





Abstract
The transcription factor c-Myb is involved in early differentiation and proliferation of haematopoietic cells, where it operates as a regulator of self-renewal and multi-lineage differentiation. Deregulated c-Myb plays critical roles in leukaemias and other human cancers. Due to its role as a master regulator, we hypothesized it might function as a pioneer transcription factor. Our approach to test this was to analyse a mutant of c-Myb, D152V, previously reported to cause haematopoietic defects in mice by an unknown mechanism. Our transcriptome data from K562 cells indicates that this mutation specifically affects c-Myb's ability to regulate genes involved in differentiation, causing failure in c-Myb's ability to block differentiation. Furthermore, we see a major effect of this mutation in assays where chromatin opening is involved. We show that each repeat in the minimal DNA-binding domain of c-Myb binds to histones and that D152V disrupts histone binding of the third repeat. ATAC-seq data indicates this mutation impairs the ability of c-Myb to cause chromatin opening at specific sites. Taken together, our findings support that c-Myb acts as a pioneer factor and show that D152V impairs this function. The D152V mutant is the first mutant of a transcription factor specifically destroying pioneer factor function.
INTRODUCTION
Pioneer transcription factors have been described as a subclass of transcription factors able to associate with closed chromatin independently of other factors and thereby capable to modulate chromatin accessibility. Upon binding, pioneer factors increase the nucleosomal accessibility of their target site and thereby allow access to other transcription factors and chromatin modifiers (1,2). The hierarchical binding of transcription factors, in which the pioneer factors bind first, has been observed in several cell types, including the haematopoietic system (3) and appears to employ a chromatin opening step prior to lineage commitment (4–7). By changing the chromatin landscape and recruiting activators or repressors that by themselves are unable to engage with silent chromatin (1,8), the pioneer factors act as master regulators able to change downstream gene regulatory networks and cell identity. In line with this, key pluripotency factors such as Oct4, Sox2 and Klf4 have been defined as pioneer factors (9). In fact, it appears that the most potent reprogramming transcription factors are pioneer factors (10,11).
The transcription factor c-Myb is highly expressed in haematopoietic progenitor cells and plays a key role in regulating expression of genes involved in differentiation and proliferation of these cells (12). c-Myb has also been found to act as a regulator in non-haematopoietic cells, such as progenitor cells in the colonic crypts and a neurogenic region in the adult brain. However, the requirement for c-Myb is most evident in the haematopoietic system (13–16). Here, c-Myb is required for the normal development of progenitor cells, and its downregulation is essential for their terminal differentiation. c-Myb appears to be involved at multiple stages of haematopoiesis, being required for the development of hematopoietic precursors rather than for their generation (17–19). In adult hematopoietic stem cells, c-Myb operates as a regulator of self-renewal and multi-lineage differentiation (20). In situations where high c-Myb levels are maintained, normal haematopoietic differentiation is suppressed and leukaemic transformation may be promoted (12,13). This is the case in many human lymphoid and myeloid acute leukaemias (21). Due to its role in lineage determination and control of other transcription factors, c-Myb has been described as a master regulator (22–25). This raises the possibility that c-Myb may in fact operate as a pioneer factor, with prospects of c-Myb biology shedding light on our understanding of pioneer factors. Likewise, pioneer properties may clarify c-Myb's role in human cancers.
Several mouse models with lowered expression or decreased activity of c-Myb have been developed to study c-Myb's role in haematopoiesis (18,26–28). One of these mouse models was generated by inducing mutations in the MYB gene and was found to have elevated levels of megakaryocytes and increased platelet production as well as decreased levels of lymphocytes (27). These mice, named Plt3 mice, harboured a mutation in the MYB gene resulting in a c-Myb protein bearing an amino acid substitution of valine for aspartate at residue 152 (D152V) within its DNA-binding domain (DBD). Overexpression of c-Myb inhibits erythroid and myeloid differentiation (29,30), whereas mice with reduced levels of c-Myb have reduced levels of cells of lymphoid origin (18). The phenotype of the Plt3 mice, therefore, suggests that this mutant represents a less active version of c-Myb, but the molecular mechanisms underlying this phenotype remain to be elucidated.
The c-Myb D152V mutant seems to influence the development of the haematopoietic system significantly. Therefore, this mutant may help to improve our understanding of how c-Myb regulates haematopoiesis and its putative role as a pioneer factor. In the present work, we show that the D152V mutation specifically affects the regulation of genes essential for differentiation and cell development, resulting in increased erythroid differentiation of K562 cells. Furthermore, this effect on gene regulation seems to be caused by an impaired ability of c-Myb D152V to induce chromatin opening due to a weakened histone interaction, suggesting a direct influence on pioneer function. Considering c-Myb's role in differentiation and lineage commitment of haematopoietic cells, we propose that c-Myb regulates transcription of genes essential for haematopoiesis by acting as a pioneer factor. The D152V mutation seems to impair specifically c-Myb's pioneer factor activity and might, therefore, represent a unique mutant and an interesting model to understand mechanisms of c-Myb pioneer factor function.
MATERIALS AND METHODS
Cell culture, transfections and reporter assays
Four cell lines were used in this work: K562 (ATCC® CCL-243™Homo sapiens bone marrow, chronic myelogenous leukaemia), HD-11 (31) (Gallus gallus macrophage), CV-1 (ATCC® CCL-70™ Cercopithecus aethiops kidney Normal) and COS-1 (ATCC® CRL-1650™ Cercopithecus aethiops kidney).
The cells were cultured as described in (32) and K562 cells stably expressing TY-tagged c-Myb variants (empty vector, 3xTY1-c-Myb, 3xTY-c-Myb D152V) were generated as described in (33). For transfection of K562 cells we used Ingenio Electroporation Solution (Mirus Bio) following the manufacture's guidelines. K562 cells were transfected with 200 pmol siGENOME (Dharmacon) or si2992 (described in (23)). The electroporation was performed in an Amaxa nuclefector II (Lonza) using program T-16. HD-11 cells, CV-1 cells and COS-1-cells were seeded and transfected with plasmids using the TransIT-LT transfection reagent (Mirus Bio) as described in (34). RNA was isolated from HD-11 cells 24 h after transfection and qRT-PCR performed as described in Supplementary materials and methods. Twenty-four hours after transfection COS-1 cells were lysed in interaction buffer (20 mM HEPES pH 7.6, 10% glycerol, 0.2% Triton-X-100, 150 mM KAc, 1 mM Dithiothreitol (DTT)) supplemented with Complete protease inhibitor cocktail (Roche) and used in GST pulldown assays. Luciferase assays were performed as described in (33) using a Wallac Victor2 multi-plate reader (Perkin Elmer). Plasmids used are described in Supplementary Materials and Methods.
RNA sequencing and analysis
Expression data from K562 cells after knockdown of c-Myb was obtained using the RNA-sequencing service at the Norwegian Sequencing Centre. RNA was isolated 24 h after siRNA transfection using the RNeasy RNA isolation kit (Qiagen). The quantity and quality of RNA were determined using a NanoDrop spectrophotometer (Thermo scientific) and an Agilent 2100 Bioanalyzer (Agilent technologies). Three biological replicates were analysed. Samples were delivered to the Norwegian Sequencing Center, Oslo, Norway, where libraries were prepared and sequenced. A total of 125-bp paired-end reads were obtained using an Illumina HiSeq 2500 sequencer.
Low quality reads and adaptors were removed using Trimmomatic (version 0.33) with recommended parameters (35). BBMap (version 34.x) (https://sourceforge.net/projects/bbmap) was further used to remove reads aligning to PhiX (RefSeq: NC_001422.1), which was added as a spike-in during sequencing. Cleaned data was aligned against the ensemble GRCh38 transcriptome using Tophat2 (version 2.0.13) (36) using ‘–library-type fr-firststrand –no-mixed –no-novel-juncs’ as parameters. Insert/Fragment size was estimated for each sample using bowtie2 (version 2.2.3) by aligning them against ensembl GRCh38 cDNA and was also provided as parameters for tophat2 alignment. The Cuffdiff (version 2.2.1) (37) pipeline was used to calculate the differential expression of the known genes described in ensembl using the tophat2 aligned bam files as input. Volcano plots showing the differentially expressed genes are found in Supplementary Figure S1. Data have been made publicly available through GEO (accession number: GSE85187).
Cell differentiation assays
Erythroid differentiation of K562 cells was induced by treating them with hemin. Cells were seeded for a density at 1 × 106 cells/3 ml IMDM in 6-well plates following transfection with siRNA. Twenty-four hours after transfection the cells were cultured for 72 more hours in the presence or absence of 30 μM hemin. Harvesting, benzidine staining and counting of the cells was performed as described in (38). Three biological replicates were analysed. Expression of MYB was analysed by qRT-PCR 72 h after induction of differentiation as described in Supplementary Materials and Methods. Megakaryocytic differentiation of K562 cells was induced by treating them with Phorbol 12-myristate 13-acetate (PMA). Cells were seeded as above following transfection with siRNA. Twenty-four hours after transfection the cells were cultured for 48 more hours in the presence or absence of 1 nM PMA. Megakaryocytic mRNA expression was evaluated by qRT-PCR with primers for ITGB3 (CD61) as described in Supplementary Materials and Methods.
Electrophoretic mobility shift assay (EMSA)
DNA binding was monitored by the electrophoretic mobility shift assay, including titration and off-rate experiments, as described in (39,40).
In vitro nucleosome binding assay
Human histone plasmids were kindly gifted from Gunnar Schotta (LMU, Munich, Germany) (H3.1) and Robert Schneider (Helmholtz Zentrum, Munich, Germany) (H2A, H2B and H4). Histones were expressed in Escherichia coli BL21(DE3pLys), purified and reconstituted into octamers as described previously (41). The 12-mer repeat 601 nucleosome positioning sequence (202 bp) was provided by Axel Imhof (LMU, Munich, Germany) (42). To obtain linearized 601 arrays, the insert was cut out with SacI and XbaI and biotinylated. To ensure only biotinylated 601 arrays, the reaction was further digested with PvuI and PstI, followed by purification of excess nucleotides by Quick Spin Sephadex G-50 columns (Roche). This results in a biotinylated 12-mer of 2437 bp and non-biotinylated 1622, 896 and 135 bp fragments derived from the pUC18 vector serving as competitor DNA in the nucleosome assembly reaction. A 12 bp PstI-XbaI fragment was purified away over the Quick Spin Sephadex G-50 column. Nucleosomes were reconstituted by salt dialysis as previously described (41). To determine the point of saturation, the 12-mer 601 array including competitor DNA was reconstituted with increasing concentrations of recombinant histone octamers and assayed by native gel electrophoresis to reach the saturation point (43). MNase and immobilization to paramagnetic streptavidin beads (M280, Invitrogen) was performed as described in (44).
For pulldown experiments, beads alone, biotinylated 12-mer 601 arrays or biotinlyated chromatinized 12-mer 601 arrays were immobilized to 20 μl streptdavidin beads and incubated on a rotating wheel with 1 μg of recombinant GST, GST-NR123 or GST-NR123D152V in EX100 (10 mM HEPES [pH 7.6], 100 mM NaCl, 1.5 mM MgCl2, 0.5 mM EGTA, 10% [vol/vol] glycerol, 0.2 mM PMSF, 1 mM DTT) containing 0.01% (vol/vol) NP-40 and 20 μg bovine serum albumin, for 1 h at 4°C. Samples were washed three times with 100 μl EX100 containing 0.01% (vol/vol) NP-40, boiled in sodium dodecyl sulphate loading dye, separated by 10–20% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and stained with Coomassie Brilliant Blue.
ATAC sequencing and analysis
Twenty-four hours after siRNA transfection of K562 cells, ATAC-seq was performed as described in (45). We used 50 000 cells per experiment. Libraries were generated using Ad1_noMX and Ad2.1–2.12 barcoded primers from (46) and were amplified for 12 total cycles. Three biological replicates were analysed. Samples were delivered to the Norwegian Sequencing Center, Oslo, Norway, where library quality was assessed and libraries were sequenced. A total of 40-bp paired-end reads were obtained using an Illumina NextSeq 500 sequencer.
Reads were aligned to the human genome assembly (hg19) using the Burrows-Wheeler-Aligner (version 0.7.5a-r405) (47). Aligned reads with poor quality (MAPQ < 20) and ambiguously aligned reads were filtered using SAMtools (version 1.3.1) (48). Peaks were called using MACS2 (version 2.1.1) (49), along with model building using the parameters ‘-m 5 50 –bw 150 –fix-bimodal –extsize 100 call summits -q 0.01’. The bdgcmp function of MACS2 was used to refine the resulting peaks using the ‘Poisson Pvalue’ (ppois) method. Aligned reads were discretized using Zerone (version 1.0) (50). The discretized profiles were then used to generate scatter plots. The scatter plots for the three biological replicates and each condition is shown in Supplementary Figure S2. One of the replicates (rep 1) was removed from further analyses at this point. The refined peaks in bedGraph format were converted to bigWig format using bedGraphToBigWig (version 4) (51). Peaks were visualized using the UCSC Genome Browser. BED formatted files were generated from the aligned BAM files using bedtools (version 2.17.0) (52). Differential peaks were identified using diffReps (version 1.55.6) (53), using the negative binomial model with p-value cut-off 0.0001 and sliding window of 1 kb and a step size of 100 bp. Significantly differential peaks (α < 0.05) were identified after Benjamini–Hochberg adjustment between two treatment conditions at a time. The differential regions obtained were filtered using log2 fold change cut-off ≥ 50% and average normalized read count of the treatment group ≥20. Motif analyses around peak regions for the intersection of differential ATAC peak regions between MYB-knock down and D152V as well as other differential peak sets were performed using the HOMER program (version 4.9) (3), findMotifsGenome.pl. The hg19 genome was used for the motif search along with the parameter ‘–size given’. Data have been publicly available through GEO (accession number: GSE92871).
GST pulldown assay
GST and GST fusion proteins were expressed in E. coli as previously described (54). GST pulldown was performed as described in (33), but after pulldown the beads were washed twice in interaction buffer with 0.5% Triton-X-100. Proteins were detected by Western blotting as described in Supplementary Materials and Methods.
RESULTS
Transcriptome sequencing following c-Myb knockdown revealed two groups of target genes in K562 cells differentially affected by D152V
The starting point of this work was a hypothesis that understanding the underlying mechanism of the D152V mutant of c-Myb might shed light on its function as a master regulator, including its putative role as a pioneer function. From the Plt3 mouse model, we knew that the D152V mutant affected haematopoietic differentiation (27,55). We first asked whether the mutant had a specific effect on the gene regulatory network controlled by c-Myb. We used the human erythroleukaemic cell line K562 to analyse the effect of the D152V mutant on global gene expression. We chose K562 cells because c-Myb is highly expressed (http://fantom.gsc.riken.jp/5/sstar/FF:10826-111C7) and has been shown to affect induced erythroid differentiation of this cell line (56). An siRNA specific for endogenous MYB mRNA (si2992 targeting its 3΄-UTR (23)) was used for knockdown of c-Myb. K562 cells stably expressing TY-tagged wild-type (WT) c-Myb or the mutant (c-Myb D152V) from cDNAs lacking the 3΄-UTRs of MYB (Supplementary Figure S3) were used to rescue the effect of the knockdown. The control K562 cells were transfected with a nonspecific siRNA (siGENOME Non-Targeting siRNA). The specific knockdown of endogenous c-Myb and the rescue by ectopically expressed c-Myb were confirmed by quantitative real-time polymerase chain reaction (qRT-PCR) and western blotting (Figure 1A). We monitored the global effect of c-Myb knockdown by sequencing the transcriptomes of K562 cells harvested 24 h after transfection with control siRNA (Ctrl) or si2992 (KD), as well as of K562 cells stably expressing TY-tagged WT or mutant c-Myb (D152V) transfected with si2992.
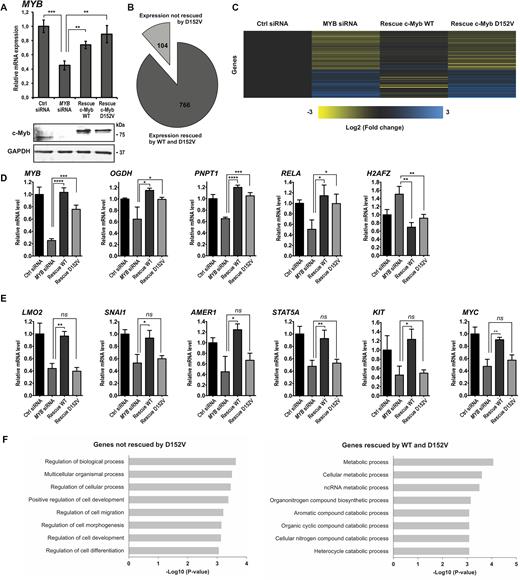
Comparison of the transcriptomes associated with c-Myb WT and D152V generated by RNA-seq following c-Myb knockdown and rescue in K562 cells. (A) Expression of MYB analysed by qRT-PCR and western blotting using anti-c-Myb and anti-GAPDH primary antibodies. RNA and cell lysates were harvested 24 h after transfection with siRNA. RNA was isolated from three independent biological replicates, delivered to high-throughput sequencing and subjected to downstream statistical processing, as described in ‘Materials and Methods’ section. (B) Number of genes differentially expressed after knockdown of c-Myb rescued by ectopic expression of TY-c-Myb and TY-c-Myb D152V (766), as well as the number of genes not rescued by TY-c-Myb D152V (104). (C) Heat map of the differential gene expression pattern after c-Myb knockdown of the genes rescued by TY-c-Myb, but not by TY-c-Myb-D152V. Yellow colour represents decreased expression while blue colour represents increased expression relative to the control (black). (D) RNA-seq data showing the expression pattern of genes differentially expressed after c-Myb knockdown rescued by expression of both TY-c-Myb and TY-c-Myb D152V. The representative genes shown are MYB, OGDH, PNPT1, RELA and H2AFZ. To illustrate the rescue for individual genes, we extracted data for each replicate to estimate mean ± SD. Significance was evaluated by unpaired, two-tailed t-tests on selected pairs and indicated with P-values (*P < 0.05; **P < 0.01; ***P < 0.001; ns P > 0.05). (E) RNA-seq data showing the expression pattern of genes differentially expressed after c-Myb knockdown rescued by expression of TY-c-Myb, but not TY-c-Myb D152V. The representative genes shown are LMO2, SNAI1, AMER1, STAT5A, KIT and MYC. P-values are indicated as in Figure 1D. (F) Gene ontology (GO) analysis of the two groups of c-Myb target genes. The top eight GO terms (based on P-value) for both groups are shown.
Analysis of the sequencing data revealed that 766 of the genes differentially expressed between the control and knockdown cell line were rescued by expression of the WT and the mutant c-Myb (Figure 1B, full heat map shown in Supplementary Figure S3B). The expression of 104 genes, however, was not rescued by c-Myb D152V (Figure 1B and C), constituting about 12% of the c-Myb target genes identified. Among the 766 genes whose expression is rescued by ectopic expression of both versions of c-Myb are OGDH, PNPT1, RELA and H2AFZ (Figure 1D). Their expression profiles resemble the expression pattern of MYB itself (Figure 1D). The expression of the target genes LMO2, SNAI1, AMER1, STAT5A, KIT and MYC, on the other hand, was rescued by WT c-Myb, but not by c-Myb D152V (Figure 1E). These are among the 104 target genes that the mutant c-Myb failed to regulate. The RNA-seq data indicates that a single amino acid substitution in the c-Myb DBD impairs its ability to regulate a subset of its target genes. Of note, the genes not regulated by c-Myb D152V generally have a lower expression level in the control cell line (without c-Myb knockdown) than the other group of genes (Supplementary Figure S3C, upper panel). Furthermore, these genes seem to be more dependent on c-Myb for their expression since their mRNA levels decrease relatively more upon c-Myb knockdown than the mRNAs of the genes regulated by both (Supplementary Figure S3C, lower left panel).
In order to investigate what distinguish the genes regulated by the D152V mutant from the ones that are not, we performed a gene ontology (GO) analysis of both groups of genes using the GO search program Gorilla (http://cbl-gorilla.cs.technion.ac.il/) (57), selecting the option that calculates GO term enrichment in a target gene list over a background list of genes. This analysis revealed that the subset of genes that the mutant c-Myb is incapable of rescuing is involved in biological process regulation and development (Figure 1F, left). The genes regulated by both versions of c-Myb, however, showed an enrichment of genes involved in metabolism (Figure 1F, right). In an analysis for enriched biological pathways using the Ingenuity Pathway Analysis (IPA) tool (Qiagen), the genes not regulated by c-Myb D152V are classified as mainly involved in processes of cell proliferation, growth and development (including differentiation), whereas the genes regulated by both WT and mutant c-Myb are involved in metabolic processes and cell survival (Supplementary Figure S4A). Interestingly, the group of genes that c-Myb D152V fails to regulate is involved in acute myeloid leukaemia (AML) signalling as the top canonical pathway (IPA analysis, Supplementary Figure S4B), a process in which c-Myb has been reported to play a critical role (58–60). Our analyses strongly suggest that these two groups of genes are involved in distinct biological pathways and processes and that c-Myb may utilize different molecular mechanisms in regulating their expression, as one group seems to be dependent on aspartate 152 whereas the other is not.
The D152V mutation leads to increased erythroid differentiation of K562 cells
c-Myb has been shown to be essential for the self-renewal of progenitor haematopoietic cells, as well as suppressing their differentiation and promote leukaemic transformation (12). To validate that the D152V mutant of c-Myb indeed affects differentiation in our model cell line, we explored its ability to suppress differentiation of K562 cells, here used as a model for myeloid differentiation.
K562 cells resemble multipotent progenitor myeloid cells and can differentiate along the erythroid lineage upon treatment with hemin (61). Overexpression of c-Myb has been shown to suppress the chemically induced differentiation of K562 cells into erythrocytes (56). In contrast, the chemically induced differentiation along the megakaryocytic lineage is not affected by c-Myb overexpression. We treated K562 cells with hemin for 72 h and counted the number of haemoglobin-positive cells after benzidine staining. A representative result of the hemin-induced erythroid differentiation is shown as colour change (Figure 2A) and benzidine-stained cells (Figure 2B), both showing the expected increase in the number of erythroid lineage cells after hemin treatment. The expression level of c-Myb itself is elevated in immature progenitor cells of different lineages and decreases upon differentiation (30,62,63). We confirmed that this was also the case in the differentiation assay used here (Figure 2C).
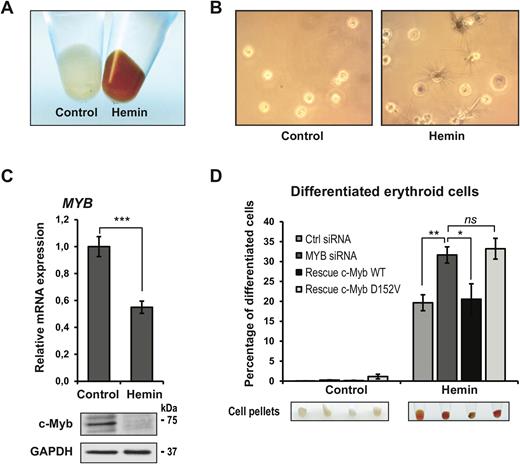
Hemin-induced erythroid differentiation of K562 cells. (A) K562 cells were induced for erythroid differentiation with 30 μM hemin for 72 h. The erythroid differentiation is evident by the red cell pellets. (B) Seventy-two hours after induction with hemin, the cells were stained with benzidine and the haemoglobin-positive cells (black, some with spikes) were measured. The number of benzidine-positive cells was estimated by manually counting triplicates of 300 cells in a light microscope. Three biological replicates were analysed (total of 900 cells). (C) Expression of MYB analysed by qRT-PCR and protein level of c-Myb analysed by western blotting using anti-c-Myb and anti-GAPDH primary antibodies 72 h after induction with hemin in the control K562 cell line. (D) The amount of haemoglobin-positive cells, counted as described above, presented as percentage of differentiated cells. Cell pellets are also shown to visualize the degree of differentiation. All cell counting and qRT-PCR results are presented as mean ± SD of three independent biological replicates. P-values are indicated as in Figure 1D.
To monitor the effect of the D152V mutant on the hemin-induced erythroid differentiation, the same stable K562 cell lines used for transcriptome analysis were transfected with control siRNA or si2992 for 24 h before treatment with hemin (Figure 2D). Knockdown of endogenous c-Myb resulted in an increased number of hemin-induced differentiated cells, as expected from c-Myb's suppression of erythrocyte differentiation (38). The effect of the c-Myb knockdown was rescued in the cell line stably expressing WT c-Myb, as the number of differentiated erythroid cells fell back close to the siRNA control level. The c-Myb mutant, however, was not able to rescue the effect of the knockdown since the number of hemin-induced differentiated cells was similar to the number upon knockdown of endogenous c-Myb. Thus, the c-Myb-mediated suppression of hemin-induced erythroid differentiation of K562 cells seems to depend on aspartate 152. Taken together with the findings from the RNA sequencing, these results suggest that the D152V mutation in the DBD of c-Myb inhibits the differentiation-blocking activity of c-Myb.
The Plt3 mice were found to have elevated megakaryocyte levels and platelet production (27). We also induced megakaryocytic differentiation of K562 by treatment with PMA for 48 h. Our result confirmed that chemically induced megakaryocytic differentiation of K562 cells is unaffected by c-Myb knockdown (56), here measured by the level of the surface marker CD61 (Supplementary Figure S5A). The expression level of c-Myb itself however, was reduced upon PMA-induced megakaryocytic differentiation to a similar degree as the erythroid differentiation of K562 cells (Supplementary Figure S5B).
The D152V mutation in the DNA-binding domain of c-Myb does not weaken its DNA binding
Since the D152V mutation is located in the DBD of c-Myb, the most obvious mechanism would be a weakened DNA interaction. The DBD of the c-Myb protein is evolutionary conserved and consists of three Myb repeats (R1, R2 and R3) (64,65), each of which is closely related to the chromatin-interacting SANT domain identified in several chromatin regulatory proteins (66,67). All three Myb repeats consist of three α-helices, which form a helix-turn-helix-related structure (68,69). The minimal DBD consists of R2R3, where the third α-helix in each of the two repeats is the recognition helix binding to DNA (Figure 3A, structure from (68) with aspartate 152 highlighted). The first α-helix in all three Myb repeats contains an acidic patch of amino acids, suggesting that these helices have other roles than binding to DNA (70).
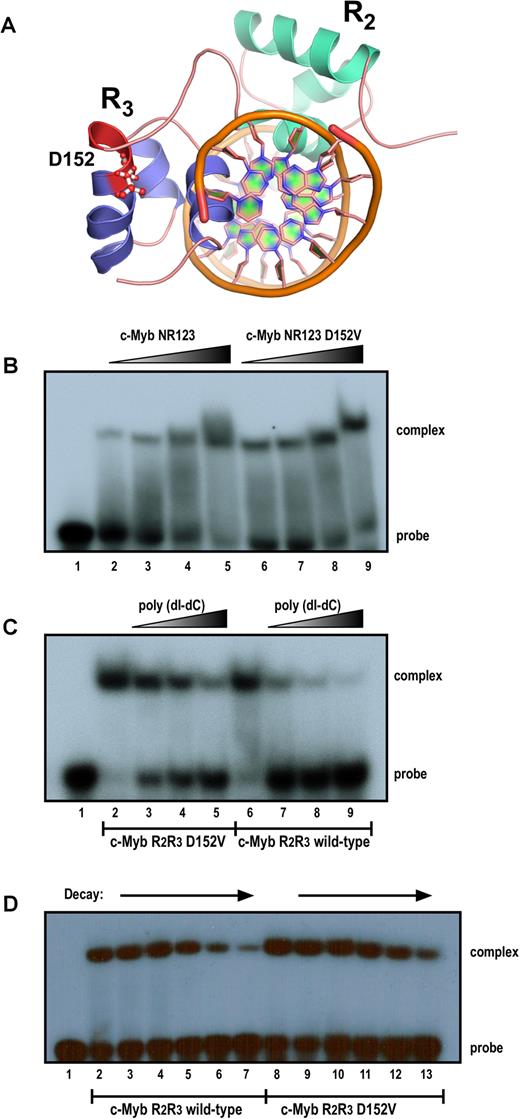
Comparison of the activity of the DNA-binding domain of c-Myb in wild-type (WT) and D152V mutant form, monitored by electrophoretic mobility shift assay (EMSA). (A) Illustration of the 3D structure of the minimal DBD (R2R3) of c-Myb bound to DNA. Graphical rendering of the structure was made with PyMOL (version 1.7.4) using the protein data bank identifier 1MSF. (B) Complex formation with increasing amounts added of purified recombinant human c-Myb protein (NR123 = amino acids 1–192) in either WT or mutant (D152V) form. A total of 5 fmol (lanes 2 and 6), 10 fmol (lanes 3 and 7), 40 fmol (lanes 4 and 8) and 100 fmol (lanes 5 and 9) of purified NR123 WT (lanes 2–5) or D152V mutant (lanes 6–9) protein were incubated with 20 fmol MRE(mim) probe at 25°C for 10 min and analysed by the electrophoretic mobility shift assay as described in ‘Materials and Methods’ section. (C) Titration of protein–DNA complexes with poly(dI-dC). A total of 30 fmol of purified Myb R2R3 proteins (WT or D152V) were incubated with 20 fmol of probe and increasing amounts of poly(dI-dC) and analysed by the electrophoretic mobility shift assay as described above. The amounts poly(dI-dC) added was 0 μg (lanes 2 and 6), 0.25 μg (lanes 3 and 7), 1 μg (lanes 4 and 8) and 2 μg (lanes 5 and 9). (D) Time course of complex dissociation upon competition. Myb R2R3–DNA complexes (WT and D152V) were allowed to form at 25°C for 10 min before they were exposed to a 75-fold excess of unlabelled specific MRE(mim) probe for t = 0, 2, 5, 10, 20 and 30 min (WT: lanes 2–7, D152V: lanes 8–13). DNA binding was monitored by the electrophoretic mobility shift assay as described above.
As shown in Figure 3B, the D152V mutant had no apparent effect in a DNA-binding assay with recombinant c-Myb DBD in WT and mutant form. Titration and decay assays indicated that the mutant, if anything, rather bound slightly stronger to the specific DNA sequence than the WT (Figure 3C and D). Thus, weakened DNA interaction cannot be the underlying mechanisms of the D152V mutant. This is also consistent with the specific effects seen in the transcriptome analysis. Here, a generally weakened DNA binding would be expected to tune down most c-Myb target genes equally.
The D152V mutation strongly affects transactivation by c-Myb at target genes where chromatin opening is required
Since the D152V mutation seems to impair c-Myb's ability to regulate specific genes in vivo, we pursued our search for a mechanism by studying how the mutation affected the transcriptional activity of c-Myb in various reporter assays. Reporter constructs containing either a synthetic (3×MRE) or a natural (mim-1) c-Myb-responsive promoter upstream of the luciferase gene were co-transfected with c-Myb expression plasmids in CV-1 cells. As shown in Figure 4A, we found that c-Myb D152V activated the reporters to the same extent as WT c-Myb. Since we could not exclude that the gene-specific D152V phenotype depends on a particular sequence arrangement, we performed the same analysis with reporter constructs driven by the MYC, STAT5A or LMO2 promoters, three of the target genes showing a clear D152V effect in vivo. However, again we observed no difference between the WT and D152V versions of c-Myb in their ability to activate these reporters (Figure 4B). Common to these reporters are that they are located on transiently transfected plasmids where transactivation primarily depends on c-Myb binding to DNA. This is because transiently transfected reporter plasmids are not fully chromatinized, exhibiting only intermediate levels of nucleosomal assembly (71).
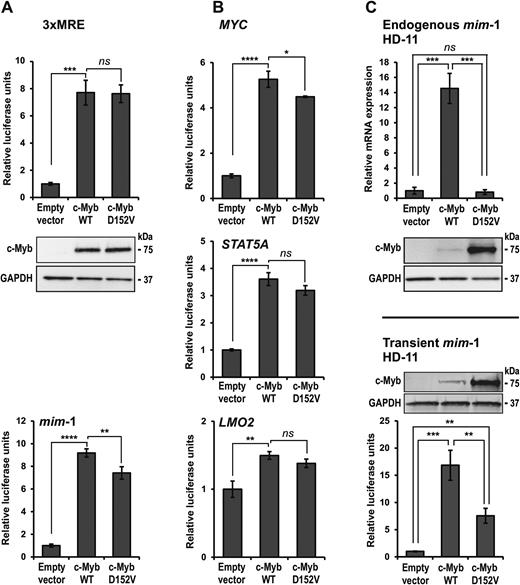
Comparison of transactivation by c-Myb in WT and D152V mutant form. (A) CV-1 cells were transfected with c-Myb responsive reporter plasmids (pGL4b-3xMRE(GG)-myc or pmim3mim) and plasmids encoding full-length HA-tagged c-Myb or c-Myb D152V. The reporter activations are presented as relative luciferase units. The western blots were analysed using anti-HA (c-Myb) and anti-GAPDH primary antibodies. (B) CV-1 cells were transfected with c-Myb responsive reporter plasmids (pGL3-MYC, pGL3-STAT5A or pGL3-LMO2) and plasmids encoding full-length HA-tagged c-Myb or c-Myb D152V. The reporter activations are presented as relative luciferase units. (C) Upper panel: HD-11 cells were transfected with plasmids encoding full-length HA-tagged c-Myb or c-Myb D152V. Total RNA was isolated and mim-1 expression was measured by qRT-PCR. Lower panel: HD-11 cells were transfected with the c-Myb responsive reporter plasmid pmim3mim and the same c-Myb plasmids as above. The reporter activations are presented as relative luciferase units. The western blots were analysed using anti-HA (c-Myb) and anti-GAPDH primary antibodies. All luciferase and qRT-PCR results are presented as mean ± SD of three independent biological replicates, each performed in triplicates (n = 3). Significance was evaluated by unpaired, two-tailed t-tests on selected pairs.
To extend this analysis to a more physiological setting, we analysed transcriptional activation of the endogenous mim-1 gene in the chicken macrophage cell line HD-11, which is an established model of c-Myb-dependent activation of a chromatin-embedded gene. This cell line does not express c-Myb endogenously, but the collaborating factor C/EBPβ, both of which are required for expression of mim-1. We found that ectopic expression of WT c-Myb resulted in high expression of endogenous mim-1, whereas c-Myb D152V failed to activate transcription of mim-1 (Figure 4C, upper panel). When the plasmid-encoded mim-1 luciferase reporter was tested in the same cell line (Figure 4C, lower panel), we observed significant activation with both variants shown (D152V: 7.5-fold; WT: 17-fold activation). Compared to the parallel analysis in CV-1 cells (Figure 4A, lower panel), D152V was less active than WT in the cellular context of HD-11 cells. However, comparison of transient versus endogenous mim-1 activation shows a major change from reduced activation to no activation with D152V c-Myb. The c-Myb dependent transcription of the endogenous mim-1 in HD-11 cells has previously been shown to involve chromatin opening at the mim-1 promoter (72). Furthermore, c-Myb is implicated in remodelling of the nucleosomal architecture at the mim-1 enhancer, which is accompanied by the transcription of a non-coding RNA from the enhancer (73). The inability of the mutated c-Myb to activate transcription of a fully chromatinized target gene, whereas the activation of its promoter in a transiently transfected plasmid remains significant, strongly suggests that the D152V mutation specifically interferes with the ability of c-Myb to activate transcription in a chromatin context. Notably, the D152V mutant seems to be highly stable in HD-11 cells, as judged from its strong western blot signal (Figure 4C). This observation seems to be cell type-specific, since equal signals are observed in CV-1 cells transfected with the same plasmids. We did not investigate this further, but it reinforces our conclusion since even enhanced levels of c-Myb D152V are incapable of activating expression of endogenous mim-1.
In search for a mechanism: altered recruitment of cooperating factors or remodelling complexes?
c-Myb is expressed in all haematopoietic lineages, while mim-1 expression is restricted to myelomonocytic cells. This restricted expression has been found to be due to a cooperation of c-Myb with C/EBPβ, which is highly expressed in the myelomonocytic lineage (63,74). The D152V mutation is located close to the region of the c-Myb DBD previously found to be involved in interaction with C/EBPβ based on a far-western analysis (74). To test the hypothesis that the inability of c-Myb D152V to activate mim-1 expression is due to a loss of interaction with C/EBPβ, we performed a GST pulldown assay using C/EBPβ expressed in COS-1 cells and the c-Myb DBD fused to GST. The interaction between c-Myb and C/EBPβ was found to be unaffected by the D152V mutant (Figure 5A), hence not supporting this loss-of-cooperation hypothesis.
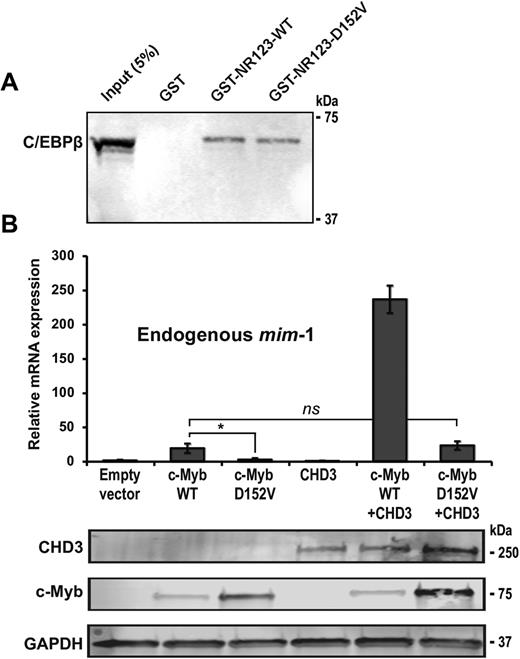
Interaction and recruitment of cooperating factors is not affected by D152V. (A) GST pulldown assay performed with pEYFP-N1-CEBPβ (COS-1 cell lysate) and GST-fused c-Myb DBD (R123) and the N-terminal region upstream of the DBD (N) expressed in Escherichia coli. The western blot was analysed by using anti-GFP (C/EBPβ) primary antibody. About 5% of the COS-1 cell lysate was loaded as reference. (B) HD-11 cells were transfected with plasmids encoding full-length HA-tagged c-Myb, c-Myb D152V and Flag-tagged CHD3. Total RNA was isolated and mim-1 expression was measured by qRT-PCR. The western blots were analysed using anti-HA (c-Myb), anti-Flag (CHD3) and anti-GAPDH primary antibodies. All qRT-PCR results are presented as mean ± SD of three independent biological replicates, each performed in triplicates (n = 3). P-values are indicated as in Figure 1D.
Another interaction candidate possibly lost by the D152V mutation is CHD3 (Mi-2α), which we previously found to associate with c-Myb DBD (34). CHD3 might, as a chromatin remodeller, be particularly relevant for explaining the chromatin-associated behaviour of D152V. CHD3 acts as a transcriptional co-activator for c-Myb and is recruited to c-Myb target genes, including mim-1 (34,38). When HD-11 cells were co-transfected with c-Myb and CHD3, we observed a significant enhancement of mim-1 expression (Figure 5B). Moreover, the inability of c-Myb D152V to activate mim-1 was partially rescued by overexpression of CHD3, suggesting that chromatin remodelling compensates for the defect. However, the relative increase in activity with CHD3 present as a co-activator was about the same for both WT and c-Myb D152V (∼10 times higher with than without CHD3) (Figure 5B), arguing against their interaction being affected by the mutation. Instead, we believe that enhanced remodelling of chromatin by the recruited CHD3 is able to partly rescue the activity of c-Myb D152V.
The interaction between c-Myb and histone H3 is weakened by the D152V mutation
In addition to binding to DNA, c-Myb also interacts with several gene regulatory proteins through its DBD (33,34,74,75), as well as with histones (70,76). Aspartate 152 is located in an acidic patch of the R3 repeat within the first α-helix of the repeat. As shown in Figure 3A, the α-helix where aspartate 152 is located seems to be pointing away from the DNA helix. In addition, the acidic nature of this region may indicate that it has other roles than DNA binding, possibly being involved in histone tail interactions.
By GST pulldown assays, we confirmed that c-Myb binds the histone tails of H2B, H3 and H4 (Figure 6A), as reported by others (70,76). The interaction with H3 has been mapped to the DBD of c-Myb and was proposed to be required for histone tail positioning and acetylation by the histone acetyltransferase p300 (76). Considering the basic nature of histone tails, we suspected that since the D152V mutation causes one acidic amino acid less in R3, it might weaken the interaction between the c-Myb DBD and histones. To address this, we generated GST-fused fragments of the c-Myb DBD (Figure 6B) and performed GST pulldown assays with recombinant H3. Both the R2 and the R3 repeats seem to interact individually with H3, suggesting that c-Myb has at least two histone-binding modules within its DBD (Figure 6C). Interestingly, the binding of R3 alone to H3 is lost when R3 is mutated, whereas the binding is intact when R2 is also present (Figure 6C). Therefore, it seems as if the D152V mutation weakens the binding to H3 by impairing the R3 histone-binding module; however, the binding of the full DBD is not completely lost due to the additional histone-binding site in the R2 repeat.
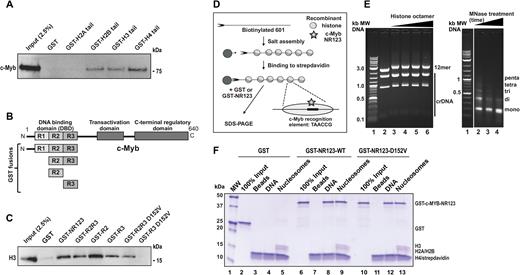
Nucleosome binding and disrupted histone binding in c-Myb D152V. (A) GST pulldown assay performed with pCIneo-3x-Flag-hcM (COS-1 cell lysate) and GST-fused histone tails expressed in Escherichia coli. The western blot was analysed by using anti-Flag (c-Myb) primary antibody. About 2.5% of the COS-1 cell lysate was loaded as reference. (B) Schematic figure of the full-length c-Myb protein and the regions fused to GST in GST pulldown assays. (C) GST pulldown assay performed with GST-fused fragments of c-Myb and 3 μg recombinant H3. The western blot was analysed by using anti-H3 primary antibody. About 2.5% (0.075 μg) of H3 was loaded as reference. (D) Binding of recombinant GST-fused c-Myb NR123 (WT and D152V) to in vitro assembled nucleosomes. Scheme of the chromatin reconstitution protocol. The DNA used is a linearized biotinylated fragment containing 12 repeats of 601 nucleosome positioning sequence. Each positioning sequence repeat contains one c-Myb binding site in each repeat. (E) Left panel: DNA and nucleosomal arrays analysed by native agarose gel electrophoresis in a 0.7% agarose gel. The upper 2437-bp band is the biotinylated (*) 12-mer 601 repeat and the lower DNA fragments (1622, 896 and 135 bp) lacking positioning sequences are non-biotinylated and serve as competitor DNA (crDNA) to bind excess histones. The DNA was reconstituted with histone octamer salt dialysis with increasing concentrations of recombinant human histone octamers to reach saturation plateau of 12-mer and an upward shift of crDNA. Addition of histone octamers showed slower migration of the array on native agarose (compare lane 2 and 3). When the 601 sequences become saturated, also the crDNA start to migrate slower (lane 6). Right panel: a micrococcal nuclease digestion pattern of salt-reconstituted linearized nucleosomes (saturation point, preparation from lane 6 in 6E) analysed by electrophoresis in a 1.3% agarose gel. (F) Pulldown of GST, GST-c-Myb-NR123-WT and GST-c-Myb-NR123-D152V with beads, DNA or nucleosomes (saturation point, preparation from lane 6 in 6E). Boiled beads were separated by 10–20% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and visualized by Coomassie Brilliant Blue staining. This figure represents one biological replicate of three experiments showing the same pattern.
Since c-Myb is able to interact with both DNA and histones, we asked whether it might also interact with assembled nucleosomes, which is one of the properties of pioneer factors (1). To investigate the binding of c-Myb and the D152V mutant to nucleosomes, we assembled 12-mer 601 nucleosome arrays on a biotinylated linearized 12-mer nucleosome positioning sequence (42) using recombinant human histone octamers (Figure 6D and E) (42). The DNA sequence (601) contains one Myb-recognition element in each 202 bp nucleosome-positioning repeat (Figure 6D). Saturation of histone octamer to linearized DNA was determined as described in the legend to Figure 6E. Pulldown with GST protein showed no binding to beads alone, linearized 601 sequence or linearized nucleosomes (Figure 6F, lanes 3–5). Both the DBD (NR123) of WT c-Myb and the D152V mutant bound DNA similarly (Figure 6F, lanes 8 and 12), supporting the results shown in Figure 3B. Moreover, both the WT and mutated proteins bound saturated 12-mer 601 nucleosomes with efficiencies similar to their DNA binding (Figure 6F, lanes 9 and 13). This ability of both the WT and the mutant c-Myb DBD to bind to assembled nucleosomes (Figure 6F) indicates that c-Myb might harbour some of the properties of pioneer factors. Furthermore, since the D152V mutant does not seem to impair the ability of c-Myb to bind to assembled nucleosomes in this in vitro assay, probably because of histone binding of R2 and unaffected DNA-binding (Figure 3), we asked whether the weakened H3 binding by D152V might have a larger effect on chromatin accessibility than on chromatin binding.
The ability of c-Myb to increase chromatin accessibility is impaired by the D152V mutation
The dual function of c-Myb binding both DNA and histones, as well as assembled nucleosomes, suggests an ability to associate with and possibly open chromatin, a property characteristic of pioneer factors. To test our hypothesis that c-Myb might increase chromatin accessibility, we applied ATAC sequencing (ATAC-seq), a recently developed method for genome-wide mapping of chromatin accessibility (45,46). We performed ATAC-seq on the same K562 cell lines as we used for the transcriptome analysis, 24 h after transfection with control siRNA or si2992, to map changes in chromatin accessibility. ATAC-seq analysis identified 964 regions having at least 50% difference in chromatin accessibility between K562 cells transfected with si2992 versus control siRNA (Figure 7A). Of these, 910 regions showed decreased chromatin accessibility after knockdown, whereas only 54 regions showed an increase in chromatin accessibility, suggesting a role for c-Myb in keeping specific regions in an open chromatin state. Comparing all peak regions that differed between the WT and the D152V rescue cell line, the D152V rescue cell line showed significantly more peak regions with decreased chromatin accessibility (2295) than increased chromatin accessibility (491) compared to the WT rescue (Figure 7B), consistent with an impaired opening activity of the mutant. A total of 692 of the 910 regions with decreased chromatin accessibility upon knockdown also had a significant difference between the WT c-Myb and the D152V cell line (Figure 7C), indicating that WT c-Myb is able to rescue a majority (692/910) of the reduced opening effect of the knockdown, whereas c-Myb D152V is not. Of the 692 regions identified, we found that a majority (499) were located in ‘other intergenic regions’ (Figure 7D). Manual inspection of these regions showed that most of them have a clear epigenetic signature indicative of enhancers. By HOMER analysis (3), we found that in this group of 692 regions, 73% contained a rather stringent Myb recognition motif (MRE) (Figure 7E).
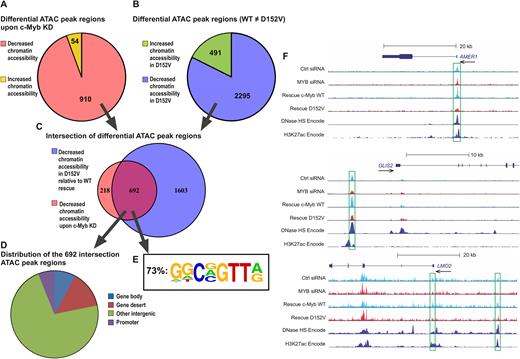
Differential chromatin accessibility revealed by ATAC-seq (A) Regions with ≥50% fold difference after knockdown of c-Myb (comparing K562 cells transfected with control siRNA and si2992) showing decreased chromatin accessibility (910) or increased chromatin accessibility (54). (B) Regions with ≥50% fold difference between WT c-Myb rescue and the D152V rescue (comparing the K562 cell lines ectopically expressing TY-tagged versions of c-Myb after knockdown of endogenous c-Myb) showing decreased chromatin accessibility (2295) in D152V compared to WT or increased chromatin accessibility (491) in D152V compared to WT. (C) Venn diagram showing the overlap (692) between the 910 regions with decreased chromatin accessibility upon c-Myb knockdown and the 2295 regions with decreased chromatin accessibility upon D152V rescue compared to WT c-Myb rescue. (D) Genomic distribution (defined by diffReps) of the 692 overlapping regions. (E) The frequency of the c-Myb binding motif shown, for the indicated subgroup (n = 692). Motif analysis around peak regions for the intersection of differential ATAC peak regions between MYB knockdown and D152V was performed using the HOMER program (3), as described in ‘Materials and Methods’ section. Values for the n = 1603 and n = 910 groups were 71 and 64% respectively. (F) UCSC Genome Browser tracks of the AMER1, GLIS2 and LMO2 loci showing ATAC-seq signal of K562 cells transfected with control siRNA, si2992 (MYB siRNA), as well as the WT c-Myb and D152V rescues. DNase I hypersensitivity (HS) mapping from K562 cells and ChIP-seq data on H3K27Ac in K562 cells (generated by the ENCODE project) is also shown.
Three specific examples of ATAC profiles are shown in Figure 7F. The AMER1 profile illustrates one of the 692 ATAC peak regions in a promoter with at least 50% difference in chromatin accessibility and with the profile of WT rescue, but less efficient D152V rescue. The GLIS2 profile illustrates similarly one ATAC peak region with an enhancer signature. The LMO2 example illustrates the profiles observed with changes in both promoter and enhancer regions. For validation, we compared our ATAC-seq data with DNase hypersensitivity regions and H3K27ac ChIP-seq data generated in K562 cells (ENCODE, https://www.encodeproject.org).
Taken together, our ATAC-seq analysis showed that knockdown of c-Myb results in decreased chromatin accessibility at specific sites, especially at putative enhancers, strongly suggesting that c-Myb has the ability to induce chromatin opening. Furthermore, the D152V mutation seems to abolish this chromatin-opening activity.
DISCUSSION
In this study, we aimed to elucidate the molecular function of a previously identified c-Myb mutant and thereby examine the hypothesis that c-Myb functions as a pioneer factor. Mice harbouring this mutant were shown to have increased megakaryocytopoiesis and impaired lymphopoiesis (27), strongly suggesting that the D152V mutation affects c-Myb's ability to regulate differentiation of haematopoietic cells in these mice. We show that this single amino acid substitution in DBD prevents c-Myb from regulating differentiation-associated genes and we define a specific role of the mutant in abrogating a pioneer function of c-Myb important for its regulatory role in haematopoiesis.
Several lines of evidence support a mechanism for the D152V-effect that is linked to chromatin function, rather than to DNA binding. First, we observed no reduction in DNA-affinity of the mutant DBD in vitro (Figure 3). Second, the effect of the mutant on target gene activation appeared to be most severe when the promoter was fully chromatin-embedded, as illustrated by the inability of c-Myb D152V to activate transcription of the endogenous mim-1 gene while it was fully able to activate the same promoter on a transfected plasmid (Figure 4). Third, a global transcriptome analysis after c-Myb knockdown and rescue revealed two different groups of genes, one rescued by both WT and mutant c-Myb, the other rescued by WT only and not by the mutant (Figure 1). When reporter assays were performed using transient transfection of promoters from the second group, the difference disappeared (Figure 4). Since the group of target genes affected by the mutant were linked to differentiation, this indicates that aspartate 152 is particularly involved when gene programs have to be altered, a process where chromatin opening and remodelling is expected to be involved. In support of this, we observed a clear effect of the mutant on hemin-induced erythroid differentiation of K562 cells (Figure 2). Finally, we were able to show that the mutant directly abrogated the histone binding of one of the Myb repeats, R3, in the DBD of c-Myb, strongly supporting a mechanism where D152V impairs the histone-binding properties of c-Myb (Figure 6). Having delimited our search for a mechanism to chromatin and histone interaction, the most obvious function abrogated would be the ability of c-Myb to access closed chromatin. This is a key element in our hypothesis that c-Myb has properties of a pioneer factor and furthermore suggests that the D152V mutant specifically abrogates this pioneer function.
The concept of pioneer factors was described in the ‘Introduction’ section. Association with closed chromatin and capability to increase the nucleosomal accessibility of their target sites are key defining features (1,2). We showed by a biochemical assay that c-Myb was able to bind nucleosomes (Figure 6) and we used ATAC-sequencing to examine whether we could find direct evidence for a defect in chromatin-opening associated with the D152V mutant (Figure 7). Knockdown of a single transcription factor is not expected to cause major alterations in the genomic packaging, but still we detected a significant number of changes at specific loci all over the genome. More specifically, a majority of the ATAC peak regions that showed reduced accessibility after c-Myb knockdown also showed a significant difference between the WT and D152V c-Myb in rescuing this reduced accessibility, strongly suggesting both an ability of c-Myb to contribute to chromatin opening and a defect in this ability associated with the D152V mutant. We also found a stringent MRE motif in a majority of these peak regions. To evaluate the link between the two global datasets (Figures 1 and 7), we had to take into consideration that the ATAC-alterations are mostly found in non-coding regions with enhancer-like epigenetic profiles. Thus, the challenge of linking the two datasets is similar to the general challenge of linking promoters and enhancers (77). However, we took advantage of public available maps of topologically associated domains (TADs) in K562 cells (78) (http://promoter.bx.psu.edu/hi-c/view.php) and inspected a key subset of the c-Myb target genes, those that differ in their dependence of D152V. Of the 104 genes in this group (Figure 1), 59 genes were downregulated upon c-Myb knockdown. We looked up the TAD-maps for all these 59 genes and identified a differential ATAC-peak within the same TAD (from our set of 692 peaks in Figure 7) in 51 of these (results not shown). We are therefore confident that the two datasets reflect related phenomena.
The exact mechanism of how pioneer factors cause chromatin opening is not fully understood. A study of the nucleosome and chromatin targeting activities of the pioneer and pluripotency factors Oct4, Sox2 and Klf4 showed that these proteins can bind nucleosomes in vitro, and in vivo they preferentially target silent sites enriched with nucleosomes (9). Mechanistically, their pioneer activities were explained by their ability to target partial motifs displayed on the nucleosome surface, a property that correlated with an apparent flexibility of their respective DBDs. We have previously reported that the DBD of c-Myb is flexible and undergoes a conformational change upon binding to DNA (79,80). It is at least conceivable that some of this flexibility, as well as its histone-binding property, are needed to target binding sites exposed on the nucleosome surface during a pioneer attack of c-Myb.
Future research has to address two key questions regarding how c-Myb contributes to chromatin openness, related to redundancy and cooperation with other chromatin modulators. Our in vitro data has revealed a certain redundancy in histone binding since both the R2 and R3 repeats are able to bind individually to H3. Hence, we see loss of histone binding of D152V only when tested in the format of R3 alone. The question is how this could explain the differences observed in vivo in the context of all three repeats. First, we assume that the in vivo conditions are much more competitive than what is the case in a pulldown with purified components in vitro, which would allow for these differences in affinities to cause an effect. Moreover, we cannot exclude that there are more elements in this histone association mechanism than simple binding. Even if we think the histone binding is a prerequisite to the pioneer function of c-Myb, there may be more requirements to cause opening of closed chromatin than there is to histone binding. It is conceivable that any of the repeats contribute to a first association of c-Myb with histones, but that a subsequent step involving specifically R3 binding is needed to contribute to the opening process. We do see binding of both WT c-Myb and D152V to assembled nucleosomes in vitro, suggesting that also the mutant is able to bind to chromatin. However, the most apparent effect of the mutant seems to be associated with chromatin opening, as seen from the ATAC experiment as well as activation of the endogenous mim-1 gene in HD-11. The second question is how a nucleosome bound c-Myb cooperates with other chromatin modulators. One may imagine the selective recognition of this complex by a chromatin remodeller, or the complex being a preferred substrate for histone modifications.
Whether the pioneer factors in general are able to perform chromatin opening fully independent of other factors, such as chromatin remodelling complexes, is still being debated (2,81). Interestingly, several studies have shown links between c-Myb and chromatin modifications; such as the recruitment of the histone methyltransferase complex Mixed Lineage Leukemia (82) and Protein arginine methyltransferase 4 (38) to target genes by c-Myb, both leading to activation of c-Myb-dependent transcription. Furthermore, the recruitment of p300 and CREB-binding protein (CBP) to target genes by c-Myb leads to histone acetylation and gene activation (24,76,83). A particularly impressive case of Myb-induced genomic reprogramming was reported recently, where a subset of T-cell acute lymphoblastic leukaemia cells was found to have acquired mutations that introduce c-Myb binding motifs upstream of the TAL1 oncogene (24). Binding of c-Myb to this mutant site resulted in recruitment of CBP, as well as core components of a major leukaemogenic transcriptional complex, generating a super-enhancer defined by extensive H3K27 acetylation and open chromatin (24). The ability of c-Myb to create a de novo super-enhancer to activate transcription of TAL1 strongly suggests it harbours pioneer factor activity, as pioneer transcription factors are typically found bound to enhancers before lineage commitment and employs a chromatin opening step to establish competence for gene activation (1,7).
Taken together, c-Myb interacts directly with both histones and DNA and with proteins responsible for chromatin modifications. Based on data presented in this work and several observations from the literature, we propose that c-Myb operates as a pioneer transcription factor being able to access closed chromatin at specific loci and thereby change gene programs during haematopoiesis. The detailed mechanism involved has to be further investigated, but we have presented strong evidence that the pioneer activity can be specifically abrogated by the D152V mutant of c-Myb. To our knowledge, this is the first pioneer-specific mutant of any transcription factor where this particular function is impaired while keeping its DNA binding intact. This mutant then represents a powerful tool to study c-Myb pioneer factor function in particular, but also to dissect the pioneer mechanism in a way that will be relevant to pioneer factors in general. Of note, the genes the D152V mutant failed to regulate, and which represents the genes that require the pioneer factor function of c-Myb intact, are involved in AML signalling (Supplementary Figure S4B). Since c-Myb has been identified as a critical mediator of oncogene addiction in AML (59), and is involved in the development and maintenance of several human cancers (12,84), this may suggest that the pioneer factor function of c-Myb is closely connected to its functions in cancer.
ACCESSION NUMBERS
GEO GSE85187 and GSE92871.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Karl-Heinz Klempnauer for providing C/EBPβ expression plasmids and mim-1 reporter plasmid, Achim Leutz for providing plasmids for expression of GST-tagged histone tails, Gunnar Schotta for H3.1 expression plasmid and Robert Schneider for plasmids to express H2A, H2B and H4. We are grateful to Marit Ledsaak for valuable help with construction of plasmids and recombinant protein expression. We also thank Mads Bengtsen for valuable discussions and assistance. The sequencing service was provided by the Norwegian Sequencing Centre (www.sequencing.uio.no), a national technology platform hosted by the University of Oslo (UiO) and supported by the ‘Functional Genomics’ and ‘Infrastructure’ programs of the Research Council of Norway and the South-Eastern Regional Health Authorities. Mapping of ATAC-seq reads were carried out at Centre de Regulació Genòmica, Barcelona, in Guillaume Fillon's lab, with valuable help from Eduard Valera i Zorita. The ATAC-seq peak calling, diffReps run and other downstream analyses were carried out on the Abel Cluster (project nn9374k), owned by the UiO and the Norwegian metacenter for High Performance Computing (NOTUR). The Abel Cluster is operated by the Department for Research Computing at USIT, the UiO IT-department (http://www.hpc.uio.no/).
FUNDING
University of Oslo PhD Grant (to B.M.F.); Norwegian Cancer Society [419436 107692-PR-2007-0148 to O.S.G., 3485238-2013 to R.E.]; Research Council of Norway [240768 to O.S.G., 231217/F20 to R.E.].
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments