





Abstract
Background. Chronic low oxygen in the tubulointerstitial area is a crucial cause of renal degradation and tubulointerstitial damage. Previous reports have suggested that the maintenance of renal blood flow plays a role in the suppression of progressive renal damage. Neovascularization is important for the maintenance of blood flow. We studied the production of angiogenic factors by culturing renal proximal tubular epithelial cells (PTEC) under hypoxic conditions.
Methods. Cultured PTEC were exposed to normal and low-oxygen conditions. The levels of angiogenin (ANG) and vascular endothelial growth factor (VEGF) in the cell supernatants were measured by enzyme-linked immunosorbent assay. The messenger RNAs (mRNAs) of ANG and VEGF in the PTEC were examined by real-time reverse transcriptase polymerase chain reaction (real-time RT–PCR). The presence of ANG, VEGF and hypoxia-inducible factor-1 (HIF-1) was studied by immunofluorescence techniques. The effect of cobalt chloride (CoCl2), which is an HIF-1 inducer, on the production of ANG and VEGF was also examined in order to elucidate the contribution of the HIF-1 pathway to the production of these cytokines.
Results. ANG and VEGF were demonstrated to exist in the cell supernatants, and ANG and VEGF mRNAs were detected in the PTEC. Hypoxic conditions stimulated the secretion of ANG (2.5-fold vs normoxia, P<0.001) and VEGF (3.2-fold vs normoxia, P<0.001) by PTEC. Hypoxic conditions increased the mRNA expression of ANG for 6 h (1.38-fold vs normoxia, P<0.05) and VEGF for 24 h (2.04-fold vs normoxia, P<0.01). Hypoxic conditions also enhanced ANG, VEGF and HIF-1 protein expression in PTEC. The CoCl2 increased the secretion of ANG (5.2-fold vs control, P<0.0001) and VEGF (2.3-fold vs control, P<0.0001) by PTEC.
Conclusion. Under hypoxic conditions, the ANG and VEGF secreted by PTEC may modulate angiogenesis and vascular remodeling in the renal interstitium via an increase in the production of HIF-1.
Introduction
It is well known that several factors are related to the progression of kidney diseases to end-stage renal failure. Histopathological studies have revealed that tubulointerstitial damage correlates with the decrease in renal function and the worsening of renal prognosis [1]. The precise mechanism by which a tubulointerstitial disorder degrades renal function is uncertain. Tubular damage may cause disorders of the tubulo-glomerular feedback, thus resulting in the damage of glomerular function [2]. Severe tubular damage causes the formation of atubular glomeruli and the decrease of functional nephrons. As tubulointerstitial damage progresses, peritubular microvascular loss increases significantly [3].
Chronic low oxygen in the tubulointerstitial area is an important cause of renal degradation and tubulointerstitial damage [4,5]. The vascular endothelial growth factor (VEGF), also known as the vascular permeability factor, is an endothelial cell-specific mitogen in vitro, and it is considered to be an important regulator of both physiological and pathological neovascularization. It has been reported that VEGF improves renal blood flow, suppresses renal fibrosis and maintains renal function [6]; this report suggests that the maintenance of renal blood flow plays a role in the suppression of progressive renal damage. Thus, neovascularization may play an important role in the maintenance of renal blood flow. Angiogenin (ANG) is one of the most potent angiogenic factors in vivo. However, little is known about ANG expression and its function in the kidney, despite the fact that ANG is present in human renal-cell carcinoma and in the kidney cells of the human embryo [7,8].
Hypoxia-inducible factor-1 (HIF-1) is a major transcription factor specifically activated during hypoxia. This transcription factor is composed of two subunits, HIF-1α and HIF-1β. HIF-1β is constitutively expressed, whereas HIF-1α is targeted for proteosome degradation by prolyl 4-hydroxylase. It is known that VEGF shows HIF-1-dependent hypoxia-inducible expression in cell cultures [9], and that the activation of the HIF-1 pathway by the addition of prolyl 4-hydroxylase inhibitors stimulates angiogenesis [10]. However, the relationship between ANG and HIF-1 has not been investigated.
In this study, we compared ANG synthesis in human PTEC under hypoxic conditions with VEGF synthesis. Furthermore, we investigated the role of HIF-1 in the synthesis of these cytokines in PTEC.
Materials and methods
Cell culture and hypoxic conditions
A primary human PTEC cell line was purchased from BioWhittaker (Walkersville, MD). The cells were cultured with renal epithelial cell based medium (REBM) supplemented with 5 µg/ml of insulin, 10 µg/ml of transferrin, 0.5 µg/ml of hydrocortisone, 6.5 ng/ml of trio-iodothyronine, 0.5 µg/ml of epinephrine, 10 ng/ml of epidermal growth factor and 5% heat-inactivated foetal calf serum (FCS) (all from BioWhittaker) at 37°C in 5% CO2. The PTEC between passages three and eight were used for the experiments.
The hypoxic condition (an atmosphere of 1% oxygen) was achieved by using an anaerobic chamber equipped with an aerating agent (Anaeropack, Mitsubishi Gas Chemical, Tokyo, Japan).
Evaluation of cytotoxicity under the hypoxic conditions
The cytotoxicity of the hypoxic condition was evaluated by assessing the lactate dehydrogenase (LDH) release from the PTEC. The PTEC were incubated in 12-well plates (Becton Dickinson, Franklin Lakes, NJ) for 24 h under hypoxic conditions, and the LDH levels in the cell supernatants were measured. After removing the cell supernatants, the PTEC were lysed by mellitin (50 µg/ml, Sigma, St Louis, MO) and the LDH levels in the cell lysates were also measured. The LDH was quantified by a colorimeter using an LDH assay kit (Sanassay LDH, Sankou-Junyaku, Inc., Tokyo, Japan).
Assay of ANG and VEGF
The confluent PTEC cultures on the 12-well plates were washed twice with Hank's balanced salt solution (HBSS, Becton Dickinson) and incubated in REBM without FCS for 24 h under normal or hypoxic conditions. Culture supernatants were collected at 6, 12 or 24 h, and stored at −80°C after centrifugation, which was carried out to remove cell debris. After removing the cell supernatants, the cells were lysed with 1 N NaOH. The cell lysates were subjected to the Lowry method to determine the protein content in each well. The concentrations of ANG and VEGF in the culture supernatants were measured using a commercially available enzyme-linked immunosorbent assay (ELISA) kit for human ANG and VEGF (R&D, O’Fallon, MO), in accordance with the manufacturer's instructions. The levels of ANG and VEGF were expressed as picograms per microgram of cell protein.
Quantitative analysis of ANG and VEGF mRNAs by real-time RT–PCR
The confluent PTEC cultured on the six-well plates (Becton Dickinson) were incubated in REBM without FCS for 24 h. Then the cells were incubated in REBM without FCS for 6 h and 24 h under normal or hypoxic conditions.
Total RNA was extracted from the cells using QIA shredder and RNeasy Protect Mini Kit (QIAGEN, Valencia, CA). The RNA was transcribed into the first-strand cDNA with the Omniscript RT kit (QIAGEN) according to the manufacturer's description. Quantitative RT–PCR was performed using an ABI PRISM 7700 Sequence Detector (Applied Biosystems, Foster City, CA) with TaqMan Universal PCR Master Mix (Applied Biosystems). The specific primers and the probes were purchased from Applied Biosystems for detecting human ANG (Assay ID: Hs02379000_s1), human VEGF (Assay ID: Hs00173626_m1) and human glyceraldehyde-3-phosphatase-dehydrogenase (GAPDH) (Assay ID: Hs99999905_m1). The ANG, VEGF and GAPDH mRNA levels were quantified separately using standard curves of diluted standard cDNA. For quantitative analysis, the relative amounts of ANG and VEGF mRNA were normalized to GAPDH as the housekeeping gene.
Immunofluorescence study of ANG, VEGF and HIF-1α
PTEC were cultured on sterile slide glasses. When the cells were grown, they were incubated in REBM without FCS for 24 h under normal or hypoxic conditions. Thereafter, they were fixed with 99.5% methanol (Wako, Tokyo, Japan) at room temperature for 5 min. Then the cells were stained for ANG, VEGF and HIF-1α. Anti-human ANG goat antibody, anti-human VEGF goat antibody and anti-human HIF-1α goat antibody were used as first antibodies in the indirect immunofluorescence technique. All antibodies were purchased from R&D Systems, Inc. The FITC-conjugated rabbit anti-goat IgG (DAKO, Copenhagen, Denmark) was used as the second antibody. The negative controls were stained with normal goat serum, followed by FITC-conjugated rabbit anti-goat IgG. The cells were observed with a Zeiss Axiophoto equipped with Axiovision software (Carl Zeiss, Jena, Germany). Axiophoto images were acquired with an AxioCam camera and processed with Axiovision software. Confocal images were processed with Adobe Photoshop.
Effect of cobalt chloride (CoCl2) on ANG and VEGF production
The HIF-1 is a major transcription factor, specifically activated during hypoxia. Most angiogenic cytokines are mediated via HIF-1, and CoCl2 is known as a chemical inducer of HIF-1, so CoCl2 (150 μM) was added to the culture medium to investigate the role of HIF-1 in the production of ANG and VEGF. The PTEC were incubated with 150 μM CoCl2 for 24 h under normoxic and hypoxic conditions. Then, the concentrations of ANG and VEGF in the supernatants were measured with ELISA kits.
Effect of ANG and VEGF on PTEC proliferation
The PTEC were placed on 96-well plates (Becton Dickinson) at a density of 5000 cells/well and a final volume of 100 µl, and were incubated with REBM without FCS including various concentrations of human ANG (10 pg/ml–10 ng/ml) or human VEGF (10 pg/ml–10 ng/ml) for 24 h. Thereafter, the proliferation of PTEC was evaluated by colorimetric assay using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) dye (Promega, Madison, WI). Briefly, the cells were incubated for 2 h after the addition of a 20 µl MTT solution to each well, and the absorbance at 550 nm with a reference filter of 650 nm was measured by an ELISA reader. The REBM with 5% FCS was used as positive control.
Statistical analysis
All data are expressed as means±standard deviation. The statistical analysis was performed with the unpaired t-tests for comparison of two groups. The analysis of variance and Fisher's PLSD were used for comparison among multiple groups. Values of P<0.05 were considered to be significant.
Results
Cytotoxicity under hypoxic conditions
The LDH activity in the culture supernatants and cell lysates was measured after 24 h exposure to normoxia and hypoxia. There was no significant change in the LDH levels in the culture supernatants and cell lysates under hypoxic conditions, as compared with normoxic conditions, indicating that cell viability was not affected by 24 h exposure to hypoxia.
ANG and VEGF secretion under hypoxic conditions
The time course of ANG secretion by PTEC is shown in Figure 1A. The secretion of ANG increased in a time-dependent manner in the cells exposed to hypoxia. After 24 h of culturing, hypoxia induced a significantly greater ANG secretion than normoxia (17.1±4.0 pg/µg protein under hypoxia and 6.9±1.5 pg/µg protein under normoxia, P<0.001).
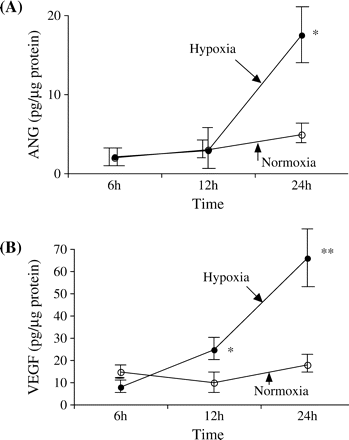
(A) The time course of angiogenin (ANG) secretion by proximal tubular epithelial cells (PTEC) is shown. The secretion of ANG increased in a time-dependent manner in the cells exposed to hypoxia. After 24 h of culturing, hypoxia (filled circles) induced a significantly greater ANG secretion than normoxia (open circles). *P<0.001 vs normoxia. Values are means±SD for four wells, and representative data from two separate experiments are shown. (B) The time course of vascular endothelial growth factor (VEGF) secretion by PTEC is shown. The secretion of VEGF increased in a time-dependent manner in the cells exposed to hypoxia. After 12 and 24 h of culturing, hypoxia (filled circles) induced a significantly greater VEGF secretion than normoxia (open circles). *P<0.05 vs normoxia, **P<0.001 vs normoxia. Values are means±SD for four wells, and representative data from two separate experiments are shown.
The time course of VEGF secretion by PTEC is shown in Figure 1B. The secretion of VEGF increased in a time-dependent manner in the cells exposed to hypoxia. After 12 and 24 h of culturing, hypoxia induced a significantly greater VEGF secretion than normoxia (12 h: 26.6±5.6 pg/µg protein under hypoxia and 10.3±4.0 pg/µg protein under normoxia, P<0.05; 24 h: 65.7±19.0 pg/µg protein under hypoxia and 20.5±6.0 pg/µg protein under normoxia, P<0.001).
ANG and VEGF mRNA expression under hypoxic conditions
The time course of ANG mRNA expression in PTEC is shown in Figure 2A. The average amount of ANG mRNA at 6 h under normoxic conditions was set to 1.0. The relative ANG mRNA expression under hypoxic conditions was significantly higher than normoxic conditions both at 6 h (normoxia: 1.00±0.16 vs hypoxia: 1.384±0.25, P<0.05) and 24 h (normoxia: 0.92±0.06 vs hypoxia: 1.24±0.31, P<0.05).
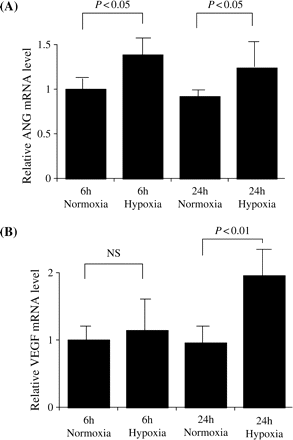
(A) Relative ANG mRNA expression in normoxic or hypoxic conditions after 6 and 24 h incubation. The average amounts of ANG mRNA at 6 h in normoxic conditions was set to 1.0. Relative ANG mRNA expression in hypoxic conditions was significantly increased both at 6 and 24 h (P<0.05) compared with the relative ANG mRNA expression in normoxic conditions at the same time points. Values are means±SD for six wells. (B) Relative VEGF mRNA expression in normoxic or hypoxic conditions for 6 and 24 h incubation. The average amount of VEGF mRNA at 6 h in normoxic conditions was set to 1.0. Relative VEGF mRNA expression in hypoxic conditions was not changed at 6 h, while it was significantly increased at 24 h (P<0.01) compared with the VEGF mRNA expression in normoxic conditions. Values are means±SD for six wells.
The time course of VEGF mRNA expression by PTEC is shown in Figure 2B. Average amount of VEGF mRNA at 6 h under normoxic conditions was set to 1.0. The relative VEGF mRNA expression under hypoxic conditions was not changed at 6 h (normoxia: 1.00±0.25 vs hypoxia: 1.14±0.63, P = NS), while it was significantly higher than normoxic conditions at 24 h (normoxia: 0.96±0.26 vs hypoxia: 1.96±0.41, P<0.01).
Immunofluorescence study of ANG, VEGF and HIF-1α
In the immunofluorescence study, the enhancement of ANG and VEGF staining was observed in the cytoplasm of PTEC exposed to hypoxia. HIF-1 was also strongly stained in the cell nuclei of PTEC exposed to hypoxia (Figure 3).
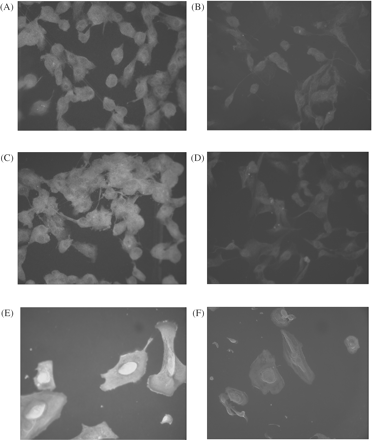
The enhancement of ANG staining was observed in the cytoplasm of PTEC exposed to (A) hypoxia in comparison with (B) normoxia. The enhancement of VEGF staining was observed in the cytoplasm of PTEC exposed to (C) hypoxia in comparison with (D) normoxia. Hypoxia enhanced hypoxia inducible factor-1α (HIF-1α) expression in the (E) cell nuclei compared with (F) normoxia. No immunoreactivity was obtained in the absence of primary antibody. (A–D): original magnification 200×. (E, F): original magnification 400×.
Effect of CoCl2 on the synthesis of ANG and VEGF
As shown in Figure 4, the secretions of ANG and VEGF increased under normoxic conditions after treatment with CoCl2 (ANG: 11.9±3.4 pg/μg protein in the controls and 61.8±5.4 pg/µg protein in the CoCl2-stimulated cells, P<0.0001; VEGF: 46.8±4.9 pg/µg protein in the controls and 109.0±16.0 pg/µg protein in the CoCl2-stimulated cells, P<0.0001). Although hypoxic conditions stimulated the secretions of ANG and VEGF compared with normoxic conditions (ANG: P<0.001, VEGF: P<0.005), CoCl2 did not further increase the production of ANG and VEGF in the PTEC exposed to hypoxia.
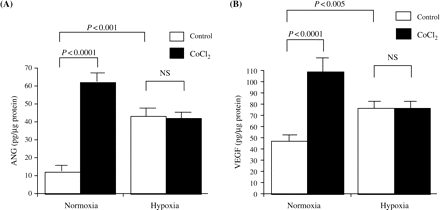
(A) The secretion of ANG increased under normoxic conditions after treatment with 150 μM CoCl2 (P<0.0001). Under hypoxia, the secretion of ANG increased compared with normoxia (P<0.001) and further enhancement by CoCl2 was not observed. Values are means±SD for three wells, and representative data from one of two experiments are shown. (B) The secretion of VEGF increased under normoxic conditions after treatment with 150 μM CoCl2 (P<0.0001). Under hypoxia, the secretion of VEGF increased compared with normoxia (P<0.005) and further enhancement by CoCl2 was not observed. Values are means±SD for three wells, and representative data from three separate experiments are shown.
Effect of ANG and VEGF on PTEC proliferation
Although 5% FCS significantly stimulated PTEC proliferation, neither ANG nor VEGF (10 pg/ml–10 ng/ml) affected PTEC proliferation (Figure 5).
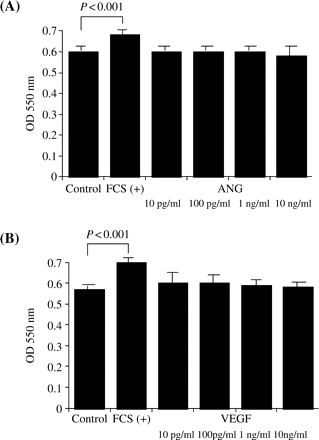
The PTEC were incubated with REBM without FCS including various concentrations of human ANG (10 pg/ml–10 ng/ml) or human VEGF (10 pg/ml–10 ng/ml) for 24 h, and the proliferation of PTEC was evaluated by colorimetric assay using MTT. Although 5% FCS significantly stimulated PTEC proliferation (P<0.001 vs control), neither (A) ANG nor (B) VEGF affected PTEC proliferation. Values are means±SD for six wells, and representative data from two separate experiments are shown.
Discussion
Hypoxic tubulointerstitial lesions play an important role in tubular and interstitial damage and the degradation of renal function [4]. There is some new evidence emerging that renal vascular changes contribute to the progression of renal disease, and that the alteration of VEGF expression might play an important role in microvascular loss or macrovascular remodelling in the kidney [4]. ANG and VEGF are cytokines known as proangiogenic factors, in addition to hepatocyte growth factor and basic fibroblast growth factor [11].
Microvascular endothelial loss in both the glomerular and the peritubular capillaries in progressive renal disease is directly linked to impaired blood flow, the development of renal ischaemia and scarring. VEGF is a proliferative survival factor for endothelial cells, which might preserve the stressed endothelium or stimulate angiogenesis, stabilizing the renal function and slowing down histologic progression, as shown by different animal models of progressive renal disease [5,12]. Recently, it has been suggested that VEGF may be a survival factor for the renal tubular epithelium in vivo [13].
The ANG, a 14.2 kDa protein originally isolated from a medium conditioned by HT-29 human colon adenocarcinoma cells and subsequently from normal serum, is one of the most potent angiogenic factors in vivo [14]. It binds to the cell-surface actin on the endothelial cells, and is then internalized and translocated to the nucleus [13]. Although its physicochemical properties and receptor-binding mechanisms have been characterized in detail, little is known about its role in neovascularization. Previous experiments have shown that monoclonal antibody against human ANG suppresses the growth of human adenocarcinoma in nude mice by reducing tumour neovascularization, supporting the idea that ANG plays an important function in angiogenesis [15]. However, the mechanisms underlying the induction of angiogenesis are still poorly understood. Recently, several endothelial growth factors with angiogenic activity have been described, including the fibroblast growth factors, VEGF, platelet-derived growth factor and ANG [16].
In the present study, we demonstrated for the first time that ANG protein is present in the culture supernatants of PTEC, and that ANG mRNA is detectable in PTEC. The expression of ANG has been previously reported in cultured newborn-hamster kidney cells [8] but not in human PTEC, although VEGF expression has been demonstrated in PTEC. In our study, hypoxia stimulated the secretion of ANG and VEGF by PTEC. Hypoxia also increased the mRNA expressions of ANG and VEGF. Our immunofluorescence study revealed that hypoxia enhanced the expression of ANG and VEGF protein in PTEC. Considering the fact that these cytokines exhibit their biological effects through different mechanisms, it seems that ANG plays as important a role in the neovascularization of the adult kidney as VEGF.
When the cells are exposed to hypoxia, the regulation of gene expression is an important adaptive response to the reduced availability of oxygen. For a number of groups of mammalian genes typically induced by hypoxia, regulatory studies have implicated an inducible transcriptional complex termed HIF-1 [17]. HIF-1α is kept at a lower or an undetectable level and HIF-1α is induced to a striking degree in hypoxia. It is known that cobalt activates the HIF-1 signal in spite of normal oxygen pressure through the inhibition of HIF-1α degradation [18].
In our experiment, it was shown that HIF-1α expression in the cell nuclei was increased by the hypoxic condition in PTEC. Increases in ANG and VEGF production were observed subsequent to the addition of CoCl2, which is a chemical inducer of HIF-1α. This shows precisely that, under hypoxia, ANG is stimulated via the HIF-1 pathway in PTEC, as is VEGF. The CoCl2 did not increase the secretion of ANG and VEGF by PTEC exposed to hypoxia. This may be explained by the hypothesis that HIF-1α was fully expressed in PTEC, and that CoCl2 did not further enhance HIF-1 activity. It seems likely that, under hypoxia, PTEC induces neovascularization in tubulointerstitial lesions via ANG and VEGF secretion. Neither ANG nor VEGF had a proliferative effect on PTEC in an autocrine or paracrine fashion. The ANG and VEGF secreted by PTEC may act mainly on the vascular endothelial cells in the tubulointerstitium.
In summary, we showed that PTEC secreted ANG and VEGF in higher quantities under hypoxia. We also demonstrated that ANG and VEGF mRNAs were increased in PTEC under hypoxic condition. The HIF-1 expression was enhanced by hypoxia, and CoCl2 also increased the secretion of ANG and VEGF. The ANG and VEGF in PTEC may play an important role in the tubulointerstitial neovascularization induced by hypoxia via the HIF-1 mechanism.
Conflict of interest statement. None declared.
References
Fine LG, Orphanides C, Norman JT. Progressive renal disease: the chronic hypoxia hypothesis.
Johnson RJ, Schreiner GF. Hypothesis: the role of acquired tubulointerstitial disease in the pathogenesis of salt-dependent hypertension.
Kim YG, Suga SI, Kang DH et al. Vascular endothelial growth factor accelerates renal recovery in experimental thrombotic microangiopathy.
Fine LG, Bandyopadhay D, Norman JT. Is there a common mechanism for the progression of different types of renal diseases other than proteinuria? Towards the unifying theme of chronic hypoxia.
Kang DH, Johnson RJ. Vascular endothelial growth factor: a new player in the pathogenesis of renal fibrosis.
Kang DH, Hughes J, Mazzali M, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function.
Moenner M, Gusse M, Hatzi E, Badet J. The widespread expression of angiogenin in different human cells suggests a biological function not only related to angiogenesis.
Kurachi K, Rybak SM, Fett JW et al. Expression of human angiogenin in cultured baby hamster kidney cells.
Forsythe JA, Jiang BH, Iyer NV et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1.
Warnecke C, Griethe W, Weidemann A et al. Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors.
Moroianu J, Riordan JF. Nuclear translocation of angiogenin in proliferating endothelial cells is essential to its angiogenic activity.
Kang DH, Kanellis J, Hugo C et al. Role of the microvascular endothelium in progressive renal disease.
Kanellis J, Fraser S, Katerelos M, Power DA. Vascular endothelial growth factor is a survival factor for renal tubular epithelial cells.
Fett JW, Strydom DJ, Lobb RR et al. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells.
Olson KA, French TC, Vallee BL, Fett JW. A monoclonal antibody to human angiogenin suppresses tumor growth in athymic mice.
Ebert BL, Firth JD, Ratcliffe PJ. Hypoxia and mitochondorial inhibitors regulate expression of glucose transporter-1 via distinct cis-acting sequences.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments