





Abstract
A-to-I RNA editing is an important post-transcriptional modification, known to be altered in tumors. It targets dozens of sites within miRNAs, some of which impact miRNA biogenesis and function, as well as many miRNA recognition sites. However, the full extent of the effect of editing on regulation by miRNAs and its behavior in human cancers is still unknown. Here we systematically characterized miRNA editing in 10 593 human samples across 32 cancer types and normal controls. We find that the majority of previously reported sites show little to no evidence for editing in this dataset, compile a list of 58 reliable miRNA editing sites, and study them across normal and cancer samples. Edited miRNA versions tend to suppress expression of known oncogenes, and, consistently, we observe a clear global tendency for hypo-editing in tumors, in strike contrast to the behavior for mRNA editing, allowing an accurate classification of normal/tumor samples based on their miRNA editing profile. In many cancers this profile correlates with patients' survival. Finally, thousands of miRNA binding sites are differentially edited in cancer. Our study thus establishes the important effect of RNA editing on miRNA-regulation in the tumor cell, with prospects for diagnostic and prognostic applications.
INTRODUCTION
A-to-I RNA editing by the ADAR enzymes is a process that alters the content of RNA molecules post-transcriptionally, by deamination of Adenosines into Inosines (1,2). In many cellular processes, including translation and splicing, inosines are read as guanosines (3). Thus, A-to-I editing can provide an additional layer of complexity to the transcriptome, diversifying the genetic information beyond the linear sequence encoded in the genome. However, the majority of editing activity targets non-coding regions of the RNA molecules (4,5), and thus has a limited impact on their function.
Nevertheless, some non-coding RNA sequences do have important functions, and their editing could therefore play a significant role in regulation. One class of functional non-coding RNAs is micro-RNAs (miRNAs). These are short (∼22nt), endogenous, RNAs molecules that regulate mRNA expression and translation. miRNA targeting is conferred by sequence complementarity between the seed region of the miRNA molecule (positions 2–7) and a recognition site within the target mRNA sequence (6–8). miRNAs are known to regulate most protein-coding genes, and their expression and regulation has been shown to have a substantial role in development and cancer (9). Accordingly, editing of the miRNA sequence (especially its seed region) or editing of its recognition sites, are expected to modify the miRNA-target interaction and have a profound effect on gene regulation (10–12).
Notably, mature miRNAs are cleaved and processed from parent RNA molecules that have a distinct double stranded RNA (dsRNA) secondary structure, the structural motif preferred by the ADAR enzymes (13,14). Moreover, DICER, one of the major players in miRNA maturation, is known to create a complex with ADAR1 (15). Thus, it may be expected that miRNAs will be extensively edited. Indeed, a decade ago it was explicitly demonstrated how A-to-I editing of a single site can alter miRNA target specificity, leading to changes in targets' expression levels (12).
Several groups have attempted to systematically map the miRNA editome. Altogether, these reports documented editing events in ∼100 human miRNAs. However, the vast majority of detected sites are edited to a very low level (<1%), which most likely has no biological implication. Furthermore, the overlap between the lists of sites reported by the different studies is rather low, suggesting insufficient specificity and/or sensitivity.
Earlier this year, Wang et al. (16) have published comprehensive analyses of 8595 miRNA-seq samples representing 20 cancer types, using a de novo detection method. This method allowed them to identify 19 editing sites within mature microRNAs. They were able to experimentally validate 15 of them and to correlate their editing levels with patients’ survival, disease state and tumor subtype. Moreover, they showed that editing in the miR-200b seed region redirects its targets to alternate miR-200b role in cancer metastasis.
Here, we take a different, complementary, approach to screen for editing events using the large scale expression dataset of The Cancer Genome Atlas (TCGA): 10 593 miRNA-seq samples representing 32 different cancer types and normal controls (17,18). We complemented the de novo approach by profiling the editing levels at 58 pre-known, reliable, miRNA editing sites. Thus, we were able to find many more cases where editing levels correlate with diagnostic and prognostic state, and, more importantly, identify some general characteristics of the cancer editome, as well as showing the predictive power of the editing profile. In contrast with mRNA editing, miRNA editing is globally suppressed in cancer. Editing profiles may be used to classify the tissue as normal or cancerous, as well as predict patients' prognosis. Furthermore, we study the effect of RNA editing on miRNA recognition sites within mRNA targets, and find thousands of sites in hundreds of different genes in which miRNA recognition sites are differentially edited in cancer. This previously neglected mode of interaction between miRNA regulation and RNA editing could therefore be as important as direct editing of miRNA molecules.
MATERIALS AND METHODS
Data retrieval
10 593 miRNA-seq samples originating from 32 human cancer types and 23 normal tissue types (Supplementary Table S1) were downloaded from ‘The Cancer Genome Atlas’ (TCGA; http://tcga-data.nci.nih.gov) via The Cancer Genomics Hub (CGHub) (19). Strand-specific 22nt-enriched sequencing libraries were constructed and sequenced as described (18).
Detection of de novo sites
Pre-processing
BAM files were converted to FASTQ using BEDtools (20), reads with low quality were filtered out using FASTX. Only reads with length between 15–28 were kept for the alignment.
Alignment
FASTQ files were aligned to the human reference genome (hg19) using Bowtie (21) with recommended parameters as described (22,23).
De novo detection
editing sites were detected using the Analyze_mutation.pl and Binomial_analysis.pl scripts (22). Briefly, for each position along a miRNA we counted the high-quality (Q ≥ 30) read-nucleotides mapped to it. We used a binomial test followed by FDR correction in order to identify locations where the nucleotide distribution cannot be explained by random sequencing errors (statistically significant mismatches).
Parameters’ Optimization
Several features might influence the detection of reliable editing sites (number of samples, sequencing-depth, miRNA expression levels and editing-enzyme expression levels). These features vary across the 55 sample types we analyzed. Hence, we optimized the parameters (minimal editing levels of a mismatch and minimal percent of samples that supports a mismatch in a groups of samples from the same type) for each tissue-type separately. For each group of samples from the same type the combination of the two parameters with the highest ratio of |$\frac{{\# \ {\rm of}\ {\rm mismatches}\ {\rm from}\ {\rm most}\ {\rm abundant}\ {\rm type}}}{{{\rm Total}\ {\rm number}\ {\rm of}\ {\rm mismatches}}}$| was used.
Calculation of editing levels of known sites across all samples
We collected a list of 129 A-to-I editing sites within mature human miRNAs from 17 different studies. In order to measure the editing levels at these sites, we used the same alignment of the TCGA data to the human genome and kept only bases with Phred score Q ≥ 30. We used mpileup for base calling for each of the known editing sites and calculated the editing levels as the ratio between the number of G bases and the sum of A and G bases.
We looked for sites showing editing at a level significantly higher than 1% (using the binomial test followed by FDR multiple testing correction) in at least one of the 55 samples types.
Significant differential editing (between cancer and normal samples) was determined using the two-tailed Mann–Whitney test followed by a Benjamini–Hochberg multiple testing correction.
Prediction of sample state
To show how the editing profile can be used to predict the sample state we used logistic regression to build the statistical model. We then applied the leave-one-out cross validation method to assess accuracy. Namely, for each tissue type we used all samples but one as a training set in order to determine the parameters of the logistic regression, and then calculated the probability of a correct prediction for the left out sample. As usual, different cutoffs result in varying specificities and sensitivities (see Figure 4D). The overall success is evaluated by the AUC (area under curve).
Correlation between editing and survival
Kaplan–Meier analysis was applied to test for differences in survival between patients with high (>median) and low (≤median) editing levels. P-values were corrected using FDR.
miRNA target prediction
We predicted the targets of both the unedited and edited seeds using TargetScan 7 (24), assuming inosines behave like guanosines. Pathway enrichment was assessed using Ingenuity Pathway Analysis (IPA, http://www.ingenuity.com).
Differential editing in miRNA targets
We analyzed RNA-seq data available from TCGA for 9 cancer types having tumor samples and matched controls from the same patients (BLCA, BRCA, COAD, HNSC, KIRC, LIHC, LUAD, PRAD, THCA, see Supplementary Table S2). Using the method described in (4) we generated a list of 70 687 RNA editing events within Alu repeats in 3′UTRs. In order to find loss of function alterations in miRNA targets, we have downloaded the human miRNA predicted targets by TargetScanHuman 7.1 (24), and used ANNOVAR (25) to find editing sites that modifies these targets. We then calculated the editing levels of these sites using REDITools (26) script, trimming 6bp on both sides of the read. Statistical significance was evaluated using Wilcoxon signed-rank test, followed by FDR multiple testing correction. To find miRNA gain alterations which reside within 3′UTR Alu sequences, we looked at perfect complementary matches between editing sites to 8-mer, 7mer-a1 and 7mer-m8 seeds, and applied the same statistical analysis as described above.
RESULTS
De novo detection lead to small number of sites
Several de novo methods for detecting miRNA editing based on small-RNA sequencing data were developed recently (22,23,27–38). Basically, they all align the miRNA-seq reads to the genome, or to a reference miRNA sequences, and then look for systematic mismatches where the reference genomic sequence reads ‘A’ for an adenosine, but the miRNA-seq reads show a ‘G’ for a guanosine. Such mismatches could implicate an inosine in the actual RNA molecule. One might have thought that the tight dsRNA structure of the primary miRNA molecules would make them a favourable target of the ADAR enzymes. Interestingly, though, only a limited number of editing sites were found in mature miRNAs, and the editing levels observed are, in most sites, rather low (see below). However, these studies were typically based on analysis of only a few samples each (36,38–41), and the overlap between different studies is generally low (Supplementary Figure S1). The low overlap could point to low sensitivity, where each study finds only a small fraction of the whole set (maybe due to the limited number of samples). In addition, it may attest for low specificity, a large number of false detections in each study, not reproduced in other studies.
In order to clarify the situation, we analyzed here the large TCGA database, including miRNA-sequencing data for 10 593 samples originating from 55 different human tissue types (32 different cancer types, and normal controls for 23 tissues, see Supplementary Table S1), in order to obtain a comprehensive picture of the miRNA editome in normal and cancerous tissues. We first employed the approach designed by Alon and Eisenberg (22,35,37), (see Materials and Methods), to detect editing sites de novo. Briefly, reads were pre-processed, mapped to the pre-miRNAs in the human genome (hg19) using the Bowtie (21) alignment tool, and mismatches were called. We applied a binomial test to remove random sequencing errors, and additional cutoffs for minimal average editing levels and minimal fractions of samples showing editing. Cutoffs were set to each of the 55 tissue types separately (see methods). Altogether, 34 154 mismatches were detected, 32 229 (94.3%) of them were A-to-G mismatches mapped to only 19 unique editing site (Supplementary Figure S2). All of these 19 locations were previously reported to be edited. The detection scheme used here is rather restrictive, and certainly misses some sites (see below), but considering the large size and wide spectrum of tissues analyzed, the result suggests that the scope of editing in mature human miRNAs is rather narrow, and that previous reports have nearly exhausted the set of miRNA editing sites. In the following we therefore focus on these previously reported sites.
Most of previously reported sites exhibits no evidence for editing in the TCGA dataset
We then tested whether previously reported miRNA editing sites are observed as edited in our large-scale database (even if undetected by the de novo approach). We collected a set of 129 editing sites in human mature-miRNA from 17 different studies (12,23,36,38–51), (Supplementary Table S3), and measured the editing levels at each site for each of the TCGA samples (see methods). Notably, only 55 sites have shown an averaged editing level above 1% in at least one tissue type (Figure 1A), suggesting that many of the previously reported miRNA editing sites are either edited to a very low editing level (possibly below biological significant), edited (or expressed) only in a rare tissue or condition that is not represented in the large cohort of the TCGA dataset, or are not edited at all (i.e. being false-positives of the detection methods used in previous studies). Three additional sites showed editing levels which were lower but very close to 1%, and we included them to create a list of 58 high-confidence miRNA editing sites (Table 1; Figure 1B). Most of these sites, 48/58 (83%), are located in bona-fide mature miRNAs as defined by the MirGene database (52), compared to only 47/71 (66%) of the sites discarded (P-value = 0.04; Fisher's exact test).
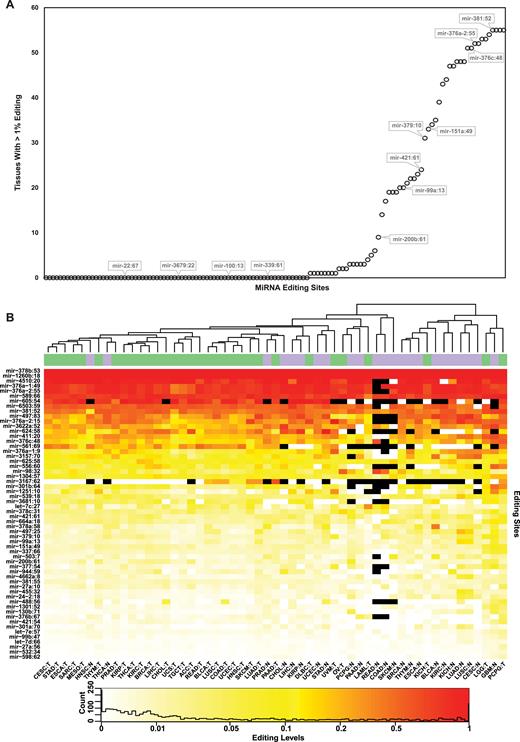
Editing profiles of the 58 reliable A-to-I editing sites. (A) The editing level per tissue was calculated for each of the previously reported 129 sites, coalescing all samples available for each of the 55 tissue-types (normal and cancer are considered separate types). For each site, we present the number of tissue types for which the editing level exceeds 1% editing. Notably, 74 sites exhibit levels lower than 1% in all 55 tissue types (Supplementary Table S3). (B) Editing levels per site and tissue type. Sites are ordered by their averaged editing levels. Unsupervised hierarchical clustering of tissues largely separates them to normal and tumors (purple and green on top bar, respectively). Black cells represents low coverage (<10 reads). See Supplementary Table S1 for tissues acronyms.
List of the 58 high-confidence sites
| miRNA | Locus | Maximal editing levels | Tissue type | In seed? | Validated by miRgene? |
|---|---|---|---|---|---|
| hsa-let-7c | 27 | 27.4 | LAML Tumor | No | Yes |
| hsa-let-7d | 66 | 0.8 | LUSC Normal | Yes | Yes |
| hsa-let-7e | 57 | 1.7 | PCPG Tumor | Yes | Yes |
| hsa-mir-1251 | 10 | 28.6 | GBM Normal | Yes | Yes |
| hsa-mir-1260b | 18 | 100.0 | GBM Normal | No | No |
| hsa-mir-1301 | 52 | 4.5 | GBM Normal | Yes | Yes |
| hsa-mir-1304 | 57 | 17.9 | GBM Normal | Yes | No |
| hsa-mir-130b | 71 | 4.6 | PCPG Normal | No | Yes |
| hsa-mir-151a | 49 | 7.8 | GBM Normal | Yes | Yes |
| hsa-mir-200b | 61 | 6.3 | PCPG Normal | Yes | Yes |
| hsa-mir-24–2 | 18 | 5.0 | LUAD Normal | Yes | Yes |
| hsa-mir-27a | 10 | 3.8 | PCPG Tumor | No | Yes |
| hsa-mir-27a | 56 | 1.4 | PCPG Tumor | Yes | Yes |
| hsa-mir-301a | 70 | 3.4 | LUAD Normal | No | Yes |
| hsa-mir-301b | 64 | 35.7 | GBM Normal | No | Yes |
| hsa-mir-3157 | 70 | 34.5 | BLCA Normal | No | No |
| hsa-mir-3167 | 62 | 25.0 | LUAD Tumor | No | No |
| hsa-mir-337 | 66 | 6.3 | GBM Normal | Yes | Yes |
| hsa-mir-3622a | 52 | 60.3 | GBM Normal | Yes | Yes |
| hsa-mir-3681 | 10 | 9.6 | LIHC Tumor | Yes | Yes |
| hsa-mir-376a-1 | 9 | 42.9 | UVM Tumor | Yes | Yes |
| hsa-mir-376a-1 | 49 | 90.0 | THCA Normal | Yes | Yes |
| hsa-mir-376a-2 | 15 | 69.2 | LUAD Normal | Yes | Yes |
| hsa-mir-376a-2 | 55 | 89.4 | PRAD Normal | Yes | Yes |
| hsa-mir-376b | 67 | 3.6 | PAAD Normal | Yes | Yes |
| hsa-mir-376c | 48 | 44.8 | PCPG Tumor | Yes | Yes |
| hsa-mir-377 | 54 | 7.5 | GBM Normal | No | Yes |
| hsa-mir-378a | 58 | 32.2 | BLCA Normal | No | Yes |
| hsa-mir-378b | 53 | 100.0 | LAML Tumor | No | No |
| hsa-mir-378c | 31 | 12.8 | OV Tumor | No | No |
| hsa-mir-379 | 10 | 8.1 | GBM Normal | Yes | Yes |
| hsa-mir-381 | 52 | 61.8 | PCPG Tumor | Yes | Yes |
| hsa-mir-381 | 55 | 1.5 | PCPG Tumor | Yes | Yes |
| hsa-mir-411 | 20 | 53.6 | GBM Normal | Yes | Yes |
| hsa-mir-421 | 54 | 3.6 | PCPG Tumor | Yes | Yes |
| hsa-mir-421 | 61 | 7.6 | PCPG Tumor | No | Yes |
| hsa-mir-4510 | 20 | 100.0 | PCPG Normal | No | No |
| hsa-mir-455 | 32 | 9.1 | LUAD Normal | No | Yes |
| hsa-mir-4662a | 8 | 6.7 | LUSC Normal | Yes | Yes |
| hsa-mir-488 | 56 | 4.9 | PCPG Tumor | Yes | Yes |
| hsa-mir-497 | 25 | 17.4 | LUAD Normal | Yes | Yes |
| hsa-mir-497 | 83 | 71.5 | LUSC Normal | No | Yes |
| hsa-mir-503 | 7 | 7.8 | BLCA Normal | Yes | Yes |
| hsa-mir-532 | 34 | 0.6 | PCPG Tumor | No | Yes |
| hsa-mir-539 | 18 | 16.2 | LUAD Normal | No | Yes |
| hsa-mir-556 | 60 | 30.3 | PCPG Tumor | Yes | Yes |
| hsa-mir-561 | 69 | 38.2 | READ Tumor | No | Yes |
| hsa-mir-589 | 66 | 93.9 | PCPG Tumor | Yes | Yes |
| hsa-mir-598 | 62 | 1.0 | PCPG Tumor | Yes | No |
| hsa-mir-605 | 54 | 83.3 | COAD Tumor | Yes | Yes |
| hsa-mir-624 | 58 | 39.3 | LIHC Normal | Yes | Yes |
| hsa-mir-625 | 58 | 16.9 | KICH Normal | Yes | Yes |
| hsa-mir-6503 | 59 | 67.8 | LIHC Normal | Yes | No |
| hsa-mir-664a | 18 | 11.3 | GBM Normal | No | No |
| hsa-mir-944 | 59 | 7.5 | KICH Normal | Yes | Yes |
| hsa-mir-98 | 32 | 55.6 | READ Normal | No | Yes |
| hsa-mir-99a | 13 | 12.4 | LUAD Normal | No | Yes |
| hsa-mir-99b | 47 | 1.3 | PCPG Tumor | Yes | Yes |
| miRNA | Locus | Maximal editing levels | Tissue type | In seed? | Validated by miRgene? |
|---|---|---|---|---|---|
| hsa-let-7c | 27 | 27.4 | LAML Tumor | No | Yes |
| hsa-let-7d | 66 | 0.8 | LUSC Normal | Yes | Yes |
| hsa-let-7e | 57 | 1.7 | PCPG Tumor | Yes | Yes |
| hsa-mir-1251 | 10 | 28.6 | GBM Normal | Yes | Yes |
| hsa-mir-1260b | 18 | 100.0 | GBM Normal | No | No |
| hsa-mir-1301 | 52 | 4.5 | GBM Normal | Yes | Yes |
| hsa-mir-1304 | 57 | 17.9 | GBM Normal | Yes | No |
| hsa-mir-130b | 71 | 4.6 | PCPG Normal | No | Yes |
| hsa-mir-151a | 49 | 7.8 | GBM Normal | Yes | Yes |
| hsa-mir-200b | 61 | 6.3 | PCPG Normal | Yes | Yes |
| hsa-mir-24–2 | 18 | 5.0 | LUAD Normal | Yes | Yes |
| hsa-mir-27a | 10 | 3.8 | PCPG Tumor | No | Yes |
| hsa-mir-27a | 56 | 1.4 | PCPG Tumor | Yes | Yes |
| hsa-mir-301a | 70 | 3.4 | LUAD Normal | No | Yes |
| hsa-mir-301b | 64 | 35.7 | GBM Normal | No | Yes |
| hsa-mir-3157 | 70 | 34.5 | BLCA Normal | No | No |
| hsa-mir-3167 | 62 | 25.0 | LUAD Tumor | No | No |
| hsa-mir-337 | 66 | 6.3 | GBM Normal | Yes | Yes |
| hsa-mir-3622a | 52 | 60.3 | GBM Normal | Yes | Yes |
| hsa-mir-3681 | 10 | 9.6 | LIHC Tumor | Yes | Yes |
| hsa-mir-376a-1 | 9 | 42.9 | UVM Tumor | Yes | Yes |
| hsa-mir-376a-1 | 49 | 90.0 | THCA Normal | Yes | Yes |
| hsa-mir-376a-2 | 15 | 69.2 | LUAD Normal | Yes | Yes |
| hsa-mir-376a-2 | 55 | 89.4 | PRAD Normal | Yes | Yes |
| hsa-mir-376b | 67 | 3.6 | PAAD Normal | Yes | Yes |
| hsa-mir-376c | 48 | 44.8 | PCPG Tumor | Yes | Yes |
| hsa-mir-377 | 54 | 7.5 | GBM Normal | No | Yes |
| hsa-mir-378a | 58 | 32.2 | BLCA Normal | No | Yes |
| hsa-mir-378b | 53 | 100.0 | LAML Tumor | No | No |
| hsa-mir-378c | 31 | 12.8 | OV Tumor | No | No |
| hsa-mir-379 | 10 | 8.1 | GBM Normal | Yes | Yes |
| hsa-mir-381 | 52 | 61.8 | PCPG Tumor | Yes | Yes |
| hsa-mir-381 | 55 | 1.5 | PCPG Tumor | Yes | Yes |
| hsa-mir-411 | 20 | 53.6 | GBM Normal | Yes | Yes |
| hsa-mir-421 | 54 | 3.6 | PCPG Tumor | Yes | Yes |
| hsa-mir-421 | 61 | 7.6 | PCPG Tumor | No | Yes |
| hsa-mir-4510 | 20 | 100.0 | PCPG Normal | No | No |
| hsa-mir-455 | 32 | 9.1 | LUAD Normal | No | Yes |
| hsa-mir-4662a | 8 | 6.7 | LUSC Normal | Yes | Yes |
| hsa-mir-488 | 56 | 4.9 | PCPG Tumor | Yes | Yes |
| hsa-mir-497 | 25 | 17.4 | LUAD Normal | Yes | Yes |
| hsa-mir-497 | 83 | 71.5 | LUSC Normal | No | Yes |
| hsa-mir-503 | 7 | 7.8 | BLCA Normal | Yes | Yes |
| hsa-mir-532 | 34 | 0.6 | PCPG Tumor | No | Yes |
| hsa-mir-539 | 18 | 16.2 | LUAD Normal | No | Yes |
| hsa-mir-556 | 60 | 30.3 | PCPG Tumor | Yes | Yes |
| hsa-mir-561 | 69 | 38.2 | READ Tumor | No | Yes |
| hsa-mir-589 | 66 | 93.9 | PCPG Tumor | Yes | Yes |
| hsa-mir-598 | 62 | 1.0 | PCPG Tumor | Yes | No |
| hsa-mir-605 | 54 | 83.3 | COAD Tumor | Yes | Yes |
| hsa-mir-624 | 58 | 39.3 | LIHC Normal | Yes | Yes |
| hsa-mir-625 | 58 | 16.9 | KICH Normal | Yes | Yes |
| hsa-mir-6503 | 59 | 67.8 | LIHC Normal | Yes | No |
| hsa-mir-664a | 18 | 11.3 | GBM Normal | No | No |
| hsa-mir-944 | 59 | 7.5 | KICH Normal | Yes | Yes |
| hsa-mir-98 | 32 | 55.6 | READ Normal | No | Yes |
| hsa-mir-99a | 13 | 12.4 | LUAD Normal | No | Yes |
| hsa-mir-99b | 47 | 1.3 | PCPG Tumor | Yes | Yes |
Sites with at least one tissue type with >1% editing levels were kept for further analyses. miRNA annotations are based on miRBase db, location represents the position of the editing site within the pre-microRNA. The reported maximal editing levels represents the highest (weighted average) editing level across the 55 tissues studied. See Supplementary Table S1 for tissues acronyms.
| miRNA | Locus | Maximal editing levels | Tissue type | In seed? | Validated by miRgene? |
|---|---|---|---|---|---|
| hsa-let-7c | 27 | 27.4 | LAML Tumor | No | Yes |
| hsa-let-7d | 66 | 0.8 | LUSC Normal | Yes | Yes |
| hsa-let-7e | 57 | 1.7 | PCPG Tumor | Yes | Yes |
| hsa-mir-1251 | 10 | 28.6 | GBM Normal | Yes | Yes |
| hsa-mir-1260b | 18 | 100.0 | GBM Normal | No | No |
| hsa-mir-1301 | 52 | 4.5 | GBM Normal | Yes | Yes |
| hsa-mir-1304 | 57 | 17.9 | GBM Normal | Yes | No |
| hsa-mir-130b | 71 | 4.6 | PCPG Normal | No | Yes |
| hsa-mir-151a | 49 | 7.8 | GBM Normal | Yes | Yes |
| hsa-mir-200b | 61 | 6.3 | PCPG Normal | Yes | Yes |
| hsa-mir-24–2 | 18 | 5.0 | LUAD Normal | Yes | Yes |
| hsa-mir-27a | 10 | 3.8 | PCPG Tumor | No | Yes |
| hsa-mir-27a | 56 | 1.4 | PCPG Tumor | Yes | Yes |
| hsa-mir-301a | 70 | 3.4 | LUAD Normal | No | Yes |
| hsa-mir-301b | 64 | 35.7 | GBM Normal | No | Yes |
| hsa-mir-3157 | 70 | 34.5 | BLCA Normal | No | No |
| hsa-mir-3167 | 62 | 25.0 | LUAD Tumor | No | No |
| hsa-mir-337 | 66 | 6.3 | GBM Normal | Yes | Yes |
| hsa-mir-3622a | 52 | 60.3 | GBM Normal | Yes | Yes |
| hsa-mir-3681 | 10 | 9.6 | LIHC Tumor | Yes | Yes |
| hsa-mir-376a-1 | 9 | 42.9 | UVM Tumor | Yes | Yes |
| hsa-mir-376a-1 | 49 | 90.0 | THCA Normal | Yes | Yes |
| hsa-mir-376a-2 | 15 | 69.2 | LUAD Normal | Yes | Yes |
| hsa-mir-376a-2 | 55 | 89.4 | PRAD Normal | Yes | Yes |
| hsa-mir-376b | 67 | 3.6 | PAAD Normal | Yes | Yes |
| hsa-mir-376c | 48 | 44.8 | PCPG Tumor | Yes | Yes |
| hsa-mir-377 | 54 | 7.5 | GBM Normal | No | Yes |
| hsa-mir-378a | 58 | 32.2 | BLCA Normal | No | Yes |
| hsa-mir-378b | 53 | 100.0 | LAML Tumor | No | No |
| hsa-mir-378c | 31 | 12.8 | OV Tumor | No | No |
| hsa-mir-379 | 10 | 8.1 | GBM Normal | Yes | Yes |
| hsa-mir-381 | 52 | 61.8 | PCPG Tumor | Yes | Yes |
| hsa-mir-381 | 55 | 1.5 | PCPG Tumor | Yes | Yes |
| hsa-mir-411 | 20 | 53.6 | GBM Normal | Yes | Yes |
| hsa-mir-421 | 54 | 3.6 | PCPG Tumor | Yes | Yes |
| hsa-mir-421 | 61 | 7.6 | PCPG Tumor | No | Yes |
| hsa-mir-4510 | 20 | 100.0 | PCPG Normal | No | No |
| hsa-mir-455 | 32 | 9.1 | LUAD Normal | No | Yes |
| hsa-mir-4662a | 8 | 6.7 | LUSC Normal | Yes | Yes |
| hsa-mir-488 | 56 | 4.9 | PCPG Tumor | Yes | Yes |
| hsa-mir-497 | 25 | 17.4 | LUAD Normal | Yes | Yes |
| hsa-mir-497 | 83 | 71.5 | LUSC Normal | No | Yes |
| hsa-mir-503 | 7 | 7.8 | BLCA Normal | Yes | Yes |
| hsa-mir-532 | 34 | 0.6 | PCPG Tumor | No | Yes |
| hsa-mir-539 | 18 | 16.2 | LUAD Normal | No | Yes |
| hsa-mir-556 | 60 | 30.3 | PCPG Tumor | Yes | Yes |
| hsa-mir-561 | 69 | 38.2 | READ Tumor | No | Yes |
| hsa-mir-589 | 66 | 93.9 | PCPG Tumor | Yes | Yes |
| hsa-mir-598 | 62 | 1.0 | PCPG Tumor | Yes | No |
| hsa-mir-605 | 54 | 83.3 | COAD Tumor | Yes | Yes |
| hsa-mir-624 | 58 | 39.3 | LIHC Normal | Yes | Yes |
| hsa-mir-625 | 58 | 16.9 | KICH Normal | Yes | Yes |
| hsa-mir-6503 | 59 | 67.8 | LIHC Normal | Yes | No |
| hsa-mir-664a | 18 | 11.3 | GBM Normal | No | No |
| hsa-mir-944 | 59 | 7.5 | KICH Normal | Yes | Yes |
| hsa-mir-98 | 32 | 55.6 | READ Normal | No | Yes |
| hsa-mir-99a | 13 | 12.4 | LUAD Normal | No | Yes |
| hsa-mir-99b | 47 | 1.3 | PCPG Tumor | Yes | Yes |
| miRNA | Locus | Maximal editing levels | Tissue type | In seed? | Validated by miRgene? |
|---|---|---|---|---|---|
| hsa-let-7c | 27 | 27.4 | LAML Tumor | No | Yes |
| hsa-let-7d | 66 | 0.8 | LUSC Normal | Yes | Yes |
| hsa-let-7e | 57 | 1.7 | PCPG Tumor | Yes | Yes |
| hsa-mir-1251 | 10 | 28.6 | GBM Normal | Yes | Yes |
| hsa-mir-1260b | 18 | 100.0 | GBM Normal | No | No |
| hsa-mir-1301 | 52 | 4.5 | GBM Normal | Yes | Yes |
| hsa-mir-1304 | 57 | 17.9 | GBM Normal | Yes | No |
| hsa-mir-130b | 71 | 4.6 | PCPG Normal | No | Yes |
| hsa-mir-151a | 49 | 7.8 | GBM Normal | Yes | Yes |
| hsa-mir-200b | 61 | 6.3 | PCPG Normal | Yes | Yes |
| hsa-mir-24–2 | 18 | 5.0 | LUAD Normal | Yes | Yes |
| hsa-mir-27a | 10 | 3.8 | PCPG Tumor | No | Yes |
| hsa-mir-27a | 56 | 1.4 | PCPG Tumor | Yes | Yes |
| hsa-mir-301a | 70 | 3.4 | LUAD Normal | No | Yes |
| hsa-mir-301b | 64 | 35.7 | GBM Normal | No | Yes |
| hsa-mir-3157 | 70 | 34.5 | BLCA Normal | No | No |
| hsa-mir-3167 | 62 | 25.0 | LUAD Tumor | No | No |
| hsa-mir-337 | 66 | 6.3 | GBM Normal | Yes | Yes |
| hsa-mir-3622a | 52 | 60.3 | GBM Normal | Yes | Yes |
| hsa-mir-3681 | 10 | 9.6 | LIHC Tumor | Yes | Yes |
| hsa-mir-376a-1 | 9 | 42.9 | UVM Tumor | Yes | Yes |
| hsa-mir-376a-1 | 49 | 90.0 | THCA Normal | Yes | Yes |
| hsa-mir-376a-2 | 15 | 69.2 | LUAD Normal | Yes | Yes |
| hsa-mir-376a-2 | 55 | 89.4 | PRAD Normal | Yes | Yes |
| hsa-mir-376b | 67 | 3.6 | PAAD Normal | Yes | Yes |
| hsa-mir-376c | 48 | 44.8 | PCPG Tumor | Yes | Yes |
| hsa-mir-377 | 54 | 7.5 | GBM Normal | No | Yes |
| hsa-mir-378a | 58 | 32.2 | BLCA Normal | No | Yes |
| hsa-mir-378b | 53 | 100.0 | LAML Tumor | No | No |
| hsa-mir-378c | 31 | 12.8 | OV Tumor | No | No |
| hsa-mir-379 | 10 | 8.1 | GBM Normal | Yes | Yes |
| hsa-mir-381 | 52 | 61.8 | PCPG Tumor | Yes | Yes |
| hsa-mir-381 | 55 | 1.5 | PCPG Tumor | Yes | Yes |
| hsa-mir-411 | 20 | 53.6 | GBM Normal | Yes | Yes |
| hsa-mir-421 | 54 | 3.6 | PCPG Tumor | Yes | Yes |
| hsa-mir-421 | 61 | 7.6 | PCPG Tumor | No | Yes |
| hsa-mir-4510 | 20 | 100.0 | PCPG Normal | No | No |
| hsa-mir-455 | 32 | 9.1 | LUAD Normal | No | Yes |
| hsa-mir-4662a | 8 | 6.7 | LUSC Normal | Yes | Yes |
| hsa-mir-488 | 56 | 4.9 | PCPG Tumor | Yes | Yes |
| hsa-mir-497 | 25 | 17.4 | LUAD Normal | Yes | Yes |
| hsa-mir-497 | 83 | 71.5 | LUSC Normal | No | Yes |
| hsa-mir-503 | 7 | 7.8 | BLCA Normal | Yes | Yes |
| hsa-mir-532 | 34 | 0.6 | PCPG Tumor | No | Yes |
| hsa-mir-539 | 18 | 16.2 | LUAD Normal | No | Yes |
| hsa-mir-556 | 60 | 30.3 | PCPG Tumor | Yes | Yes |
| hsa-mir-561 | 69 | 38.2 | READ Tumor | No | Yes |
| hsa-mir-589 | 66 | 93.9 | PCPG Tumor | Yes | Yes |
| hsa-mir-598 | 62 | 1.0 | PCPG Tumor | Yes | No |
| hsa-mir-605 | 54 | 83.3 | COAD Tumor | Yes | Yes |
| hsa-mir-624 | 58 | 39.3 | LIHC Normal | Yes | Yes |
| hsa-mir-625 | 58 | 16.9 | KICH Normal | Yes | Yes |
| hsa-mir-6503 | 59 | 67.8 | LIHC Normal | Yes | No |
| hsa-mir-664a | 18 | 11.3 | GBM Normal | No | No |
| hsa-mir-944 | 59 | 7.5 | KICH Normal | Yes | Yes |
| hsa-mir-98 | 32 | 55.6 | READ Normal | No | Yes |
| hsa-mir-99a | 13 | 12.4 | LUAD Normal | No | Yes |
| hsa-mir-99b | 47 | 1.3 | PCPG Tumor | Yes | Yes |
Sites with at least one tissue type with >1% editing levels were kept for further analyses. miRNA annotations are based on miRBase db, location represents the position of the editing site within the pre-microRNA. The reported maximal editing levels represents the highest (weighted average) editing level across the 55 tissues studied. See Supplementary Table S1 for tissues acronyms.
The 58 sites in the restricted set show the well characterized 5′-UAG-3′ ADAR preference motif (23,43,45) (Figure 2A). Interestingly, editing in miRNAs is overrepresented in 3p miRNAs, compared with 5p miRNAs (P = 0.036; Figure 2B). Possibly, this might be related to the stronger dependence of 3p miRNAs on DICER (53), making them more exposed to ADAR1 which is in complex with DICER (15).
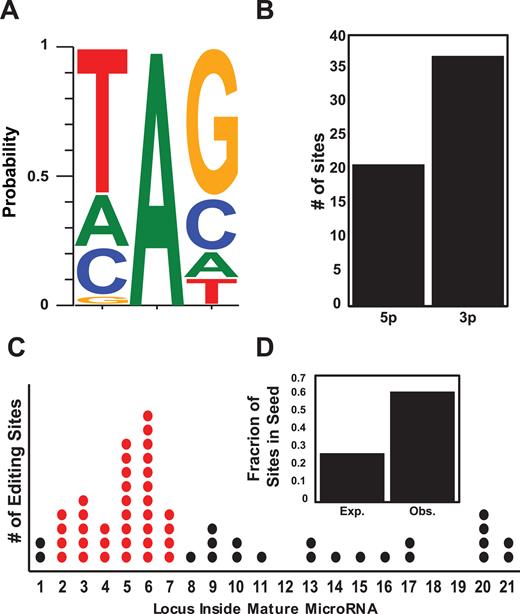
Characterizing miRNA editing sites. (A) Local sequence preference: distribution of nucleotides one position up- and down-stream of the 58 reliable miRNA editing sites exhibits the familiar ADAR motif. (B) Editing sites are more prevalent in 3′ microRNAs (P = 0.03, proportion test). (C) Editing sites distribution along the different positions within mature microRNAs. Positions 2–7 are the seed region, responsible for target recognition. (D) Expected and observed ratio of the 58 editing sites within the seed region (positions 2–7; O/E = 2.3, P = 2.7 × 10−8, χ2 test).
More importantly, the majority of these sites (36/58, 62%) reside in the seed region (Observed/Expected = 2.3, P = 2.7 × 10−8, χ2 test, Figure 2C and D). Target recognition of miRNA requires Watson–Crick pairing between the ‘seed’ region of a miRNA (nucleotides 2–7 in the mature sequence) and the targeted transcript (7,8,54,55). Hence, this region is highly conserved and mutations within it are rare (52,56). In contrast, editing events are enriched in the seed (33,57), strongly suggesting a functional role. In the following, we consider only these reliable 58 sites, and quantify their editing levels (hereafter, the editing profile) across samples. Note that even these are typically edited to low editing levels (Figure 1B).
The editing profile is markedly different in tumor tissues
In order to quantify the normal-cancer differences in the editing profile, we measured the editing level in each of the 58 editing sites for all samples of a specific cancer type, and compared it to the editing level observed in the normal controls (where available). Of the 1276 comparisons, we found 305 cases (24%) of significant (FDR < 0.05) differential editing (Figure 3A; Supplementary Table S4). Many of the comparisons not yielding a significant results are those with a very small number of normal samples, resulting in a poor statistical power. In most of the significantly different cases editing was lower in the cancer tissue (213/305 cases, 70%, P = 3.3 × 10−12, exact binomial test). In 34 cases (13 unique sites), the difference in editing levels between normal and tumors was larger than 10% (absolute difference), 29 of which having higher editing in normal tissues (Figure 3B). One can speculate that the differential editing reflects inversed differential expression of the edited miRNAs. However, no correlation was found between them (Supplementary Figure S3).
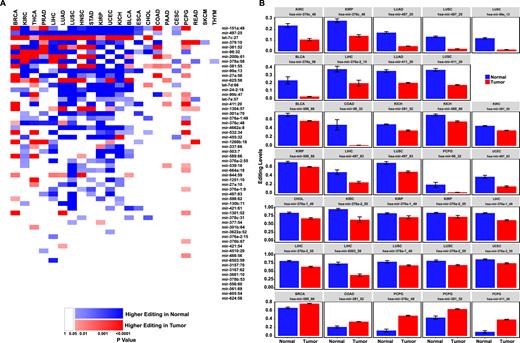
miRNA editing is suppressed in human cancers. (A) Editing levels were calculated for each site in each of the 22 tissues for which tumor and normal samples are available. Cases where editing levels are significantly higher in normal samples are colored in blue while cases where editing levels are significantly higher in tumor samples are colored in red. Darker shades of red/blue correspond to higher statistical significance (P value calculated by two-tailed Mann–Whitney test followed by a Benjamini–Hochberg multiple testing correction). Cases with P > 0.05 are colored in white. Tissue types are ordered from left to right by ascending number of normal samples (see Supplementary Table S1). Sites are ordered from top to bottom by ascending number of significant differences. In most sites and most cancer types, editing is higher in the normal tissue. (B) Editing levels of sites with significant and sizable (>10%) difference between normal and tumors. P < 0.05 Mann–Whitney test followed by a Benjamini Hochberg multiple testing correction. Error bars denote ± SEM. See Supplementary Table S1 for tissues acronyms.
The large fraction of sites showing differential editing suggests that the global editing profile might distinct between normal and cancer tissues. For each of the 55 cancer and normal tissue types we calculated the averaged editing level in each of the 58 sites, and clustered the tissues according to these averaged editing profiles. Interestingly, two large clusters appear. One which is pre-dominantly normal and the other pre-dominantly composed of cancer tissues (Figure 1B). In other words, the averaged editing profile for normal breast tissue is more similar to other normal tissues than it is to the averaged breast cancer profile. Taken together, these results point to a global tendency towards lower miRNA-editing in cancer tissues. To further demonstrate that, we looked at the averaged editing level over the 58 sites as a single measure of miRNA activity per sample, in all normal and tumor samples. In 19 of the 22 cancer types with matched normal controls the averaged editing level per sample was lower, on average (over samples) than that measured in the normal controls (significant difference in 13) (Figure 4A). Furthermore, we looked for site-site correlations between the editing profiles across all samples. Out of the 1653 site-pairs, we found significant positive correlation in 780 cases, compared to only 55 cases of negative correlation (Figure 4B). The same behavior emerges when correlations are calculated for each tissue type separately (Figure 4C). These observations again suggest that the differences in editing levels are controlled by a global tendency (towards reduction) in cancer samples.
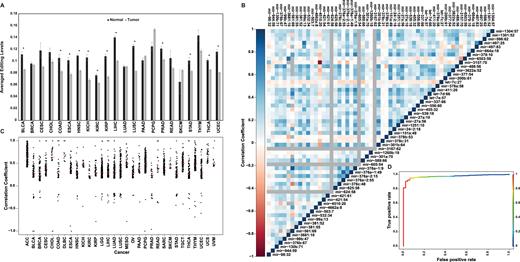
Global characterization of miRNA editing suppression in tumors. (A) As a global measure of miRNA editing, we looked at the editing level averaged over the 58 editing sites. In all but 3 of the 22 tissues shown, this average was higher in normal samples. Significant differential averaged editing (*P < 0.05, two-tailed t test) was found in 13/22 tissues, in all of which higher levels are observed in the normal samples. Error bars denote ±SEM. (B and C) Site-site Pearson's correlations of editing levels are almost all positive, reflecting the global nature of miRNA editing regulation. (B) Correlation matrix for all site pairs across all samples. (C) Significant Pearson's correlation coefficients per cancer (corrected P value ≤ 0.05). The vast majority (∼97%) of the significant correlations (3088 out of 3187) are positive. (D) The editing profile can be used to predict whether a sample is normal or cancerous. Here we present the ROC (receiver operating characteristic) curve for predicting the sample state (normal or tumor) of 422 Liver Hepatocellular Carcinoma (LIHC) samples. The AUC (area under curve) is 0.97. Coloring refers to the different cutoffs used to achieve the various false positive and negative rates (scale at the side). Similar results are observed for other tissues (see Supplementary Table S6). See Supplementary Table S1 for tissues acronyms.
The above results, showing a global and correlated change in the editing profile in cancer, raise the possibility that a combined global measure, taking into account changes in multiple sites, might better capture the normal-tumor differences. We used logistic regression analysis to fit a model that predicts whether a given editing profile describes a normal or a tumor sample. The fitted model was statistically significant for all cancer types (FDR < 0.05), providing high accuracy classification (average accuracy 0.97; Supplementary Table S5). In order to assess the predictive power, we used leave-one-out cross validation method (i.e. we fit the model with the full dataset leaving only one sample out, and then test for this single sample, repeating the procedure such that each sample is left out once). We found high accuracy in all cancer types (overall accuracy 0.94; see Supplementary Table S6 for specific cancers, and Figure 4D for a concrete example).
Higher editing levels are correlated with longer survival
If editing dysregulation is relevant to cancer progression, one may hypothesize that the editome profile correlates with patients’ survival. For each of the 58 sites in each of the 32 cancers-types in our database we divided the patients into two equal-sized groups based on their editing level—a group of patients with editing level higher than the median, and another one with levels below the median. Using Kaplan-Meier analysis we found 56 cases (26 editing sites; 15 cancer types) with significant (FDR < 0.2) difference in survival between the two groups (Supplementary Table S7). As expected from the previously described tendency for lower miRNA editing in tumors, we found better prognosis for the patients with higher editing levels in 40 out of the 56 cases (Supplementary Table S7). For example, Head and Neck squamous cell carcinoma (HNSC) patients with high editing levels in mir-376a-2 had more than 3-fold longer survival (median) compared to those with low editing levels (Figure 5).
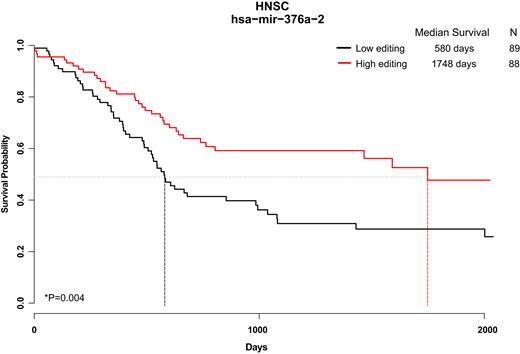
Editing levels correlate with clinical outcome. We present one example (out of 56 significant results, see Supplementary Table S7) where Kaplan-Meier analysis shows a significantly different clinical outcome depending on the editing level at a given site. Here, head and neck sarcoma (HNSC) patients are separated to two groups, higher and lower than median, based on the editing levels of mir-376a-2 in position 55 (Median editing level 73.8%). Median survival for the groups of patients are 1748 days (editing higher than median) and 580 days (lower editing). n = 177 patients; P = 0.00462.
Targets of edited miRNAs
Editing in the seed region of a miRNA will modify the set of targeted genes (12,41). For each of the eight miRNAs that are edited in the seed region and show a sizable (>10%) difference in editing level between normal and tumors in at least one cancer type (Figure 3B), we used TargetScan (24) to predict the targets for both the edited and unedited version. On average, the unedited and edited version share 36.4% of their target genes (Supplementary Table S8). Editing of miRNAs is only partial, and the unedited molecules still bind those targets that are exclusive to the unedited version. We thus focused on targets genes that are exclusive to the edited version, and used ingenuity pathway analysis (IPA) to look for association with diseases and pathways. Strikingly, for all the above eight edited miRNAs, the disease that was most significantly associated with genes targeted by the edited miRNAs was cancer (P < 0.001; Supplementary Figures S4–S11). In addition, cellular functions related to cell proliferation, growth or survival (Supplementary Tables S9–S16) were enriched. Interestingly, there are two genes that are targeted by the edited version of all of the above eight miRNAs (but not by their unedited version) (Supplementary Table S17). One is the growth and differentiation factor Activin receptor type-1B (ACVR1B) that was found to regulate cell proliferation and death. Somatic mutations in this gene were found in pancreatic carcinoma (58). The second target of these eight differentially edited miRNAs is CD30 (TNFRSF8), a positive regulator of apoptosis, a tumor marker, and a target of the drug Brentuximab vedotin which is used to treat patients with Hodgkin lymphoma (Figure 6A). Taken together, these observations further strengthen the link between differential miRNA editing and cancer.
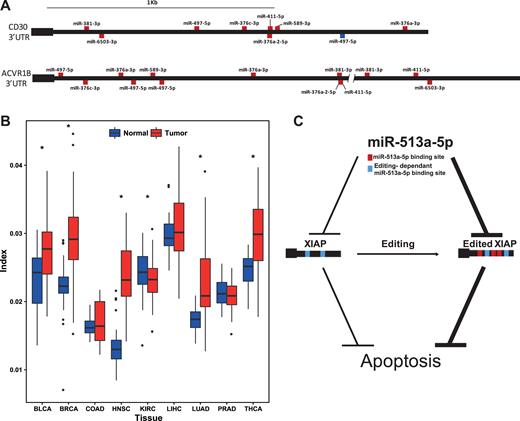
Suppressed miRNA-editing and elevated targets-editing affects miRNA-dependent gene regulation. (A) Two examples of genes, the cancer related CD30 and ACVR1B, gaining multiple new miRNA targets due to miRNA editing. Binding sites of unedited miRNAs are marked in blue, and those of edited-miRNAs are marked in red. (B) Editing of microRNA targets is elevated in tumors. Boxplot of editing index per sample for matched samples in 9 different cancers. Targets-editing is significantly (P < 0.05 Wilcoxon signed-rank test) higher in 5 cancers and lower in 1. See Supplementary Table S1 for tissues acronyms. (C) Suggested mechanism for activation of apoptosis by editing in 3′utr of the apoptosis inhibitor XIAP. Two target sites of miR-513a-5p are present in the XIAP transcript, enabling inhibition of XIAP, resulting in apoptosis. Alu editing of XIAP creates three additional target sites of the same miRNA, further inhibiting XIAP and inducing apoptosis.
A cross talk between editing in miRNAs and their targets
In addition to editing the miRNA sequence, RNA editing can affect miRNA regulation in yet another way, by editing and modifying miRNA-targets in 3′UTRs of mRNAs, destroying targets or creating new ones (11,59). Virtually, all human editing sites reside in Alu sequences, and thus we focused here on editing sites in Alu elements, within 3′UTRs, and studied 357 tumor RNA-seq samples from 9 cancer types and their matched normal samples (93% overlap with the miRNA-seq samples, see Methods and Supplementary Table S2). We looked for sites where editing creates a novel target of any miRNA seed (7mer-1A, 7mer-m8 and 8mer) or destroys complementarity between a miRNA and the UTR of the target. We found 63 308 such editing sites (out of 70 687 sites detected in Alu, see Methods) in 2687 genes. Thus, editing in Alu elements might be involved in the miRNA regulation of transcription of ∼10% of the genes in the human genome. Previous studies have found that Alu editing is globally higher in cancer (60–62). Accordingly, the weighted average level of the editing in the sites affecting miRNA recognition was higher in tumors than in their matched normal samples (0.026 and 0.022 respectively P = 3.55 × 10−22, Wilcoxon signed-rank test; Figure 6B and Supplementary Figure S12). Examining each of the 63 308 sites separately, we find 7982 sites in 583 genes that were significantly (FDR < 0.05) differentially edited between tumors and their matched normal samples (Supplementary Tables S18 and S19).
Some of the miRNAs gain multiple new targets. Interestingly, the miRNA hsa-miR-513a-5p gains the highest number of targets (214 new targets in 157 genes) as a result of target editing, as was noted by Borchert et al. (11) This miRNA is known to induce apoptosis via down-regulation of the apoptosis inhibitor XIAP (63). Three novel targets of miR-513a-5p are created by editing in the 3′ UTR of XIAP, editing levels of which is elevated in tumors. Thus, it seems editing plays a role in enabling XIAP inhibition by miR-513a-5p, thereby promoting apoptosis. In cancer, editing of the miRNA target is stronger, inhibition of XIAP increases, and the cell is further pushed towards apoptosis (Figure 6C). In addition, the conserved miRNA with highest number of lost targets (146 targets in 60 genes) is miR-129–5p, regulating cell proliferation, death, invasion and migration. This miRNA was found to be a biomarker for prognosis and diagnosis of dozens of cancers (64).
Editing of miRNA targets may have a sizable impact of the affected genes. The number of targets a specific mRNA transcript gains/losses due to editing may be comparable to the number of miRNA targets in the unedited transcript, and even higher (see Supplementary Table S20). Finally, there are cases in which editing creates both a novel miRNA isoform as well as its targets (Supplementary Table S21). Two genes were found to gain the highest number of novel edited target of an edited-miRNA: XIAP, which was mentioned before, and MAVS. Noticeably, MAVS is known to be tightly connected to ADAR1. The MDA5-MAVS RNA sensing pathway is negatively regulated by ADAR1 (65), and MAVS knockout is known to rescue ADAR1 knockouts (66), suggesting a delicate balance between the two (67). Possibly, editing of the miRNAs and their target serves as one of the feedback mechanisms required for maintaining this balance.
DISCUSSION
pri-miRNAs are known to form a rather tight dsRNA stem-loop structure, making them a perfect ADAR target. Thus, it may have been anticipated that they would be extensively edited, and that these editing events would result in edited mature miRNAs. Here we analyzed a large scale miRNA expression data and found that most of the previously reported sites are not edited in our dataset, suggesting they are either very weakly edited, edited under rather specific conditions, or not edited at all. Furthermore, even the 58 sites we found to be edited exhibit, mostly, very weak editing - the majority of them are edited to less than 5% in all tissues studied. Certainly our screen could have missed sites that are edited in tissues or conditions not represented in our samples. Nevertheless, it seems the big picture is that miRNAs are rather weakly edited, in general. Given the underlying dsRNA structure, one may wonder why would that be the case. Possibly, the proteins involved in miRNA processing block the ADAR enzymes and prevent, to most extent, editing of the pri-miRNAs. Alternatively, pri-miRNAs might be extensively edited, but editing suppressed their processing into mature miRNAs. Future experiments looking directly at pri-miRNA editing could help resolve this question.
We point out that computational prediction of miRNAs' re-targeting following editing are of limited accuracy. Accordingly, our results concerning any particular target should be taken as putative, and need to be confirmed experimentally. However, the statistical properties of the predicted targets as a group, are likely robust.
While this work has been prepared for publication, another work studying miRNA editing in the TCGA dataset has been published (16). Wang et al. have analyzed the 19 sites that were identified de novo based on the TCGA data alone, while we chose to look at all previously reported sites. Focusing on the 58 reliable ones, we were able to find a higher number of differentially edited sites and sites associated with survival. In addition, the larger number of sites allowed us to look at the data globally, identifying the general hypo-miRNA-editing occurring in tumors.
Interestingly, the editing sites we do observe tend to occur in the seed region, responsible for the miRNA target recognition. In contrast, genomic mutations are suppressed in the critical seed region (52,56), which is highly conserved across species. This stark difference may be understood in terms of an important difference between editing and genomic mutations - a mutation affects all copies of the miRNA, changing dramatically its target set and the associated regulation mechanisms. Thus, a random mutation in the seed region is usually quickly removed by purifying selection. miRNA editing, on the other hand, usually affects only a small fraction of the miRNA copies, allowing for the majority of the copies to keep their original function while presenting the cell with a new version of the molecule that can be exapted for new processes. Therefore, adaptation of editing sites in the seed region is a route for miRNA diversification that entails a lower evolutionary barrier compared with a genomic mutation.
The similarities and differences between A-to-I editing and genomic mutations carry over from the evolutionary considerations to the analysis of pathologies. Altered editing profiles may be viewed as ‘RNA mutations’ that, unlike genomic mutations, are highly dynamic and could be easier to exapt and adapt. It has been previously shown that mRNA editing activity is indeed altered in tumors. Intriguingly, unlike the global mRNA editing indices, editing in miRNAs is actually suppressed in most of human cancers. Either way, these alterations of editing may contribute to cancer transformation, an idea supported by the present results associating differential miRNA editing with cancer and its prognosis. Furthermore, miRNA profiling is currently being used clinically to identify the tissue of origin in cancer (68). Adding the miRNA editing information to the expression levels data could be used to improve identification power of liquid biopsy.
The global hypo-editing of miRNAs in tumors is surprising, as it was previously shown (using the same samples) that, generally speaking, ADAR1 expression levels and mRNA editing levels are elevated in many tumor types (61,62). This discrepancy seems to suggest a specific dysregulation mechanism relevant to miRNA editing, independent of the overall increase of ADAR1 levels. Alternatively, the difference may be explained by the activity of ADAR2, which is known to edit many miRNAs (2). Unlike ADAR1, ADAR2 was found to be downregulated in many cancers (Supplementary Figure S13). In addition, the increase in Alu editing coupled by decreased of miRNA editing may be related to a change in the balance between the interferon-induced isoform of ADAR1-p150 and the constitutively expressed ADAR1-p110 isoform. Unfortunately, it is difficult to measure the expression level of each variant from mRNA-seq data.
In addition to editing of miRNAs, we highlight here another way in which editing affects miRNA-mediated gene regulation, namely editing of miRNA targets. We show that this phenomenon is quite extensive, affecting hundreds of human genes, some of which gained or lost a sizable fraction of their miRNA targets, thus changing dramatically their miRNAs regulation by Alu editing within 3′ UTR human genes. In some cases the 3′ exon harbors two inverted Alu repeats, or more, that bind together to form a tight dsRNA structure. These are extensively edited, but do not make effective miRNA targets, as the mature miRNA cannot compete with the ∼300 bp-long inverted repeat (69). However, the majority of 3′ UTR Alus are edited in the pre-mRNA stage, where they pair with an intronic repeat, while the Alu in the mature mRNA is unpaired and may bind miRNAs.
As many of the human edited targets reside within Alu elements, their editing is, by and large, primate-specific, introduced by the recent Alu invasion to the primates genome. Thus, editing of miRNA targets is very much different from editing of the miRNAs themselves, which is often conserved across mammals. Accordingly, it is possible that many of the modified targets have no biological implication and may be considered ‘noise’. However, the few cases in which multiple targets are affected (some of them highlighted above) might have been adapted to provide a functional role, providing a primate-specific fine-tuning of the miRNA regulation pathway. Of particular interest is the MAVS gene. The primary role of ADAR1 is preventing MAVS activation by endogenous dsRNAs (65–67). Fascinatingly, MAVS is also regulated by ADAR1 through editing of miRNA targets within its 3′ UTR.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Tsviya Olender for helping with the analysis of the effect of editing in mRNAs 3′UTR and for using her scripts. We also want to thank Sivan Pinto for graphical help.
FUNDING
European Research Council [311257]; The Israel Science Foundation [1380/14 to E.Y.L., 379/12 to E.E.]; I-CORE Program of the Planning and Budgeting Committee in Israel [1796/12]. Funding for open access charge: The Israel Science Foundation.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments