microhaematuria); (iii) parental consanguinity.
The autosomal dominant form of ATS has been described more recently (ADAS, OMIM 104200). In 1997, this form had been linked to the COL4A3/COL4A4 locus in a large family from Northern Ireland and 3 years later there came the first report of a COL4A3 heterozygous mutation segregating in a family with ADAS [11,12]. Till now, only nine families with ADAS and a proven COL4A4 or COL4A3 mutation have been reported. Both female and male patients showed a high clinical variability with a renal phenotype ranging from isolated haematuria to late onset ESRD, associated in few instances with hearing loss [11–15].
Heterozygous mutations in COL4A4 or COL4A3 genes have been found also in the subset of patients with ‘Benign Familial Hematuria’ (BFH, OMIM 141200). BFH is clinically defined by persistent glomerular haematuria and by the absence of extra-renal findings [16–18]. The typical ultrastructural findings are diffuse thinning of GBM.
Although the term ‘Thin Basement Membrane Nephropathy’ (TBMN) is a pure morphological definition, it is still erroneously used to describe patients with the diagnosis of BFH. In fact, the finding of isolated thinning of the basement membrane can represent the only lesion identified in patients with ATS [19].
In this study, we present eight ATS families with autosomal dominant transmission and heterozygous mutations in the COL4A4 gene. The aim of this study is to better define the natural clinical history of ADAS in order to help in the differential diagnosis with the other known forms of ATS and with BFH.
Subjects and methods
Patients
Thirty-eight patients belonging to eight families with a clinical suspect of ADAS were enrolled in this study. Patients were diagnosed by experienced nephrologists and clinical geneticists. Urinalysis and renal function evaluations were performed in all patients. Audiological and ophthalmic examinations were performed in several patients. Whenever indicated, a renal biopsy with ultrastructural study by electron microscopy was performed.
Mutation analysis
Blood samples were collected from patients after informed consent. Genomic DNA was extracted using a QIAamp DNA blood maxi kit, according to the manufacturers’ protocol (Quiagen, Hilden, Germany). All the COL4A4 exons were amplified using the primers and PCR condition already described [14]. Mutation analysis was performed by denaturing high performance liquid chromatography (DHPLC) using Transgenomic WAVETM (Transgenomic, San Jose, CA, USA) and by subsequent genomic sequencing analysis. PCR products were denatured at 95°C, re-annealed at 65°C for 10 min and cooled at 4°C to generate heteroduplex [15]. The optimal column temperature for fragments analysis was calculated using the WaveMaker Software (Transgenomic, San Jose, CA, USA). DHPLC analysis was performed at the melting temperature of 60°C for exon 25, 60.2°C for exon 22, 60.6°C for exon 28, 57.7°C for exon 46, 55.3°C for exon 35 and 54°C for exon 16. PCR products resulting in abnormal DHPLC profiles were purified and sequenced on both strands by using a PE Big dye terminator cycle sequencing kit on an ABI Prism 310 genetic analyser (PE Applied Biosystems, Foster City, CA, USA). Segregation analysis was performed by direct sequencing in all families.
Results
A COL4A4 mutation in eight families with ADAS has been identified by DHPLC analysis (Figure 1). Detailed clinical information of a total of 38 patients belonging to these eight families has been collected (Table 1). The molecular diagnosis was confirmed in 29/38 patients. The remaining nine patients were either deceased or unavailable.
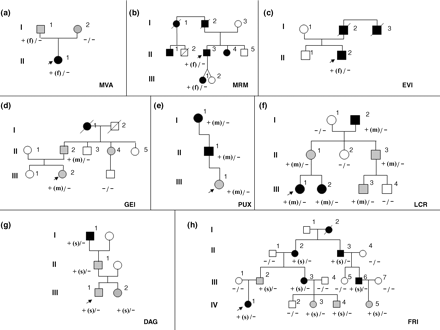
Pedigree of families. The figure represents the pedigree of family 1 (a), 2 (b), 3 (c), 4 (d), 5 (e), 6 (f), 7 (g), 8 (h).  Males,
Males,  females. Filled grey symbols are individuals with isolated microhaematuria. Filled black symbols indicate individuals with microhaematuria plus macrohaematuria or proteinuria or hypoacusia or renal failure. White symbols indicate individuals without clinical sings of the disease. An oblique bar indicates a deceased individual. The arrows indicate the index patients. The genotype at the COL4A4 locus is indicated below each symbol as follows: −, wild type allele; +, mutated allele. The type of mutation is indicated in brackets: f = frameshift mutation; m = missense mutation; s = splice site mutation. In (a) case I-2 has microhaematuria probably not related to ATS.
females. Filled grey symbols are individuals with isolated microhaematuria. Filled black symbols indicate individuals with microhaematuria plus macrohaematuria or proteinuria or hypoacusia or renal failure. White symbols indicate individuals without clinical sings of the disease. An oblique bar indicates a deceased individual. The arrows indicate the index patients. The genotype at the COL4A4 locus is indicated below each symbol as follows: −, wild type allele; +, mutated allele. The type of mutation is indicated in brackets: f = frameshift mutation; m = missense mutation; s = splice site mutation. In (a) case I-2 has microhaematuria probably not related to ATS.
Clinical and molecular data of reported patients
| Family/subject | Sex | Age | Urinalysis | Renal function | GBM abnormalities | Ophthalmic examination | Audiological examination | COL4A4 heterozygous mutation | COL4A4 heterozygous polymorphisms | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | Effect on coding | Exon/intron | |||||||||
| sequence | |||||||||||
| MVA/I1 | M | 55 | Microhaematuria | Normal | n.d. | Normal | Mild h.l. | 1884–1886del C | P629fsX652 | Exon 25 | 1, 4, 5, 11 |
| MVA/II1 | F | 25 | Microhaematuria proteinuria | Normal | Thinning, thickening | Normal | Normal | 1884–1886del C | P629fsX652 | Exon 25 | 1, 4, 6, 7, 8, 9, 10, 12 |
| MRM/II3 | M | 43 | Microhaematuria proteinuria | CRF at 40 year | Thickening | Normal | Normal | 1579–1581del G | G527fsX652 | Exon 22 | None |
| MRM/III1 | F | 15 | Microhaematuria proteinuria | Normal | n.d. | Normal | Normal | 1579–1581del G | G527fsX652 | Exon 22 | None |
| MRM/I1 | F | 50 | Microhaematuria | ESRD at 46 year | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| MRM/I2 | M | 68 | Microhaematuria | ESRD at 67 year | n.d. | Normal | s.h.l. | n.a. | n.a. | n.a. | n.a. |
| MRM/II1 | M | 44 | Microhaematuria proteinuria | Normal | n.d. | Normal | n.d. | n.a. | n.a. | n.a. | n.a. |
| MRM/II4 | F | 40 | Microhaematuria proteinuria | Normal | n.d. | Normal | n.d. | n.a. | n.a. | n.a. | n.a. |
| EVI/II2 | M | 33 | Microhaematuria proteinuria | Normal | Thinning | Normal | c.h.l. | 4493–4495 del G | G1498fsX1551 | Exon 46 | 5, 6, 7, 8, 9, 10, 11, 12 |
| EVI/I2 | M | 46 | Microhaematuria proteinuria | ESRD at 42 year | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| EVI/I3 | M | 60 | Microhaematuria proteinuria | n.d. | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| GEI/III2 | F | 7 | Microhaematuria | Normal | n.d. | Normal | Normal | 1837G>A | G613R | Exon 25 | 3, 4, 6, 7, 11, 12 |
| GEI/II2 | M | 45 | Microhaematuria | Normal | n.d. | Normal | Mild h.l. | 1837G>A | G613R | Exon 25 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| GEI/I1 | F | 60^ | n.d. | ESRD | n.d. | n.d. | n.d. | n.a. | n.a. | n.a. | n.a. |
| GEI/II4 | F | 41 | Microhaematuria | Normal | n.d. | n.d. | n.d. | 1837G>A | G613R | Exon 25 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| PUX/I1 | F | n.d. | n.d. | ESRD | n.d. | n.d. | n.d. | 940G>T | G314C | Exon 16 | None |
| PUX/II1 | M | 48 | Microhaematuria proteinuria | Normal | Thinning, splitting | n.d. | n.d. | 940G>T | G314C | Exon 16 | 6, 7, 8, 9, 10, 11, 12 |
| PUX/III1 | F | 7 | Microhaematuria | Normal | n.d. | Normal | Normal | 940G>T | G314C | Exon 16 | None |
| LCR/I2 | M | 76 | Microhaematuria | Normal | n.d. | Normal | s.h.l. | 1579G>T | G527C | Exon 22 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| LCR/II1 | F | 47 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | None |
| LCR/II3 | M | 40 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| LCR/III1 | F | 26 | Micro-macrohaematuria proteinuria | Normal | Thinning, thickening, splitting | Normal | Normal | 1579G>T | G527C | Exon 22 | 8, 9, |
| LCR/III2 | F | 21 | Micro-macrohaematuria | Normal | Thinning, thickening, splitting | Normal | Normal | 1579G>T | G527C | Exon 22 | None |
| LCR/III3 | M | 11 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | 2, 6, 7, 8, 9, 10, 11, 12 |
| DAG/I1 | M | 73 | Microhaematuria proteinuria | ESRD at 60 year | n.d. | Normal | Normal | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 4, 6, 7, 8, 9, 10, 11, 12 |
| DAG/II1 | M | 49 | Microhaematuria proteinuria | CRF | Thinning | n.d. | Normal | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 2, 4, 5, |
| DAG/III1 | M | 20 | Microhaematuria | Normal | n.d. | n.d. | n.d. | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 4, 6, 7, 8, 9, 10, 11, 12 |
| DAG/III2 | F | 11 | Microhaematuria | Normal | n.d. | n.d. | n.d. | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 5 |
| FRI/I2 | F | 66 | Microhaematuria proteinuria | ESRD | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| FRI/II2 | F | 64 | Microhaematuria proteinuria | ESRD at 60 year | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | None |
| FRI/II3 | M | 55 | Microhaematuria proteinuria | ESRD at 45 year | Thinning, thickening, splitting | Cataract post-tx | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III2 | M | 42 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III3 | F | 40 | Microhaematuria proteinuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III6 | M | 34 | Microhaematuria proteinuria | CRF | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | None |
| FRI/IV1 | F | 16 | Microhaematuria proteinuria | Normal | n.d. | n.d. | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV3 | F | 11 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV4 | M | 9 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV5 | F | 6 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 6, 7, 10, 11, 12 |
| Family/subject | Sex | Age | Urinalysis | Renal function | GBM abnormalities | Ophthalmic examination | Audiological examination | COL4A4 heterozygous mutation | COL4A4 heterozygous polymorphisms | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | Effect on coding | Exon/intron | |||||||||
| sequence | |||||||||||
| MVA/I1 | M | 55 | Microhaematuria | Normal | n.d. | Normal | Mild h.l. | 1884–1886del C | P629fsX652 | Exon 25 | 1, 4, 5, 11 |
| MVA/II1 | F | 25 | Microhaematuria proteinuria | Normal | Thinning, thickening | Normal | Normal | 1884–1886del C | P629fsX652 | Exon 25 | 1, 4, 6, 7, 8, 9, 10, 12 |
| MRM/II3 | M | 43 | Microhaematuria proteinuria | CRF at 40 year | Thickening | Normal | Normal | 1579–1581del G | G527fsX652 | Exon 22 | None |
| MRM/III1 | F | 15 | Microhaematuria proteinuria | Normal | n.d. | Normal | Normal | 1579–1581del G | G527fsX652 | Exon 22 | None |
| MRM/I1 | F | 50 | Microhaematuria | ESRD at 46 year | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| MRM/I2 | M | 68 | Microhaematuria | ESRD at 67 year | n.d. | Normal | s.h.l. | n.a. | n.a. | n.a. | n.a. |
| MRM/II1 | M | 44 | Microhaematuria proteinuria | Normal | n.d. | Normal | n.d. | n.a. | n.a. | n.a. | n.a. |
| MRM/II4 | F | 40 | Microhaematuria proteinuria | Normal | n.d. | Normal | n.d. | n.a. | n.a. | n.a. | n.a. |
| EVI/II2 | M | 33 | Microhaematuria proteinuria | Normal | Thinning | Normal | c.h.l. | 4493–4495 del G | G1498fsX1551 | Exon 46 | 5, 6, 7, 8, 9, 10, 11, 12 |
| EVI/I2 | M | 46 | Microhaematuria proteinuria | ESRD at 42 year | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| EVI/I3 | M | 60 | Microhaematuria proteinuria | n.d. | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| GEI/III2 | F | 7 | Microhaematuria | Normal | n.d. | Normal | Normal | 1837G>A | G613R | Exon 25 | 3, 4, 6, 7, 11, 12 |
| GEI/II2 | M | 45 | Microhaematuria | Normal | n.d. | Normal | Mild h.l. | 1837G>A | G613R | Exon 25 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| GEI/I1 | F | 60^ | n.d. | ESRD | n.d. | n.d. | n.d. | n.a. | n.a. | n.a. | n.a. |
| GEI/II4 | F | 41 | Microhaematuria | Normal | n.d. | n.d. | n.d. | 1837G>A | G613R | Exon 25 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| PUX/I1 | F | n.d. | n.d. | ESRD | n.d. | n.d. | n.d. | 940G>T | G314C | Exon 16 | None |
| PUX/II1 | M | 48 | Microhaematuria proteinuria | Normal | Thinning, splitting | n.d. | n.d. | 940G>T | G314C | Exon 16 | 6, 7, 8, 9, 10, 11, 12 |
| PUX/III1 | F | 7 | Microhaematuria | Normal | n.d. | Normal | Normal | 940G>T | G314C | Exon 16 | None |
| LCR/I2 | M | 76 | Microhaematuria | Normal | n.d. | Normal | s.h.l. | 1579G>T | G527C | Exon 22 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| LCR/II1 | F | 47 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | None |
| LCR/II3 | M | 40 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| LCR/III1 | F | 26 | Micro-macrohaematuria proteinuria | Normal | Thinning, thickening, splitting | Normal | Normal | 1579G>T | G527C | Exon 22 | 8, 9, |
| LCR/III2 | F | 21 | Micro-macrohaematuria | Normal | Thinning, thickening, splitting | Normal | Normal | 1579G>T | G527C | Exon 22 | None |
| LCR/III3 | M | 11 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | 2, 6, 7, 8, 9, 10, 11, 12 |
| DAG/I1 | M | 73 | Microhaematuria proteinuria | ESRD at 60 year | n.d. | Normal | Normal | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 4, 6, 7, 8, 9, 10, 11, 12 |
| DAG/II1 | M | 49 | Microhaematuria proteinuria | CRF | Thinning | n.d. | Normal | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 2, 4, 5, |
| DAG/III1 | M | 20 | Microhaematuria | Normal | n.d. | n.d. | n.d. | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 4, 6, 7, 8, 9, 10, 11, 12 |
| DAG/III2 | F | 11 | Microhaematuria | Normal | n.d. | n.d. | n.d. | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 5 |
| FRI/I2 | F | 66 | Microhaematuria proteinuria | ESRD | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| FRI/II2 | F | 64 | Microhaematuria proteinuria | ESRD at 60 year | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | None |
| FRI/II3 | M | 55 | Microhaematuria proteinuria | ESRD at 45 year | Thinning, thickening, splitting | Cataract post-tx | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III2 | M | 42 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III3 | F | 40 | Microhaematuria proteinuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III6 | M | 34 | Microhaematuria proteinuria | CRF | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | None |
| FRI/IV1 | F | 16 | Microhaematuria proteinuria | Normal | n.d. | n.d. | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV3 | F | 11 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV4 | M | 9 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV5 | F | 6 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 6, 7, 10, 11, 12 |
ESRD: end-stage renal disease; GBM: glomerular basement membrane; c.h.l.: conductive hearing loss; s.h.l.: sensoryneuronal hearing loss; n.d.: not defined; n.a.: DNA not available; : deceased; M: male; F: female, GBM: glomerular basement membrane.
Numbers in the last column refer to polymorphisms numbers of the Table 2.
Clinical and molecular data of reported patients
| Family/subject | Sex | Age | Urinalysis | Renal function | GBM abnormalities | Ophthalmic examination | Audiological examination | COL4A4 heterozygous mutation | COL4A4 heterozygous polymorphisms | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | Effect on coding | Exon/intron | |||||||||
| sequence | |||||||||||
| MVA/I1 | M | 55 | Microhaematuria | Normal | n.d. | Normal | Mild h.l. | 1884–1886del C | P629fsX652 | Exon 25 | 1, 4, 5, 11 |
| MVA/II1 | F | 25 | Microhaematuria proteinuria | Normal | Thinning, thickening | Normal | Normal | 1884–1886del C | P629fsX652 | Exon 25 | 1, 4, 6, 7, 8, 9, 10, 12 |
| MRM/II3 | M | 43 | Microhaematuria proteinuria | CRF at 40 year | Thickening | Normal | Normal | 1579–1581del G | G527fsX652 | Exon 22 | None |
| MRM/III1 | F | 15 | Microhaematuria proteinuria | Normal | n.d. | Normal | Normal | 1579–1581del G | G527fsX652 | Exon 22 | None |
| MRM/I1 | F | 50 | Microhaematuria | ESRD at 46 year | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| MRM/I2 | M | 68 | Microhaematuria | ESRD at 67 year | n.d. | Normal | s.h.l. | n.a. | n.a. | n.a. | n.a. |
| MRM/II1 | M | 44 | Microhaematuria proteinuria | Normal | n.d. | Normal | n.d. | n.a. | n.a. | n.a. | n.a. |
| MRM/II4 | F | 40 | Microhaematuria proteinuria | Normal | n.d. | Normal | n.d. | n.a. | n.a. | n.a. | n.a. |
| EVI/II2 | M | 33 | Microhaematuria proteinuria | Normal | Thinning | Normal | c.h.l. | 4493–4495 del G | G1498fsX1551 | Exon 46 | 5, 6, 7, 8, 9, 10, 11, 12 |
| EVI/I2 | M | 46 | Microhaematuria proteinuria | ESRD at 42 year | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| EVI/I3 | M | 60 | Microhaematuria proteinuria | n.d. | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| GEI/III2 | F | 7 | Microhaematuria | Normal | n.d. | Normal | Normal | 1837G>A | G613R | Exon 25 | 3, 4, 6, 7, 11, 12 |
| GEI/II2 | M | 45 | Microhaematuria | Normal | n.d. | Normal | Mild h.l. | 1837G>A | G613R | Exon 25 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| GEI/I1 | F | 60^ | n.d. | ESRD | n.d. | n.d. | n.d. | n.a. | n.a. | n.a. | n.a. |
| GEI/II4 | F | 41 | Microhaematuria | Normal | n.d. | n.d. | n.d. | 1837G>A | G613R | Exon 25 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| PUX/I1 | F | n.d. | n.d. | ESRD | n.d. | n.d. | n.d. | 940G>T | G314C | Exon 16 | None |
| PUX/II1 | M | 48 | Microhaematuria proteinuria | Normal | Thinning, splitting | n.d. | n.d. | 940G>T | G314C | Exon 16 | 6, 7, 8, 9, 10, 11, 12 |
| PUX/III1 | F | 7 | Microhaematuria | Normal | n.d. | Normal | Normal | 940G>T | G314C | Exon 16 | None |
| LCR/I2 | M | 76 | Microhaematuria | Normal | n.d. | Normal | s.h.l. | 1579G>T | G527C | Exon 22 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| LCR/II1 | F | 47 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | None |
| LCR/II3 | M | 40 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| LCR/III1 | F | 26 | Micro-macrohaematuria proteinuria | Normal | Thinning, thickening, splitting | Normal | Normal | 1579G>T | G527C | Exon 22 | 8, 9, |
| LCR/III2 | F | 21 | Micro-macrohaematuria | Normal | Thinning, thickening, splitting | Normal | Normal | 1579G>T | G527C | Exon 22 | None |
| LCR/III3 | M | 11 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | 2, 6, 7, 8, 9, 10, 11, 12 |
| DAG/I1 | M | 73 | Microhaematuria proteinuria | ESRD at 60 year | n.d. | Normal | Normal | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 4, 6, 7, 8, 9, 10, 11, 12 |
| DAG/II1 | M | 49 | Microhaematuria proteinuria | CRF | Thinning | n.d. | Normal | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 2, 4, 5, |
| DAG/III1 | M | 20 | Microhaematuria | Normal | n.d. | n.d. | n.d. | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 4, 6, 7, 8, 9, 10, 11, 12 |
| DAG/III2 | F | 11 | Microhaematuria | Normal | n.d. | n.d. | n.d. | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 5 |
| FRI/I2 | F | 66 | Microhaematuria proteinuria | ESRD | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| FRI/II2 | F | 64 | Microhaematuria proteinuria | ESRD at 60 year | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | None |
| FRI/II3 | M | 55 | Microhaematuria proteinuria | ESRD at 45 year | Thinning, thickening, splitting | Cataract post-tx | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III2 | M | 42 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III3 | F | 40 | Microhaematuria proteinuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III6 | M | 34 | Microhaematuria proteinuria | CRF | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | None |
| FRI/IV1 | F | 16 | Microhaematuria proteinuria | Normal | n.d. | n.d. | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV3 | F | 11 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV4 | M | 9 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV5 | F | 6 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 6, 7, 10, 11, 12 |
| Family/subject | Sex | Age | Urinalysis | Renal function | GBM abnormalities | Ophthalmic examination | Audiological examination | COL4A4 heterozygous mutation | COL4A4 heterozygous polymorphisms | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide change | Effect on coding | Exon/intron | |||||||||
| sequence | |||||||||||
| MVA/I1 | M | 55 | Microhaematuria | Normal | n.d. | Normal | Mild h.l. | 1884–1886del C | P629fsX652 | Exon 25 | 1, 4, 5, 11 |
| MVA/II1 | F | 25 | Microhaematuria proteinuria | Normal | Thinning, thickening | Normal | Normal | 1884–1886del C | P629fsX652 | Exon 25 | 1, 4, 6, 7, 8, 9, 10, 12 |
| MRM/II3 | M | 43 | Microhaematuria proteinuria | CRF at 40 year | Thickening | Normal | Normal | 1579–1581del G | G527fsX652 | Exon 22 | None |
| MRM/III1 | F | 15 | Microhaematuria proteinuria | Normal | n.d. | Normal | Normal | 1579–1581del G | G527fsX652 | Exon 22 | None |
| MRM/I1 | F | 50 | Microhaematuria | ESRD at 46 year | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| MRM/I2 | M | 68 | Microhaematuria | ESRD at 67 year | n.d. | Normal | s.h.l. | n.a. | n.a. | n.a. | n.a. |
| MRM/II1 | M | 44 | Microhaematuria proteinuria | Normal | n.d. | Normal | n.d. | n.a. | n.a. | n.a. | n.a. |
| MRM/II4 | F | 40 | Microhaematuria proteinuria | Normal | n.d. | Normal | n.d. | n.a. | n.a. | n.a. | n.a. |
| EVI/II2 | M | 33 | Microhaematuria proteinuria | Normal | Thinning | Normal | c.h.l. | 4493–4495 del G | G1498fsX1551 | Exon 46 | 5, 6, 7, 8, 9, 10, 11, 12 |
| EVI/I2 | M | 46 | Microhaematuria proteinuria | ESRD at 42 year | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| EVI/I3 | M | 60 | Microhaematuria proteinuria | n.d. | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| GEI/III2 | F | 7 | Microhaematuria | Normal | n.d. | Normal | Normal | 1837G>A | G613R | Exon 25 | 3, 4, 6, 7, 11, 12 |
| GEI/II2 | M | 45 | Microhaematuria | Normal | n.d. | Normal | Mild h.l. | 1837G>A | G613R | Exon 25 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| GEI/I1 | F | 60^ | n.d. | ESRD | n.d. | n.d. | n.d. | n.a. | n.a. | n.a. | n.a. |
| GEI/II4 | F | 41 | Microhaematuria | Normal | n.d. | n.d. | n.d. | 1837G>A | G613R | Exon 25 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| PUX/I1 | F | n.d. | n.d. | ESRD | n.d. | n.d. | n.d. | 940G>T | G314C | Exon 16 | None |
| PUX/II1 | M | 48 | Microhaematuria proteinuria | Normal | Thinning, splitting | n.d. | n.d. | 940G>T | G314C | Exon 16 | 6, 7, 8, 9, 10, 11, 12 |
| PUX/III1 | F | 7 | Microhaematuria | Normal | n.d. | Normal | Normal | 940G>T | G314C | Exon 16 | None |
| LCR/I2 | M | 76 | Microhaematuria | Normal | n.d. | Normal | s.h.l. | 1579G>T | G527C | Exon 22 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| LCR/II1 | F | 47 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | None |
| LCR/II3 | M | 40 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| LCR/III1 | F | 26 | Micro-macrohaematuria proteinuria | Normal | Thinning, thickening, splitting | Normal | Normal | 1579G>T | G527C | Exon 22 | 8, 9, |
| LCR/III2 | F | 21 | Micro-macrohaematuria | Normal | Thinning, thickening, splitting | Normal | Normal | 1579G>T | G527C | Exon 22 | None |
| LCR/III3 | M | 11 | Microhaematuria | Normal | n.d. | Normal | Normal | 1579G>T | G527C | Exon 22 | 2, 6, 7, 8, 9, 10, 11, 12 |
| DAG/I1 | M | 73 | Microhaematuria proteinuria | ESRD at 60 year | n.d. | Normal | Normal | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 4, 6, 7, 8, 9, 10, 11, 12 |
| DAG/II1 | M | 49 | Microhaematuria proteinuria | CRF | Thinning | n.d. | Normal | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 2, 4, 5, |
| DAG/III1 | M | 20 | Microhaematuria | Normal | n.d. | n.d. | n.d. | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 4, 6, 7, 8, 9, 10, 11, 12 |
| DAG/III2 | F | 11 | Microhaematuria | Normal | n.d. | n.d. | n.d. | IVS28+2 T>G | IVS28+2 T>G | Intron 28 | 5 |
| FRI/I2 | F | 66 | Microhaematuria proteinuria | ESRD | n.d. | Normal | Normal | n.a. | n.a. | n.a. | n.a. |
| FRI/II2 | F | 64 | Microhaematuria proteinuria | ESRD at 60 year | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | None |
| FRI/II3 | M | 55 | Microhaematuria proteinuria | ESRD at 45 year | Thinning, thickening, splitting | Cataract post-tx | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III2 | M | 42 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III3 | F | 40 | Microhaematuria proteinuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 8, 9, 10, 11, 12 |
| FRI/III6 | M | 34 | Microhaematuria proteinuria | CRF | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | None |
| FRI/IV1 | F | 16 | Microhaematuria proteinuria | Normal | n.d. | n.d. | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV3 | F | 11 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV4 | M | 9 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 5, 6, 7, 10, 11, 12 |
| FRI/IV5 | F | 6 | Microhaematuria | Normal | n.d. | Normal | Normal | IVS35+1G>A | IVS35+1G>A | Intron 35 | 2, 6, 7, 10, 11, 12 |
ESRD: end-stage renal disease; GBM: glomerular basement membrane; c.h.l.: conductive hearing loss; s.h.l.: sensoryneuronal hearing loss; n.d.: not defined; n.a.: DNA not available; : deceased; M: male; F: female, GBM: glomerular basement membrane.
Numbers in the last column refer to polymorphisms numbers of the Table 2.
COL4A4 mutations
The eight identified pathogenic mutations were all in a heterozygous state, private and previously not described (Supplementary Figure 1). Seven mutations were distributed in the collagenic domain, and one mutation was localized in the C-terminal domain (Figure 2a). Three were frameshift mutations leading to a protein truncation within the collagenous domain (families MVA and MRM) or in the C-terminal domain in one family (family EVI). Three were missense mutations leading to glycine substitutions in the collagenous domain (families GEI, PUX and LCR). Two were splice-site mutations localized in intron 28 and 35 (families DAG and FRI).
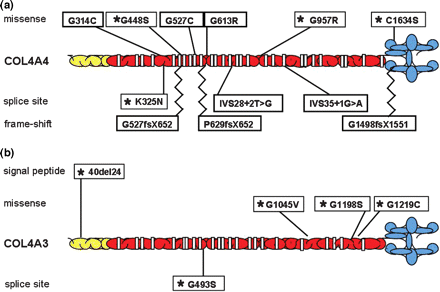
Distribution of pathogenetic mutations along COL4A4 (A) and COL4A3 (B) genes in ADAS patients. *mutations previously reported as associated with ADAS. All the others are novel mutations reported here.
During the DHPLC screening, we also identified 11 previously reported polymorphisms and one rare variant (Table 2).
Polymorphisms in the COL4A4 gene
| Nucleotide change | Effect on coding sequence | Exon/intron | |
|---|---|---|---|
| 1 | IVS5+86C>Ta | IVS5+86C>Ta | Intron 5 |
| 2 | 1444C>T | P482S | Exon 21 |
| 3 | 1634G>C | G545A | Exon 23 |
| 4 | IVS28–5C>T | IVS28–5C>T | Intron 28 |
| 5 | 3011T>C | L1004P | Exon 33 |
| 6 | 3594G>A | G1198G | Exon 39 |
| 7 | 3684G>A | K1228K | Exon 39 |
| 8 | 4080A>G | P1360P | Exon 42 |
| 9 | 3979A>G | M1327V | Exon 42 |
| 10 | 4204C>T | P1402S | Exon 44 |
| 11 | 4548A>G | V1516V | Exon 47 |
| 12 | 4932C>T | F1644F | Exon 48 |
| Nucleotide change | Effect on coding sequence | Exon/intron | |
|---|---|---|---|
| 1 | IVS5+86C>Ta | IVS5+86C>Ta | Intron 5 |
| 2 | 1444C>T | P482S | Exon 21 |
| 3 | 1634G>C | G545A | Exon 23 |
| 4 | IVS28–5C>T | IVS28–5C>T | Intron 28 |
| 5 | 3011T>C | L1004P | Exon 33 |
| 6 | 3594G>A | G1198G | Exon 39 |
| 7 | 3684G>A | K1228K | Exon 39 |
| 8 | 4080A>G | P1360P | Exon 42 |
| 9 | 3979A>G | M1327V | Exon 42 |
| 10 | 4204C>T | P1402S | Exon 44 |
| 11 | 4548A>G | V1516V | Exon 47 |
| 12 | 4932C>T | F1644F | Exon 48 |
aNot reported as polymorphism (rare variant).
Polymorphisms in the COL4A4 gene
| Nucleotide change | Effect on coding sequence | Exon/intron | |
|---|---|---|---|
| 1 | IVS5+86C>Ta | IVS5+86C>Ta | Intron 5 |
| 2 | 1444C>T | P482S | Exon 21 |
| 3 | 1634G>C | G545A | Exon 23 |
| 4 | IVS28–5C>T | IVS28–5C>T | Intron 28 |
| 5 | 3011T>C | L1004P | Exon 33 |
| 6 | 3594G>A | G1198G | Exon 39 |
| 7 | 3684G>A | K1228K | Exon 39 |
| 8 | 4080A>G | P1360P | Exon 42 |
| 9 | 3979A>G | M1327V | Exon 42 |
| 10 | 4204C>T | P1402S | Exon 44 |
| 11 | 4548A>G | V1516V | Exon 47 |
| 12 | 4932C>T | F1644F | Exon 48 |
| Nucleotide change | Effect on coding sequence | Exon/intron | |
|---|---|---|---|
| 1 | IVS5+86C>Ta | IVS5+86C>Ta | Intron 5 |
| 2 | 1444C>T | P482S | Exon 21 |
| 3 | 1634G>C | G545A | Exon 23 |
| 4 | IVS28–5C>T | IVS28–5C>T | Intron 28 |
| 5 | 3011T>C | L1004P | Exon 33 |
| 6 | 3594G>A | G1198G | Exon 39 |
| 7 | 3684G>A | K1228K | Exon 39 |
| 8 | 4080A>G | P1360P | Exon 42 |
| 9 | 3979A>G | M1327V | Exon 42 |
| 10 | 4204C>T | P1402S | Exon 44 |
| 11 | 4548A>G | V1516V | Exon 47 |
| 12 | 4932C>T | F1644F | Exon 48 |
aNot reported as polymorphism (rare variant).
Clinical data
Clinical features of the 38 affected individuals are described in Table 1. Their mean age is 34 years (range from 6 to 76 years). Six patients died with ESRD at the mean age of 58.3 years (range, 46–68 years). The main clinical manifestation was microscopic haematuria, which was present in 100% of patients. Gross haematuria occurred in 2/37 patients (5.4%). Proteinuria was present in 18/36 patients with known urinalysis (50%). Renal failure occurred in 12/37 patients (32.4%). Among these, three patients aged 34, 43 and 49 years presented CRF (creatinine 1.48 mg/dl and 1.53 mg/dl in the last two cases). The remaining nine patients developed ESRD: six patients reached ESRD after the age of 40 years, at a mean age of 51.2 year (range, 42–67 years), two died at 60 and 66 years of age and for the last patient no clinical data are available.
An ultrastructural examination of the GBM was performed in 8 patients (range of age at renal biopsy from 14 to 43 years) belonging to seven families. Alterations of GBM were identified in all. In six patients, the ultrastructural alterations were clearly diagnostic for ATS, showing a combination of thinning, thickening and splitting of the GBM, while in two patients isolated GBM thinning was identified (EVI-II2, age at biopsy 26 years and DAG II1, age at biopsy 29 years).
Bilateral hearing deficit in the 2000–8000 HZ range, with an onset after 40 years, developed in 4 of 30 tested patients (13.3%). Ocular changes were found in 1 of 30 tested patients (3.3%): this patient, aged 55 years, presented bilateral cataract post-renal transplantation attributed to steroid therapy.
Discussion
ATS and BFH are type IV collagen inherited disorders associated with heterozygous mutations in COL4A3 and COL4A4 genes. Till now, only 43 patients belonging to nine families with ADAS have been reported with either COL4A3 or COL4A4 mutations [11–15].
In this study, we showed the clinical outcome and the molecular data of 38 patients with ADAS belonging to eight families in whom a pathogenic COL4A4 mutation was identified (Table 1).
In this series of patients, haematuria, usually microscopic, was present in all cases with known urine analysis, while proteinuria was present in nearly 40% of patients. The development of renal failure was progressive with the age (Figure 3): only one patient (7.1%) younger than 40 years (1/14) presented CRF; two patients among those aged between 41 and 50 years (2/10; 20%) showed renal failure (one patient with ESRD and one patient with CRF); among patients aged between 51 and 60 years, four developed ERSD (4/6; 67%) and four out of five patients older than 60 years developed ESRD (80%).
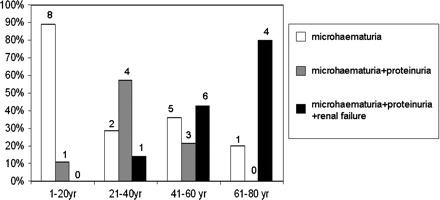
Graphic representing the phenotype of our 35 patients at different ages. Patients have been divided in four classes of age (0–20; 21–40; 41–60; 61–80) and they have been classified according to their renal involvement. The white bar represents patients with isolated microhaematuria, the grey bar represents cases with microhaematuria and proteinuria and the black bar represents patients with renal failure (both IRC and ESRD). Above each column, the absolute number of patients is reported.
GBM alterations were identified in all patients in whom a renal biopsy was performed. Only for one family, renal biopsy has not been performed. However, the hypothesis of a diagnosis of ATS is propped by the clinical manifestation of affected members of the family and by the identification of a disease segregating mutation in the COL4A4 gene leading to a glycine substitution in the collagenous domain of the protein [5]. The ultrastructural study of renal biopsy in almost all patients revealed a combination of thickening, splitting and thinning of the GBM. It is worth noting that two unrelated patients (aged 26 and 29 years) showed isolated thinning of GBM. This observation emphasizes the concept that thinning of the GBM is a non-specific finding and can be predictable of either BFH or ATS [15,19]. Recently, Voskarides K. et al. reported eight families with heterozygous COL4A3 mutations and a phenotype that in some patients was present in isolated haematuria while in older relatives progressed to CRF/ESRD [20]. The only ultrastructural finding in tested patients was a thinning of the GBM, and the authors defined these families as affected by TBMN [20]. Since TBMN is a term often associated with a benign prognosis, these patients could be rather classified as ADAS.
The extra-renal manifestations typical of ATS have not been frequently observed in our cohort of patients. Only 13% of patients developed sensorineural hearing loss that was slowly progressive. The only patient with ocular abnormalities in our series was a 55-year-old male patient who developed bilateral cataract post-renal transplantation, probably due to steroid treatment. The occurrence of congenital or early onset cataract has been already reported although in a minority of patients with a definitive diagnosis of ATS [5].
In all our patients, a heterozygous mutation in the COL4A4 gene has been identified. These mutations are of different type and are scattered throughout the gene. Heterozygous mutations in COL4A3 and COL4A4 are associated with a wide spectrum of phenotypes ranging from isolated microhaematuria to ESRD, as highlighted also by the intra- and inter-clinical variability reported in ADAS [16,17,21–23]. The intra-familial clinical variability observed in our cohort of patients was mainly due to the age at examination. However, some relatives showed a different clinical outcome at nearly the same age as in the case of family FRI where patient II3 developed ESRD at the age of 45 years while patient III2 showed isolated microhaematuria at the age of 42 years. The intra-familial clinical variability was more evident in families previously reported where even non-penetrant cases have been observed [15]. An inter-familial clinical variability was also present in our cases; in fact, while in nearly all families, older patients developed renal failure, in family LCR a male patient aged 76, and showed normal blood creatinine.
In order to explain such variability, we can hypothesize that the type and/or site of the mutation may be critical, even if genotype–phenotype correlations are made difficult by the fact that the mutations in these genes are often private and there are not mutational hot spots. However, analysing our cases and those already reported, it seems that there is not a clear correlation between type of mutation and phenotype. In fact, mutations of each type (missense, splice site and frameshift) and distributed throughout the genes are reported both in isolated microhaematuria segregating families and in families with progression towards ESRD (ADAS phenotype) (Figure 2a and b). The lack of a genotype–phenotype correlation is also highlighted by the different clinical outcome of patients of the same age, within a family, and thus bearing the same mutation. If the mutation alone cannot explain the clinical variability, we can suppose that the effect of a pathogenic mutation could be influenced by the presence of certain polymorphisms in the same genes and/or that functional variants in other proteins that are key players in renal filtration may act as modifiers.
We have compared the frequency of each sign among our patients and ADAS cases already reported in the literature, considering together patients with COL4A3 or COL4A4 gene mutations, given the low number of families reported with mutations in each gene (Table 3) [5,6,12–15]. Haematuria, usually microscopic, is the cardinal feature of the disease and was present in all patients with known urinanalysis, both in our patients and in patients previously reported. Proteinuria was present in about half of cases. Furthermore, our observations confirmed previously reported data about the slow progression towards ESRD and the lower occurrence of extra-renal signs [12–15].
| Present study | ADAS literature | ADAS total | XLAS males | XLAS females | |
|---|---|---|---|---|---|
| Number of patients | 38 | 43 | 79 | 218 | 349 |
| Microhaematuria | 100% (38/38) | 94.3% (33/35) | 97.3% (71/73) | 100% | 95.5% |
| Proteinuria | 50% (18/36) | 41.2% (14/34) | 45.7% (32/70) | 95% | 75.20% |
| Hearing loss | 13.3% (4/30) | 27% (10/37) | 20.9% (14/67) | 79% | 28% |
| Ocular lesions | 0/29 | 0 | 0 | 35.2% | 15% |
| ERSD | |||||
| Onset: <31 year | 0% (0/6) | 0% (0/8) | 0% (0/14) | 76.5% | 24% |
| Onset: 31–40 years | 0% (0/6) | 12.5% (1/8) | 7.1% (1/14) | 17.5% | 31% |
| Onset:>40 year | 100% (6/6) | 87.5% (7/8) | 92.8% (13/14) | 6% | 41% |
| Present study | ADAS literature | ADAS total | XLAS males | XLAS females | |
|---|---|---|---|---|---|
| Number of patients | 38 | 43 | 79 | 218 | 349 |
| Microhaematuria | 100% (38/38) | 94.3% (33/35) | 97.3% (71/73) | 100% | 95.5% |
| Proteinuria | 50% (18/36) | 41.2% (14/34) | 45.7% (32/70) | 95% | 75.20% |
| Hearing loss | 13.3% (4/30) | 27% (10/37) | 20.9% (14/67) | 79% | 28% |
| Ocular lesions | 0/29 | 0 | 0 | 35.2% | 15% |
| ERSD | |||||
| Onset: <31 year | 0% (0/6) | 0% (0/8) | 0% (0/14) | 76.5% | 24% |
| Onset: 31–40 years | 0% (0/6) | 12.5% (1/8) | 7.1% (1/14) | 17.5% | 31% |
| Onset:>40 year | 100% (6/6) | 87.5% (7/8) | 92.8% (13/14) | 6% | 41% |
| Present study | ADAS literature | ADAS total | XLAS males | XLAS females | |
|---|---|---|---|---|---|
| Number of patients | 38 | 43 | 79 | 218 | 349 |
| Microhaematuria | 100% (38/38) | 94.3% (33/35) | 97.3% (71/73) | 100% | 95.5% |
| Proteinuria | 50% (18/36) | 41.2% (14/34) | 45.7% (32/70) | 95% | 75.20% |
| Hearing loss | 13.3% (4/30) | 27% (10/37) | 20.9% (14/67) | 79% | 28% |
| Ocular lesions | 0/29 | 0 | 0 | 35.2% | 15% |
| ERSD | |||||
| Onset: <31 year | 0% (0/6) | 0% (0/8) | 0% (0/14) | 76.5% | 24% |
| Onset: 31–40 years | 0% (0/6) | 12.5% (1/8) | 7.1% (1/14) | 17.5% | 31% |
| Onset:>40 year | 100% (6/6) | 87.5% (7/8) | 92.8% (13/14) | 6% | 41% |
| Present study | ADAS literature | ADAS total | XLAS males | XLAS females | |
|---|---|---|---|---|---|
| Number of patients | 38 | 43 | 79 | 218 | 349 |
| Microhaematuria | 100% (38/38) | 94.3% (33/35) | 97.3% (71/73) | 100% | 95.5% |
| Proteinuria | 50% (18/36) | 41.2% (14/34) | 45.7% (32/70) | 95% | 75.20% |
| Hearing loss | 13.3% (4/30) | 27% (10/37) | 20.9% (14/67) | 79% | 28% |
| Ocular lesions | 0/29 | 0 | 0 | 35.2% | 15% |
| ERSD | |||||
| Onset: <31 year | 0% (0/6) | 0% (0/8) | 0% (0/14) | 76.5% | 24% |
| Onset: 31–40 years | 0% (0/6) | 12.5% (1/8) | 7.1% (1/14) | 17.5% | 31% |
| Onset:>40 year | 100% (6/6) | 87.5% (7/8) | 92.8% (13/14) | 6% | 41% |
Overall, the clinical outcome of ADAS seems to be close to XLAS in carrier females (Table 3). However, carriers of the XLAS have a higher incidence of proteinuria as presenting feature (75% versus 40–50%), while they have a lower progression towards ESRD than patients with ADAS, being reported in about 18% of females with XLAS [6]. Concerning the extra-renal manifestations, the development of hearing loss is comparable between the two groups (20–30%), while ocular lesions have never been reported in ADAS patients and are present in about 15% of XLAS carriers.
ADAS is suggested by a similar clinical outcome in males and females and vertical transmission of clinical signs. Clinically it is characterized by the presence of microhaematuria, ESRD onset usually after 40 years, ultrastructural changes of the GBM from isolated thinning to a combination of thinning, thickening and splitting, and presence of slowly progressive hearing loss in about 20% of cases. Even if ocular signs have never been reported, a low prevalence in ADAS cannot be excluded.
In conclusion, it is very difficult to predict the prognosis in a patient with a heterozygous mutation in either the COL4A3 or the COL4A4 gene. A correct diagnosis and prognosis is based on a combination of a comprehensive clinical investigation of all family members, including examination of renal and extra-renal signs of ATS in older members, associated with a broadly formal genetic analysis of the pedigree.
This work was supported by a FIRB grant (RBIP00PMF2) to AR. The authors thank Viviana Sanza for technical support.
Conflict of interest statement. None declared.


{kind=link}
{kind=link}
{kind=link}
Comments