





Abstract
The RNA structure and long-range interactions of the SL9266 cis -acting replication element located within the NS5B coding region of hepatitis C virus (HCV) were determined using selective 2′-hydroxyl acylation analysed by primer extension. Marked differences were found in the long-range interactions of SL9266 when the two widely used genotype 2a JFH-1 (HCVcc) and genotype 1b Con1b sub-genomic replicon systems were compared. In both genomes, there was evidence for interaction of the sub-terminal bulge loop of SL9266 and sequences around nucleotide 9110, though the replication phenotype of genomes bearing mutations that disrupted this interaction was fundamentally different. In contrast, a ‘kissing loop’ interaction between the terminal loop of SL9266 and sequences in the 3′-untranslated X-tail was only detectable in JFH-1-based genomes. In the latter, where both long-range interactions are present, they were independent, implying that SL9266 forms the core of an extended pseudoknot. The presence of the ‘kissing loop’ interaction inhibited the formation of SL9571 in the 3′-X-tail, an RNA structure implicated in genome replication. We propose that, SL9266 may contribute a switch function that modulates the mutually incompatible translation and replication events that must occur for replication of the positive-strand RNA genome of HCV.
INTRODUCTION
The positive-sense single-stranded RNA genome of hepatitis C virus (HCV), a flavivirus of the genus Hepacivirus that infects greater than 130 million people worldwide ( 1 ), is a template for two processes essential for virus propagation. First, following cell infection, the 9.6 kb genome functions as a template for translation, a process initiated at the internal ribosome entry site (IRES) in the 5′-non-coding region (5′-NCR), yielding a single polyprotein which is co- and post-translationally processed to yield the proteins necessary for genome replication and particle formation. Secondly, the genome is a template for replication, initially by synthesis of a complementary negative-strand and consequent formation of a double-stranded replicative intermediate, and then by the synthesis of new positive-sense genomes ( 2 ). RNA viruses have evolved a multitude of mechanisms to control these key events in their replication cycle, often involving genomic RNA secondary or higher-order structures ( 3–5 ). These provide binding sites for viral or cellular proteins which, together with RNA–RNA interactions, influence the temporal and spatial control of replication. The identification and functional analysis of RNA secondary structures is therefore of particular interest to our understanding of viral replication.
Two types of RNA secondary structure have been described in HCV. The first, designated genome-scale ordered RNA structure, extends throughout the genome and has an as-yet poorly understood role in virus persistence ( 6 , 7 ). In addition, there are a series of well-defined phylogenetically conserved structures, predominantly occupying the 5′ and 3′ extremities of the genome, extending from the untranslated regions into the open reading frame (ORF) ( 8–12 ). Although our understanding of these is incomplete, several are critical for genome replication and infectivity and so constitute cis -acting replication elements (CRE). At the 5′ end of the genome, the IRES contains four stem–loops (designated domains I–IV) totalling ∼340 nt with functions in ribosome recruitment, translation initiation and genome replication ( 13 ). The HCV IRES extends into the ORF, with recent studies suggesting that highly conserved structures (domains V and VI) in the region encoding the core protein are important for translation (10, 14 ). The observed suppression of synonymous sequence variation and phylogenetic analysis of thermodynamically predicted RNA structure supports the presence of at least two additional stem–loop elements within the core-coding region, though functions for these have yet to be determined ( 9 ).
The ∼200 nt HCV 3′-NCR is also extensively structured. Three discrete stem–loops (previously designated SLI–III, numbered from the 3′ end) together form the X-tail and are separated from the polyprotein-coding region by a hypervariable domain and pyrimidine-rich tract of variable length and sequence. Deletion and mutagenesis analysis shows the 3′ NCR has roles in genome replication and virion infectivity ( 12 , 15–17 ). The region 5′ proximal to the 3′ NCR, encoding the NS5B RNA-dependent RNA polymerase (RdRp) contains five additional phylogenetically conserved RNA stem–loop structures ( 8 , 9 , 11 , 18 ) designated, by the position of the 5′ nt in the H77 complete genome sequence (GenBank accession # AF011753), as SL9033, SL9132, SL9217, SL9266 and SL9324 (19 and see ′Materials and Methods' section). Of these, SL9266 (elsewhere designated 5BSL3.2 or SL-V) forms a hairpin, topped by a 12-nt terminal loop and interrupted on the 3′ side by an 8-nt bulge loop. Both unpaired loop regions of SL9266 are phylogenetically conserved and exhibit low levels of synonymous site sequence variation between divergent HCV genotypes, implying likely functional constraints on the evolution of these sequences out with their protein coding function (see Figure 4 in 11 ). Reverse genetic analysis, using either sub-genomic replicons or the JFH-1 replicating virus system, demonstrate that disruption (by synonymous substitution) of either the upper or lower duplex of SL9266 inhibits replication—SL9266 is therefore a CRE. SL9266 occupies a central position in a cruciform structure (3BSL3) involving the adjacent SL9217 (5BSL3.1) and SL9324 (5BSL3.3) stem–loops ( 11 , 17 , 18 ). However, mutagenesis or biophysical analyses have yet to demonstrate the existence of this higher-order structure, or to define SL9217 and SL9324 as CREs. SL9266 does not function in isolation; the structure forms at least two long-range RNA–RNA interactions which are critical for replication. Sequences in the apical loop of SL9266 are complementary to the terminal loop of the central stem–loop structure SL9571 (SLII) of the X-tail, and genetic studies confirm that this ‘kissing loop’ interaction is necessary for genome replication ( 17 , 20 , 21 ). More recently, we have used bioinformatic and reverse-genetic analysis to demonstrate a long-range interaction of the sub-terminal bulge loop of SL9266 with complementary sequences centred around nucleotide 9110, an unusually unstructured region of the genome flanked by conserved stem–loops (SL9033, SL9132) of untested function ( 22 ). Our study could not distinguish between the simultaneous or mutually exclusive interaction of SL9266 with the upstream and X-tail regions, or, considering the relatively weak bioinformatic prediction of SL9266 per se ( 8 , 22 ), a situation in which the simultaneous interaction with the 5′ and 3′ sequences resulted in the partial or complete unfolding of SL9266. Of these, we favoured the former and proposed that SL9266 formed the core of an extended pseudoknot which we designated SL9266/PK ( 22 ). The possible interactions of SL9266 have recently been further complicated by the demonstration that the sub-terminal bulge loop may bind sequences in domain IIId of the HCV IRES in biophysical assays ( 23 ).
The dissection of HCV replication was initially hampered by the inability to reproducibly grow the virus in cell culture. The development of culture-adapted sub-genomic replicon systems ( 24 ) in which genome replication confers a selectable advantage (e.g. antibiotic resistance), or allowed the quantifiable expression of a reporter gene (e.g. luciferase), enabled the reverse genetic analysis of HCV replication. More recently, the availability of genomes derived from the JFH-1 virus isolate has enabled the entire replication cycle to be analysed ( 25 ). Determining the phenotypic consequences of modification of the proposed SL9266/PK structure in our previous analysis was undertaken in a genotype 1b sub-genomic replicon ( 22 , 24 ). In this study, we have extended this analysis to JFH-1-based genomes.
In addition to genetic studies, previous NMR analysis ( 17 ) and RNA–RNA binding studies ( 23 ) have investigated the long-range interactions of SL9266 in trans (e.g. by seeding reactions with separate molecules containing the interacting 5′ and 3′ sequences). We reasoned that this situation was, at least for the genome that initiated cell infection, not representative and that the interactions observed may not reflect events in the cell due to the absence of other regions of the highly structured virus genome. We have therefore analysed the interplay of these regions by biophysical mapping of SL9266 using selective 2′-hydroxyl acylation analysed by primer extension (SHAPE) analysis ( 26 ). Our results support the long-range interaction we predicted bioinformatically in the Con1b-based replicon system. In addition, they demonstrate similar interactions occur in JFH-1, although the relative contributions of the 5′ and 3′ sequences differ. We extended this study to analyse the steady-state RNA structure of mutants that are known, or could be predicted, to influence RNA–RNA interactions of SL9266, and investigated the phenotype conferred by these mutations in the JFH-1 system.
We demonstrate that there are fundamental differences in the RNA–RNA interactions of SL9266 between the two virus genotypes. Additionally, we show that the phenotype caused by identical mutations in the two replication systems may also differ. Significantly, we show that the interactions with SL9266 directly influence the folding structure of parts of the X-tail region. We propose that long-range interactions of SL9266 determine the structure of cis -acting replication determinants in the X-tail and that the previously reported ‘kissing loop’ interaction may form a temporal switch required for the control of virus replication. These studies have implications for our understanding of HCV replication and for the identification of therapeutic targets to which novel anti-virals could be directed.
MATERIALS AND METHODS
Stem–loop nomenclature
Stem–loops are designated by the position of the first 5′ paired nucleotide in the structure ( 19 ) aligned and referenced to the H77 complete genome sequence (GenBank Accession #AF011753), this standardized naming scheme facilitates reference to structures in coding or non-coding regions, is independent of higher-order structures and can be logically extended as additional structures are discovered. Of relevance to this report, stem–loop structures named 5BSL3.1–3.3 ( 11 ), SLIV–VII ( 18 ) or SL8828, SL8926, SL9011, SL9061 and SL9118 ( 8 , 9 , 11 , 18 , 22 ) are designated here SL9033, SL9132, SL9217, SL9266 and SL9324, respectively. Likewise, the 5′ NCR stem–loop IIId ( 27 , 28 ) is SL253 and the three structures that together form the X-tail [5′-SLIII, SLII and SLI−3′; ( 29 )] are SL9548, SL9571 and SL9601. The same naming scheme was used in our previous report of an extended pseudoknot containing SL9266 ( 22 ).
Cell culture
Monolayers of the human hepatoma cell line Huh 7.5 (a generous gift from Charlie Rice) were maintained in Dulbecco’s modified minimal essential medium supplemented with 10% (v/v) fetal bovine serum (Invitrogen), 1% non-essential amino acids, 2 mM l -glutamine and 100 U penicillin/100 µg streptomycin/ml (DMEM P/S). Cells were passaged after trypsin/EDTA treatment and seeded at dilutions of 1:3 to 1:5.
HCV cDNA plasmids and mutagenesis
The parental firefly luciferase-encoding Con1b replicon, designated pFKnt341-sp-P1-lucEI3420-9605/5.1 (for convenience designated here as Con1b-luc-rep) was a generous gift from Ralf Bartenschlager and has been described previously ( 13 ). Mutations were introduced, using the Stratagene QuikChange™ system, to the unique SpeI–XhoI [nucleotide 5611–8031 (numbering refers to plasmid sequence, not that of the H77 reference virus, as some positions are within the vector backbone.)] sub-fragment of this plasmid cloned into pBluescript II SK (+) (Stratagene), their presence confirmed by DNA sequencing, and rebuilt into the parental plasmid. The HCVcc pFK-J6/JFH-1-C-846 (for convenience designated here as J6/JFH-1; generously provided by Takaji Wakita and NIH) full-length cDNA has previously been fully described ( 30 ). Mutations were introduced to the unique HindIII–SspI (nucleotides 8208–10128) fragment sub-cloned into pUC18, confirmed by DNA sequencing and the modified region rebuilt into the parental cDNA on a HindIII – MluI fragment (nucleotides 8208–9903). Replication-incompetent derivatives of both the replicon and HCVcc systems were generated by a GDD > GND substitution, within the active site of the NS5B polymerase as described previously ( 22 ).
In vitro RNA transcription
One microgram of either J6/JFH-1 plasmid cDNA, which includes a 3′ cis -acting ribozyme, or ScaI-linearized Con1b–luc–rep cDNA was used as template for the production of RNA in vitro using a T7 MEGAscript kit (Ambion), according to the manufacturers’ instructions. After transcription, the DNA template was removed by DNase 1 (Ambion) treatment and the RNA purified with an RNeasy mini-kit column (Qiagen). RNA integrity was confirmed by denaturing agarose gel electrophoresis and quantified by NanoDrop spectroscopy.
RNA modification for SHAPE
Templates for SHAPE reactions, either 40 pmol of a sub-genomic RNA transcript (nucleotide 9005 to the 3′ terminus) or 10 pmol of full-length J6/JFH-1 or Con1b–luc–rep RNA transcripts in 10 µl 0.5× Tris–EDTA (pH8.0) (TE), were denatured at 95°C for 3 min, incubated for 3 min on ice before addition of 6 µl of folding buffer [330 mM HEPES (pH 8.0), 20 mM MgCl 2 and 330 mM NaCl] and allowed to refold at 37°C for 20 min. Samples were then divided in half and incubated with either 1 µl of 100 mM N-methylisatoic anhydride (NMIA) dissolved in DMSO or 1 µl of DMSO for 45 min at 37°C. Each reaction was terminated by ethanol precipitation following the addition of 100 µl of EDTA (100 mM), 4 µl of NaCl (5 M) and 2 µl of glycogen (20 mg/ml). Samples were re-suspended in 0.5× TE containing RNA secure (Ambion) and heated to 65°C for 10 min before use in the primer extension reaction.
5′-[ 32 P]-primer labelling
A total of 60 µM of primer (Invitrogen) was incubated with 10 units of T4 polynucleotide kinase (New England Biolabs), 2 µl of supplied 10× buffer and 12.5 µl γ-[32p]-CTP (PerkinElmer) at 37°C for 20 min and stopped by incubation at 65°C for 20 min. Radiolabelled primers were purified through a 20% PAGE gel (7 M urea) and passively eluted from a gel slice overnight into water, ethanol precipitated and re-suspended in 100 µl 1 mM HEPES (pH 8.0) before use.
Primer extension reactions for SHAPE
NMIA- or control DMSO-treated RNA was mixed with 3 µl of 30 µM radiolabelled primer, denatured at 95°C for 5 min, annealed at 35°C for 5 min and chilled on ice for 2 min. About 6 µl of RT mix was added (5× Superscript III buffer, 17 mM DTT and 1.7 mM dNTPs; Invitrogen), the sample incubated at 55°C for 1 min before the addition of 1 µl of Superscript III (Invitrogen) and a further incubation at 55°C for 30 min. The DNA template was degraded with 1 µl of 4 M NaOH and incubation at 95°C for 5 min before the addition of 29 µl of acid stop mix (160 mM un-buffered Tris–HCl, 73% formamide, 0.43× TBE, 43 mM EDTA [pH 8.0], bromophenol blue and xylene cyanol tracking dyes) followed by a further 5 min incubation at 95°C. Dideoxynucleotide triphosphate (ddNTP) sequencing markers were generated by the extension of unmodified RNA after the addition of 2 µl of 20 mM ddNTP (Fermentas) prior to the addition of the RT mix. The cDNA extension products were separated by denaturing electrophoresis [7% (19:1) acrylamide:bisacrylamide, 1× TBE, 7 M urea] at 70 W for between 1 and 5 h (the duration being dependent on the fragment sizes analysed) and visualized with a phosphorimager (Fujitsu). Images were analysed for average band intensity/pixel in the NMIA- and DMSO control-reactions at every nucleotide position using TotalLab 1-D gel analysis software. Data was analysed as recently described ( 31 ). Briefly, after subtraction of the signal from the DMSO-control reaction, band intensity in the reaction was ranked, the top 2% discarded as being outliers and the total reactivity was normalized to the average of the next 8% of products.
Replicon analysis, virus recovery and quantification
Huh 7.5 cells were transfected by electroporation. Briefly, trypsinized and washed Huh 7.5 cells were re-suspended in phosphate buffered saline (PBS) at 3 × 10 7 cells/ml, mixed with 5 µg of in vitro transcribed RNA in a pre-chilled 4 mm cuvette, pulsed once (square wave pulse, 250 V for 25 ms) using a Bio-Rad Gene Pulser Xcell unit, before incubation on ice for 5 min and final resuspension in 10 ml of DMEM-P/S.
Luciferase expression by the Con1b–luc–rep replicon was determined at 4, 24, 48 and 72 h post-transfection from 2.5 ml of the transfected re-suspended cells transferred to a six-well plate, washed twice with PBS, lysed with 0.5 ml Glo-Lysis Buffer (Promega) and stored frozen prior to analysis using Bright-Glo luciferase substrate (Promega) and a Turner TL-20 luminometer.
The infectivity of J6/JFH-1 cDNA was determined with or without passage. Briefly, transfected Huh 7.5 cells (∼90% transfection efficiency) were incubated in a T75 flask until they reached ∼90% confluence at which point passage zero (p0) supernatant media was harvested, clarified by centrifugation and filtered through a 0.20 µM filter. The remaining cell monolayer was split 1:3, incubated for a further 3 days and the passage one (p1) supernatant harvested as previously. Virus in a 10-fold dilution series of the clarified, filtered, supernatant was quantified by immunofluorescence 2 days after infection of fresh Huh 7.5 monolayers in six-well plates. Monolayers were washed twice with PBS, fixed with 1 ml 4% paraformaldehyde for 20 min and washed twice again in PBS before permeabilization with 0.1% Triton PBS for 7 min with constant agitation. After a subsequent PBS wash infected cells were detected using a polyclonal sheep antibody to NS5A (αNS5A; generously supplied by Mark Harris) diluted 1:5000 in 10% foetal bovine serum (FBS). After incubation for 1 h, the primary antibody was detected using an AlexaFluor594-conjugated secondary anti-sheep antibody (1:500 in 10% FBS; Invitrogen), washed in PBS and stored under PBS containing 0.1% VECTASHIELD DAPI (Vector Laboratories) before analysis by UV microscopy. Virus titre was expressed in focus forming units per millilitre of supernatant (ffu/ml).
RESULTS
SHAPE analysis uses chemical modification of the unpaired bases in a folded RNA molecule to render them uncopyable during a primer extension reaction ( 26 ). As a consequence, by judicious choice of primer-binding sites, it is possible to map both local and long-range interactions in an RNA molecule. Additionally, by analysis of individual or compensatory point mutations, it is possible to determine the influence on RNA structure of mutations that, in a replicating genome, exert a phenotypic effect. SHAPE offers two distinct advantages over more traditional methods of RNA structure mapping, such as the V1 and T1 single- or double-strand specific ribonucleases, we have previously used to analyse HCV RNA structures ( 9 ). The resolution with which unpaired bases are targeted is improved, usually with sequential ‘hits’ on single-stranded regions, and the relative exposure of a nucleotide can be quantified by comparison of chemically treated and, untreated samples. Together this enables the fine-mapping of unpaired regions of RNA structure and, by analysis of sequence variants, allows the structural alterations underlying phenotypic changes to be determined.
SHAPE mapping of SL9266
We investigated the structure of SL9266 by SHAPE analysis of RNA transcripts derived from Con1b–luc–rep or J6/JFH-1. In all cases, two different positive-sense transcripts were used; a full-length transcript generated from the bacteriophage T7 polymerase promoter in the plasmid vector or a sub-genomic transcript generated from a PCR product spanning nucleotide9005 to the extreme 3′ end of the genome ( Figure 1 A). Without exception, for either Con1b–luc–rep ( Figure 1 B) or J6/JFH-1 ( Supplementary Figure S1 ) templates, the results obtained were highly reproducible and were indistinguishable within the regions analysed ( Figure 2 A). This implied that RNA structures present in the shorter NS5B-3′NCR template were not fundamentally influenced by sequences elsewhere in the virus genome.
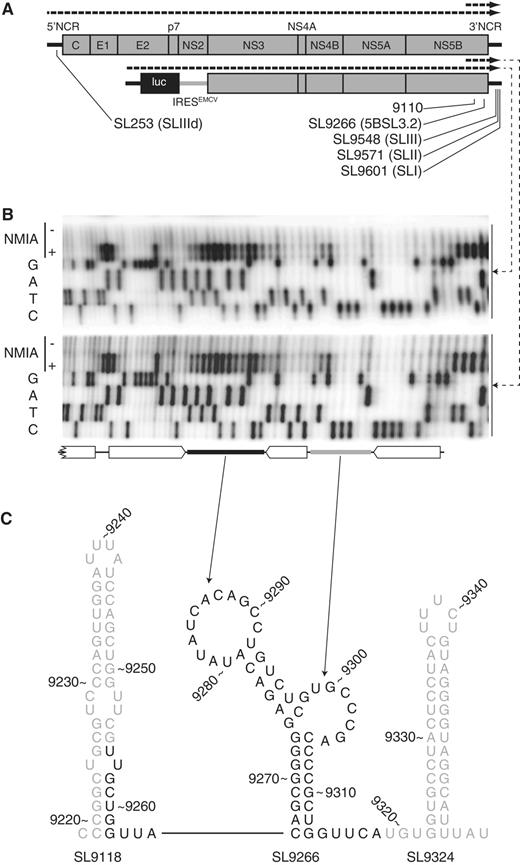
SHAPE mapping RNA structures in the HCV genome. ( A ) Schematic diagram of the genome of HCV J6/JFH-1 (top) and Con1b-luc-rep (below) indicating the location of relevant stem–loop structures (SL) using both standardized positional references ( 19 ) and naming schemes from previous publications ( 11 , 18 , 22 ). The dotted lines above each genome diagram indicate the in vitro transcripts used as templates for SHAPE mapping. ( B ) Representative SHAPE gels generated from full-length (top) and truncated (bottom) Con1b–luc–rep templates. In each case, a conventional Sanger sequencing reaction is adjacent and below primer extension reactions of templates treated (+) or untreated (−) with NMIA. Open block arrows indicate the bioinformatically predicted duplexed regions with filled rectangles (terminal loop = black, sub-terminal bulge loop = grey) representing unpaired regions. Note that when interpreting SHAPE autoradiographs the primer extension product terminates at the base before the uncopyable, chemically acylated and exposed nucleotide. ( C ) Schematic representation of the structure and context of Con1b–luc–rep SL9266. Numbering relates to reference H77 sequence ( 19 ). Bases in black are the area shown in the autoradiographs above.
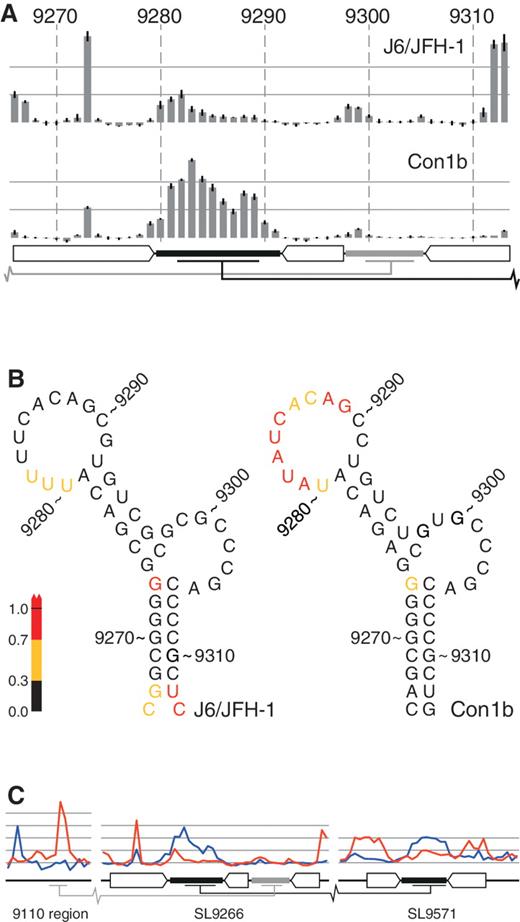
Comparative SHAPE analysis of native RNA structures. ( A ) SHAPE mapping of J6/JFH-1 and Con1b–luc–rep SL9266. The bars represent the relative exposure of the numbered nucleotides across SL9266 with the standard error of four independent gels (two short templates, two long) indicated. Duplexed and loop regions of SL9266 are indicated using the schematic representation used in Figure 1 B. Long-range interacting regions are indicated with grey (upstream) and black (‘kissing loop’) bars. ( B ) RNA structure representation of the data presented in Figure 2 A, colour coded to highlight relative exposure of the terminal loop of Con1b–luc–rep. ( C ) Comparative analysis of the 9110 region, SL9266 and SL9571 (left to right, respectively) of native unmodified templates of Con1b–luc–rep (blue) and J6/JFH-1 (red). Duplexed and loop regions of RNA structures indicated as described in Figure 2 A.
In Con1b–luc–rep, with the exception of G 9273 , the predicted duplexed stem regions of SL9266 exhibited little reactivity with the acylating agent NMIA, correlating well with the expected structure of the CRE ( Figure 1 B and C). Similarly, the reactivity of nucleotides forming the flanking stem–loops, SL9118 and SL9324, agreed well with the bioinformatically predicted structures (data not shown and Figure 1 C). In SL9266, G 9273 located opposite the sub-terminal bulge-loop and forming the closing nucleotide of the predicted lower duplex, exhibited considerable NMIA reactivity despite the fact that the nucleotide it is predicted to pair with (C 9306 ) was not accessible to NMIA ( Figure 1 B and C). The predicted sub-terminal bulge loop of Con1b–luc–rep, nucleotides 9298–9305, showed little reactivity to NMIA. In contrast, the terminal loop, nt 9280–9291, exhibited considerable reactivity with the chemical modifier ( Figure 1 B). Since the SHAPE-mapped template also contained the X-tail sequences implicated in the genetically defined ‘kissing loop’ interaction ( 17 ), we were surprised that our analysis indicated that this region was predominantly unpaired. This prompted us to undertake a comparative analysis with SL9266 of J6/JFH-1 and to investigate the influence of mutations in these regions on the RNA structure.
Full-length and truncated templates from J6/JFH-1 and Con1b–luc–rep were analysed by SHAPE, the reactivity of individual nucleotides quantified by densitometry and, after data normalization as described in the ‘Materials and Methods’ section, plotted to allow comparison ( Figure 2 ). Differences were apparent in two regions of SL9266. The nucleotides contributing to the base of the lower duplex of SL9266 in J6/JFH-1 (in particular, UC 9312–9313 ) were highly NMIA reactive, in agreement with the absence of a Watson–Crick base pair between C 9266 and C 9313 and the relatively weak pairing between G 9267 and U 9312 . Second, the reactivity of the terminal loop of SL9266 in J6/JFH-1 was markedly reduced when compared with Con1b–luc–rep ( Figure 2 A and B). In contrast, with the exception of a low signal from nucleotides 9298/9299, sequences within the sub-terminal bulge loop exhibited little NMIA reactivity in either the genotype 1b or 2a templates ( Figure 2 A). Therefore, comparison of SL9266 structure in J6/JFH-1 and Con1b–luc–rep suggested there were differences between the two genotypes. Since the RNA templates contained distal sequences involved in the long-range interactions of SL9266, these differences prompted us to also investigate the NMIA exposure of these proposed interacting regions as well. Initial comparison of the SHAPE reactivity of the 5′ and 3′ interacting regions of Con1b–luc–rep and J6/JFH-1 templates highlighted further differences between the two genotypes (shown in blue and red lines respectively in Figure 2 C). In addition to the differences in exposure of the terminal loop of SL9266, there were obvious discrepancies between the NMIA accessibility of nucleotides in both the 9110 and SL9571 regions ( Figure 2 C). In particular, the latter region was strikingly divergent in reactivity, only exhibiting a profile consistent with a stem–loop structure (NMIA-unreactive duplexed stem and -reactive terminal loop) in the Con1b–luc–rep template. We therefore undertook a detailed analysis of sequence changes that disrupt the documented interactions between SL9266 and these distal regions.
SHAPE mapping of mutants that influence the interactions of SL9266
Alone or in combination, we investigated the influence of Four nucleotide substitutions on the SHAPE-mapped structure of SL9266 and its predicted interacting regions. The mutations (all synonymous if coding), were in the terminal (C 9287 U) and sub-terminal (C 9302 A) loops of SL9266, in the upstream region centred on nucleotide 9110 (G 9110 U) and in the terminal loop of SL9571 in the X-tail region (G 9583 A). These mutations were selected as there is genetic evidence supporting the pairing of G 9110 to C 9302 ( 22 ) and, indirectly, C 9287 and G 9583 ( 17 , 20 , 21 ). Additionally, the sequence identity in these regions of J6/JFH-1 and Con1b–luc–rep meant, we could compare the influence of the same substitutions in two different genotypes. Results are presented in Figures 3 and 4 which show the SHAPE reactivity of mutants in SL9266, SL9571 and the 9110 region as bar graphs, with the reactivity of an unmodified template superimposed as a line graph. A schematic representation of the predicted structure and interactions of the regions is shown alongside to aid interpretation of the data.
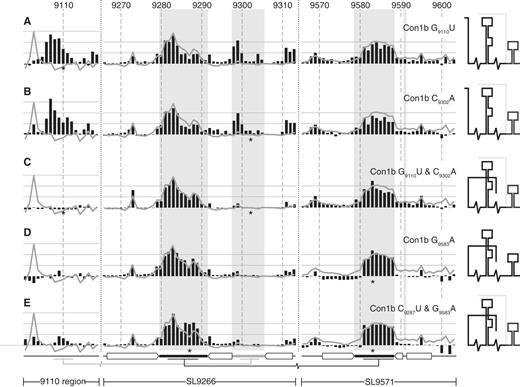
SHAPE analysis of mutagenized Con1b–luc–rep templates. ( A ) Con1b–luc–rep G 9110 U, ( B ) Con1b–luc–rep C 9302 A, ( C ) Con1b-luc-rep G 9110 U and C 9302 A, ( D ) Con1b-luc-rep G 9583 A and ( E ) Con1b-luc-rep C 9287 U and G 9583 A. Black bars show the normalized reactivity of SHAPE-mapped templates bearing the mutations indicated (position highlighted with an asterisk). The normalized exposure of an unmodified template is indicated by the superimposed line graph. The shaded background bars represent the proposed unpaired nucleotides in SL9266 and SL9571 stem–loops. The duplexed stem regions and long-range base pairing is indicated below the graphs using the format described previously (see Figures 1 B and 2 C). A cartoon interpretation of the structure of each mutant is shown on the right. Where two conformations occur simultaneously the predominant structure is shown with darker lines.
Con1b–luc–rep SHAPE mapping
In Con1b–luc–rep, the G 9110 U or C 9302 A substitutions caused four obvious changes in NMIA reactivity. Exposure of the 9110 region markedly increased ( Figure 3 A and B) as did exposure of the CUG 9311–9313 at the 3′ base of the lower stem of SL9266. In addition, exposure of the sub-terminal bulge loop also increased, though this was most marked in the GUG 9298–9301 triplet, and exposure of G 9103 was much reduced. Within SL9571 in the 3′ X-tail, there was very little change in NMIA reactivity; nucleotides forming the duplexed stem of SL9571 exhibited much lower reactivity than the predicted terminal loop, entirely consistent with it forming a simple stem–loop structure, albeit one in which the terminal loop is not paired elsewhere. The simultaneous presence of G 9110 U and C 9302 A restored NMIA reactivity of SL9266 to levels seen in the unmodified template, had little if any effect on sequences within SL9571 and reduced exposure of sequences in the 9110 region to undetectable levels ( Figure 3 C). In this double mutant, only G 9103 differed markedly in exposure when compared with the unmodified template. The NMIA reactivity of the terminal loops of SL9266 and SL9571 were very similar to the native Con1b–luc–rep template whether G 9110 U, C 9302 A or both covariant mutations were present. These results support our bioinformatic prediction of an interaction between the 9110 region with the sub-terminal bulge loop of SL9266. In addition, they provide biophysical support that mutations associated with replication defects in Con1b-based replicons cause disruption of this long-range RNA–RNA interaction ( 22 ).
Mutations introduced to disrupt/restore the proposed ‘kissing loop’ interaction in Con1b–luc–rep caused only very subtle alteration of the RNA structure ( Figure 3 D and E). Since the predicted interacting sequences, the terminal loops of SL9266 ( Figure 1 B) and SL9571, were already largely exposed any further increase was inevitably difficult to quantify. Indeed, SHAPE analysis of the SL9266 region in isolation, provides little support for a stable ‘kissing loop’ interaction in Con1b–luc–rep. However, within the X-tail, the G 9583 A substitution did subtly decrease the reactivity of the sequences that form the duplexed stem of SL9571, though this was not restored by inclusion of the complementary C 9287 U substitution. It should be noted that we did not investigate the structure of C 9287 U alone as it would still be predicted to pair with G 9583 .
J6/JFH-1 SHAPE mapping
In J6/JFH-1 substitutions introduced to disrupt the two proposed long-range interactions generally had a more pronounced influence on the SHAPE-mapped RNA structure. The independent substitution of G 9110 U or C 9302 A markedly increased the exposure of the sub-terminal bulge loop of SL9266 while also altering, though to a lesser extent, the profile of exposed nucleotides in the region around nucleotide 9110. For example, in the latter region, the exposure of GG 9109–9110 increased when compared to the unmodified template. The other marked change was the reduction in the NMIA reactivity of G 9273 . The reactivity of the remaining duplexed regions of SL9266 and across SL9571 was predominantly unaltered ( Figure 4 A and B) implying there were no gross changes in RNA structure. However, in contrast to the situation with Con1b–luc–rep, introduction of G 9110 U and C 9302 A together did not restore the structure of SL9266 or the 9110 region to the situation seen in the unmodified template. Instead, NMIA reactivity in the 9110- and SL9266 regions was almost indistinguishable from that seen with either mutation individually. As before, there was no change in the reactivity of sequences in SL9571 ( Figure 4 C) implying that the interactions of SL9266 are largely independent.
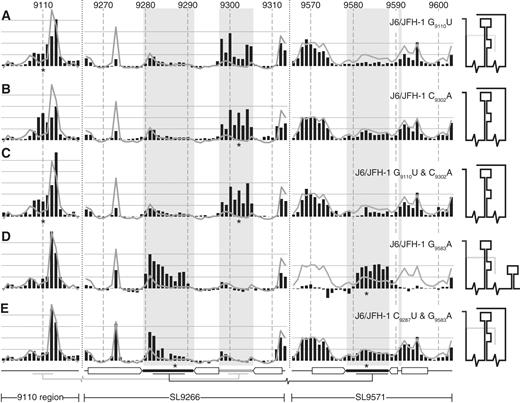
SHAPE analysis of mutagenized J6/JFH-1 templates. ( A ) J6/JFH-1 G 9110 U, ( B ) J6/JFH-1 C 9302 A, ( C ) J6/JFH-1 G 9110 U and C 9302 A, ( D ) J6/JFH-1 G 9583 A and ( E ) J6/JFH-1 C 9287 U and G 9583 A. Black bars show the normalized reactivity of SHAPE mapped templates bearing the mutations indicated (position highlighted with an asterisk). The normalized exposure of an unmodified template is indicated by the superimposed line graph. The shaded background bars represent the proposed unpaired nucleotides in SL9266 and SL9571 stem–loops. The duplexed stem regions and long-range base pairing is indicated below the graphs using the format described previously (see Figures 1 B and 2 C). A cartoon interpretation of the structure of each mutant is shown on the right. Where two conformations occur simultaneously the predominant structure is shown with darker lines.
Substitution of G 9583 A in J6/JFH-1 caused two very clear alterations to the NMIA reactivity of the RNA template ( Figure 4 D). First, sequences forming the terminal loops of SL9266 and SL9571 became exposed and highly NMIA reactive. This reactivity extended through the entire unpaired regions of the terminal loops. Secondly, the NMIA reactivity of sequences that form the duplexed stem of SL9571 was strikingly reduced ( Figure 4 D). These alterations of NMIA reactivity are consistent with profound structural changes in which an unfolded sequence in the X-tail, paired to SL9266, simultaneously adopts a stem–loop conformation and dissociates from SL9266. The covariant substitution of C 9287 U and G 9583 A restored the structure of the SL9571 region to that seen in the unmodified template. The exposure of nucleotides forming the terminal loop of SL9571 (9579–9588) was reduced as the exposure of the flanking sequences increased ( Figure 4 E). At the same time, the NMIA reactivity of the complementary sequences in the terminal loop of SL9266 were reduced, although the flanking U-rich region remained more exposed than the corresponding region in the unmodified template.
In summary, SHAPE mapping of sequences involved in forming the extended pseudoknot around SL9266 ( 22 ) supported the proposed RNA structure but also demonstrated fundamental differences, both in the native structures between J6/JFH-1 and Con1b–luc–rep, and in the structural perturbations resulting from mutagenesis of the region.
Phenotypic consequences of mutagenesis of the SL9266 pseudoknot
The interaction of the sub-terminal bulge loop of SL9266 was predicted bioinformatically and supported with genetic studies using Con1b-based sub-genomic replicons ( 22 ). The predicted interacting sequences are identical in the genotype 2a J6/JFH-1 genome and SHAPE analysis of G 9110 U and C 9302 A mutants demonstrated that either substitution increased the exposure of the complementary regions ( Figure 4 A and B). We extended this analysis by investigating the replication phenotype of J6/JFH-1 with mutations that were known or predicted to modify these interacting RNA structures.
The J6/JFH-1 genome was modified by introduction of substitutions of G 9110 U, C 9302 A or both covariant mutations together. In vitro transcribed RNA was transfected into Huh-7.5 cells and supernatant virus was harvested and quantified 72 h later and after passage ( Figure 5 , dark and light bars, respectively). In these studies, the control unmodified J6/JFH-1 genome generated ∼10 5 focus forming units per ml (ffu/ml), whereas a negative control genome bearing a well-characterized mutation (GND) within the active site of the NS5B polymerase did not replicate and generated no progeny virus, even after passage. Genomes with the G 9110 U substitution replicated indistinguishably from the parental virus. Genomes carrying single substitution of C 9302 A, or the double substitutions of C 9302 A and C 9303 G or G 9110 U and C 9302 A all yielded approximately 0.5 log 10 less virus than the positive control. The virus yield did not increase with further passage of cells transfected with these RNA genomes.
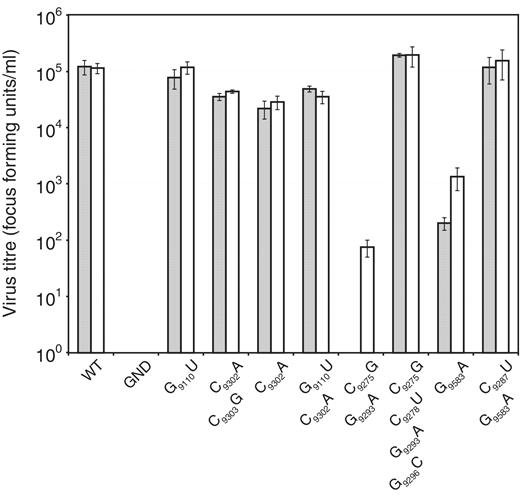
Recovery and infectivity in Huh 7.5 cells of J6/JFH-1 bearing mutations in SL9266 and interacting regions. RNA generated in vitro was electroporated into Huh 7.5 cells. HCV-infected cells were quantified following immunofluorescence staining for NS5A 72 h post-transfection (shaded bars) or 72 h after passage of transfected cell supernatant to a fresh cell monolayer of Huh 7.5 cells (open bars). Results are expressed as focus forming units per millilitre (ffu/ml) and represent the average of three independent assays (error bars indicate the standard error from the mean). Mutations present in the genome are indicated on the X axis. WT = an unmodified J6/JFH-1 template, GND = a template bearing a GDD to GND mutation within the active site of the NS5B RNA-dependent RNA polymerase.
Due to the striking divergence of the observed virus phenotype from those expected from our genotype 1b replicon-based studies ( 22 ), we then investigated the phenotypes resulting from mutagenesis of the core structure of J6/JFH-1 SL9266, or the structurally mapped substitutions involved in the ‘kissing-loop’ interaction with SL9571. The double substitution of C 9275 G and G 9293 A, both of which disrupt the upper duplex of SL9266, did not yield detectable virus upon RNA transfection but, with further passage, generated ∼10 2 ffu/ml presumably reflecting the selection of revertant or covariant genomes with a restored ability to replicate. The inclusion of additional substitutions of C 9278 U and G 9296 C to a genome already bearing C 9275 G and G 9293 A, thereby restoring the integrity of the upper duplex of SL9266, completely restored the ability to replicate at parental J6/JFH-1 levels. Finally, the single point mutation of G 9583 A in the terminal loop of SL9571 significantly reduced virus yield which was only restored to a limited extent with further passage. The addition of the covariant substitutions of C 9287 U and G 9583 A restored replication to wild-type levels, in agreement with published studies ( 17 ).
We additionally investigated the phenotype of Con1b–luc–rep replicons bearing the G 9583 A or combined C 9287 U and G 9583 A mutations as these had not previously been tested. Alone, the G 9583 A substitution prevented replication and yielded a phenotype indistinguishable from a control sub-genomic replicon with an inactive RdRp. In contrast, addition of the C 9287 U covariant substitution restored replication of the sub-genomic replicon to levels seen with an unmodified template ( Supplementary Figure S2 ).
DISCUSSION
An improved understanding of HCV replication is required if novel viral or cellular targets for therapeutic intervention are to be identified. In turn, this necessitates the study of systems that allow the replication processes of the virus to be dissected. Genotype 1b sub-genomic replicons ( 24 , 32–37 ) and the genotype 2a JFH-1 (or chimeric derivatives thereof) HCVcc system ( 25 , 30 , 38 ) have been widely used for HCV replication studies. Notwithstanding certain differences in adaptive mutations or the ability to generate progeny virions, the underlying assumption has been that the basic mechanism(s) of genome replication, and by extension the CRE structures and functions involved, are broadly conserved in these systems. This is supported by analysis of chimeras used to map the determinants that confer the full replication phenotype to JFH-1-based genomes ( 20 , 21 ). It underpinned the bioinformatic prediction of phylogenetically conserved RNA structures in the genome ( 8 , 9 , 29 , 39–41 ) and is further supported by studies that demonstrated the absolute requirement for the structural integrity of the X-tail and the polypyrimidine tract located adjacent and 5′ to the X-tail ( 12 , 15 , 42 ) in both systems. Additional commonality is a requirement for a ‘kissing loop’ interaction between the central stem–loop of the X-tail (SL9571) and SL9266, a CRE located within the NS5B-coding region. Disruption of this interaction abrogates replication, but is restored by the introduction or selection of compensatory covariant substitutions ( 17 , 20 , 21 ). SL9266 is also involved in a further long-range interaction with sequences located ∼200nt upstream between the conserved stem–loops SL9033 and SL9132, the prediction of which is supported by genetic studies in the Con1b-based replicon system ( 9 , 22 ). These studies could not distinguish whether the 5′ and 3′ long-range interactions of SL9266 were mutually exclusive, in which case the SL9266 CRE could form a ‘switch’ alternating between two conformations. Alternatively, if both long-range interactions occurred simultaneously, it would place SL9266 at the core of an extended pseudoknot, a possibility we favoured and so designated the element SL9266/PK ( 22 ). In this study, we define the structure and long-range interactions of native and mutagenized SL9266 by RNA SHAPE mapping of Con1b and J6/JFH-1 templates. In addition, we also investigated the phenotype of J6/JFH-1 genomes bearing mutations that influenced the structure and interactions of SL9266.
The core structure of SL9266 of both Con1b and J6/JFH-1 was well supported by SHAPE mapping ( Figures 1 B,C and 2). This structure was also in agreement with published studies that analysed the structure/function using reverse genetic approaches ( 11 , 18 , 22 , 43 ). Minor differences between the two systems were apparent in the exposure of the G 9273 , which may be related to the interactions of the sub-terminal bulge loop of SL9266 (see below), and the NMIA reactivity of nucleotides at the base of the lower duplex ( Figure 2 B). The structure of SL9266 was not influenced by the length of the template used in the SHAPE analysis ( Figures 1 A and 2 B; Supplementary Figure S1 ) implying that the shorter template, spanning from nucleotide 9005 to the end of the genome, is likely to contain all the sequences that influence the structure of this element.
Despite the similarities in the duplexed core structure of SL9266, there were striking differences in the interactions exhibited by the terminal and sub-terminal loops when the two genotypes were compared. These were very obvious in the analysis of unmodified templates ( Figure 2 C) and were subsequently supported by mutagenesis of the interacting regions ( Figures 3 and 4 ). In addition, the phenotypic consequences of disrupting these long-range interactions differed markedly when tested in the context of the two experimental systems. These results indicate that there are significant differences in the structure and function of cis -acting replication elements of the two widely used HCV model replication systems.
The existence of an upstream interaction between the sub-terminal bulge loop of SL9266 and the region around nucleotide 9110 was at least partially supported in both Con1b and J6/JFH-1. In the former, mutations of G 9110 U or C 9302 A were almost indistinguishable by SHAPE mapping ( Figure 3 A and B), with increased exposure of sequences centred around both mutations but little or no influence on the remainder of SL9266 or the other regions analysed. Together, the covariant mutations restored NMIA reactivity to levels seen in the unmodified template ( Figure 3 C). With our previous analysis of similarly modified replicon templates ( 22 ), we conclude that replication of Con1b requires an interaction between the sub-terminal bulge loop of SL9266 and the region centred on nucleotide 9110.
In J6/JFH-1 substitution of G 9110 U or C 9302 A again produced similar structural changes to SL9266; the sub-terminal bulge loop became more NMIA reactive and was paralleled by a reduction in exposure of G 9273 ( Figure 4 A and B). Since exposure of C 9306 (which is predicted to pair with G 9273 ) was unchanged, we interpret the decreased reactivity of G 9273 may reflect subtle conformational alterations at the top of the lower duplex, occluding G 9273 , that result from the increased exposure of the bulge loop. In contrast to Con1b, we did not observe markedly increased exposure of the region around 9110 in J6/JFH-1, though the profile of reactivity of nucleotides 9108–9113 did change. Introduction of both covariant substitutions at G 9110 U and C 9302 A did not restore the structure of SL9266 to that seen in the unmodified template ( Figure 4 C). We interpret this as indicating a minor or weak interaction between the sub-terminal bulge loop of SL9266 and the 9110 region in J6/JFH-1, but that replacement of G 9110 /C 9302 with a UA pair was incompatible with restoration of the interaction. Whether this is due to alterations in localized base stacking or the reduction in the number of hydrogen bonds remains to be determined. Analysis of sequenced HCV genomes suggests there is an absolute selection against purines at position 9302. In an alignment of 76 representative HCV sequences of seven genotypes (available at http://www.hcvdb.org/msa.asp ), only G-C (63%), A-U (36%) and G-U (1%) pairings are present between 9110 and 9302, a distribution in broad agreement with the C (89%) and U (11%) reported at position 9302 in 208 unspecified aligned HCV sequences ( 11 ). Despite this apparent bias for pyrimidine at position 9302 it is notable that, in the Con1b replicon, we have previously demonstrated that, as long as covariant substitutions are also present at nucleotide 9110, purines are fully compatible with genome replication [Figure 7 ( 22 )] whereas the non-Watson–Crick base pair of G 9110 –U 9302 was not tolerated.
The relatively high exposure of the 9110 region in an unmodified J6/JFH-1 template might argue for an interaction of the SL9266 bulge loop with a region elsewhere in the template. However, we conclude that since G 9110 U leads to the significant exposure of the SL9266 bulge loop, these regions are likely to interact. Whatever form this interaction takes, genomes bearing G 9110 U or C 9302 A mutations exhibited near wild-type virus yields ( Figure 5 ), indicating that, in marked contrast to the situation in Con1b ( 22 ), the upstream pairing is not critical for replication.
Further differences between J6/JFH-1 and Con1b systems were obvious when the proposed ‘kissing loop’ between SL9266 and SL9571 was analysed. The terminal loop of Con1b SL9266 was predominantly exposed in the native template ( Figure 2 A and C) as were the complementary sequences in SL9571 ( Figure 2 C). In contrast, sequences predicted to form the stem of SL9571 exhibited only weak NMIA reactivity in Con1b, indicating they were largely paired. We interpret this as indicating that the favoured structure in Con1b–luc–rep templates is one in which SL9266 and SL9571 form discrete and independent stem–loop structures. In J6/JFH-1 templates, the situation was very different. In this instance, the terminal loop of SL9266 and the complementary sequences in the X-tail were poorly exposed. In contrast, in the latter region, the sequences that should form the duplexed stem region of SL9571 exhibited high levels of NMIA reactivity, incompatible with them being paired. This situation was reversed by the introduction of the G 9583 A mutation. Therefore, in contrast to the Con1b templates, we propose that the native structure in J6/JFH-1 involves a relatively stable ‘kissing loop’ pairing between eight residues in the terminal loop of SL9266 (nucleotides 9282–9289) and the X-tail (nucleotides 9581–9588; Figure 6 ), supporting previous genetic studies of this interaction ( 17 , 20 , 21 ). Furthermore, we interpret the data as indicating that SL9571 sequences do not form a stem–loop structure when paired, via the ‘kissing loop’ interaction, with SL9266.

The ‘kissing loop’ interaction. The sequence of the terminal loop of SL9266 (nucleotides 9279–9292) of J6/JFH-1 (genotype 2a), Con1b (genotype 1b) and H77 (genotype 1a). Underlined characters represent nucleotides occupying the duplexed regions of SL9266 or SL9571. Nucleotides potentially involved in ‘kissing loop’ interactions are indicated depending whether SHAPE-mapping studies support (|) or do not support (:) the potential pairing.
These results suggest that in J6/JFH-1, SL9266 forms the core of an extended pseudoknot in which both long-range interactions occur simultaneously [as previously proposed and designated SL9266/PK ( 22 )]. Furthermore, the long-range interactions in this extended structure appear mutually independent; disruption of one does not significantly influence the other ( Figures 3 and 4 ). In contrast, the structure that predominates in Con1b–luc–rep is one in which the ‘kissing loop’ interaction between SL9266 and the X-tail is largely absent, though it remains necessary for replication [ Supplementary Figure S2 and ( 17 )]. Other than the differences between the steady-state native structures in the two genotypes, we noted that the absence of the ‘kissing loop’ interaction, either in the native Con1b template or in the presence of G 9583 A mutation in J6/JFH-1, appears to be a pre-requisite for the formation of a stem–loop structure by SL9571 in the X-tail. When conventionally paired, the SL9571 stem–loop resembles that determined by NMR analysis ( 17 ). It is not clear why the adoption of a stem–loop conformation by SL9571 is incompatible with formation of the ‘kissing loop’, a conclusion in agreement with the previous NMR analysis where no interaction was detected between SL9266- and SL9571-derived templates seeded in trans ( 17 ). Although the potential region of complementarity between SL9266 and SL9571 covers 12 contiguous nucleotides in J6/JFH-1 ( Figure 6 ), our SHAPE mapping indicates that the G 9583 A substitution increases exposure of only eight of these in the X-tail region (nucleotides 9581–9588; Figure 4 D). Specifically, exposure of GGU 9589–9591 does not increase indicating that the pairing does not extend into the top of the 3′ duplex of SL9571; the SL9266 and SL9571 structures are not de facto mutually exclusive. One possibility is that there are steric incompatibilities in the structure of the terminal loop of SL9571 that prevent pairing with SL9266. Although previous studies have proposed that the ‘kissing loop’ interaction may be mediated by a protein, possibly the viral NS5B ( 17 ), it is detectable in the absence of both cellular or viral proteins ( Figures 3 and 4 ). We have additionally repeated the SHAPE mapping in the presence of extracts from J6/JFH-1 infected Huh-7.5 cells and detected no differences in the mapped structures (data not shown).
Although outside the primary focus of this study, we also investigated the NMIA reactivity of domain IIId [designated SL253 ( 19 )] of the 5′ NCR which is proposed to interact with the bulge loop of SL9266 and mediate genome cyclization and translational control ( 23 , 44 ). Our SHAPE mapping was unable to detect this proposed interaction; the NMIA reactivity of the complementary sequences in SL253 (nt 254–279) remained unchanged when comparing either the native full-length J6/JFH-1 or Con1b HCV template, or templates bearing a C 9302 A substitution (or indeed any of the mutants used in our studies in the presence or absence of Huh 7.5 cell extracts; data not shown). Although the Romero-López and Berzal-Herranz study used truncated RNA transcripts that omitted the 9110 region ( 23 ), the presence of the latter in our template does not explain the failure to observe the interaction with SL253; templates bearing a G 9110 U mutation, demonstrated to prevent the interaction with SL9266 ( Figure 3 A), also exhibited no change in NMIA reactivity within SL253 when compared with the unmodified template. We therefore have no evidence for steady-state binding in genome-length templates between these regions of SL9266 and the 5′ NCR, though we cannot exclude the possibility of a transient or temporal interaction.
In a preliminary attempt to determine whether the structural differences, we observe are genotype-specific, or whether either the Con1b- or JFH-1-like structures are more typical, we also mapped the structure of SL9266 in the genotype 1a H77 strain of HCV. This was essentially indistinguishable from Con1b–luc–rep (data not shown). With exception of the pairing of nucleotides 9287 and 9587 [ Figure 6 ; ( 20 , 21 )], the complementary sequences forming the long-range interactions in JFH-1, Con1b and H77 are conserved, as is the entirety of SL9571, and further studies will be required to determine whether this alone accounts for the structural differences.
An unresolved aspect of this study is why mutations that disrupt the upstream interaction in Con1b and J6/JFH-1 ( Figures 3 a, b and 4a, b) exert markedly different phenotypes in the two replication systems ( Figure 5 ) ( 22 ). We suspect that this may reflect differences in the overall integrity of the extended pseudoknot structure, in which the upstream interaction in Con1b is required to stabilise SL9266 in a conformation compatible with function, for example, that allows the ‘kissing loop’ interaction to form as required. Although the overall stability of SL9266 is similar in Con1b and JFH-1 (ΔG of −20.0 and −20.5 for Con1b and J6/JFH-1, respectively), the contribution of the upper and lower duplexes, as determined by Loop Free-Energy composition analysis using MFOLD or UNAfold ( 45 , 46 ), differs. J6/JFH-1 exhibits a more stable upper helix (δG = −13.9, cf . −11.2 in Con1b) and a less stable lower helix (δG = −17.8, cf . −19.9 for Con1b). To this end, it is notable that introduction of G 9110 U or C 9302 A substitutions to J6/JFH-1 had little impact on the NMIA exposure of nucleotides in the duplexed regions of SL9266, whereas either substitution in the Con1b template increased the NMIA reactivity of nucleotides contributing to the upper duplex of SL9266 (nucleotides 9274–9279 and 9292–9297) and to nucleotides 9311–9313 at the 3′ base of the lower duplex ( Figure 3 a and b). Despite the differences in phenotype, and the inability of covariant substitutions to restore pairing to J6/JFH-1 ( Figure 4 c), the striking phylogenetic conservation of the interacting sequences ( 22 ) suggests there are functional constraints operating on both these regions.
Of course, speculation regarding differences between RNA structures in HCV genotypes provides little insight into the functional roles these fulfil in genome replication. There is compelling genetic data supporting the importance of the ‘kissing loop’ interaction for HCV replication ( 17 , 20–22 ). However, we demonstrate that this interaction is incompatible with SL9571 forming a stem–loop structure. The integrity of the stem region of SL9571 is required to bind the pyrimidine tract binding protein (PTB) which may be involved in alleviating translational repression mediated by PTB binding to the 5′ NCR ( 47 , 48 ). SL9571 stem integrity, and possibly the primary sequence as well, also appears critical for HCV replication ( 42 ). Considering the differences in structure of SL9571 in the presence or absence of a ‘kissing loop’ interaction, we propose that SL9266 may contribute a switch function that modulates the mutually incompatible translation and replication events that must occur for replication of the positive-strand RNA genome of HCV. Precedents already exist for the involvement of RNA structures, including pseudoknots, in temporal control of the termination of translation and initiation of genome replication. In most well-characterized examples, a feedback mechanism operates where translation is repressed in response to the accumulation, directly or indirectly, of an interacting viral or cellular protein. For example, a cloverleaf structure at the 5′ terminus of the poliovirus genome interacts with cellular poly(rC) binding protein (PCBP), an interaction that enhances translation. The subsequent accumulation of the viral 3CD protein, which also binds the 5′ cloverleaf structure, inhibits translation and is a pre-requisite for negative-strand synthesis ( 49 ). Similarly, turnip yellow mosaic virus possesses a pseudoknot, designated the tRNA-like structure, which competitively binds newly synthesized viral polymerase to mediate the switch between genome translation and replication ( 50–52 ). It should be noted that recent studies have also suggested a role for SL9266 in translational control ( 44 ), though our SHAPE mapping did not support the proposed interaction of the sub-terminal bulge loop of SL9266 and the 5′ NCR (see above).
There is evidence that HCV NS5B binds sequences within the 3′ end of the virus genome ( 18 , 53 ) though our preliminary studies have been unable to demonstrate a specific interaction with SL9266 (unpublished results). Further studies will therefore be required to determine whether the binding of cellular or viral proteins is involved in modifying the interactions of SL9266 to initiate replication events dependent upon SL9571 forming a stem–loop. The demonstration that the two widely used systems for HCV replication studies exhibit strikingly different functional RNA structures provides valuable comparative information that will contribute to both defining the function and determining whether the interactions of SL9266/PK forms a tractable target for therapeutic intervention.
FUNDING
UK Medical Research Council [G08901197 to D.J.E. and P.S.]. Funding for open access charge: Medical Research Council.
Conflict of interest statement . None declared.
ACKNOWLEDGEMENTS
We gratefully acknowledge Ralf Bartenschlager, Mark Harris, Charlie Rice, Takaji Wakita and NIH for the generous provision of reagents used in this study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments