





Abstract
RNA interference through expression of short hairpin (sh)RNAs provides an efficient approach for gene function analysis in mouse genetics. Techniques allowing to control time and degree of gene silencing in vivo, however, are still lacking. Here we provide a generally applicable system for the temporal control of ubiquitous shRNA expression in mice. Depending on the dose of the inductor doxycycline, the knockdown efficiency reaches up to 90%. To demonstrate the feasibility of our tool, a mouse model of reversible insulin resistance was generated by expression of an insulin receptor (Insr)-specific shRNA. Upon induction, mice develop severe hyperglycemia within seven days. The onset and progression of the disease correlates with the concentration of doxycycline, and the phenotype returns to baseline shortly after withdrawal of the inductor. On a broad basis, this approach will enable new insights into gene function and molecular disease mechanisms.
INTRODUCTION
The application of Cre/loxP recombination has refined the tools for manipulating the mouse genome, allowing to determine site and timing of gene alteration in the living animal (1,2). An inherent feature of these recombinase-based approaches, however, is a non-reversible gene switch that does not allow modulating gene expression in a given cell. In addition, the derivation of conditional mouse mutants is costly and time consuming due to extensive vector construction, ES cell manipulation and breeding. The finding that stably expressed short hairpin (sh) RNAs can mediate potent gene knockdown in transgenic animals opened the opportunity to reduce time and effort for the generation of genetically modified mouse models significantly (3–5). Conditional, RNAi-mediated gene knockdown in mice has been introduced by using Cre/loxP for tissue-specific control of shRNA expression (6–8). In contrast, temporal control of shRNA expression in transgenic animals has not been demonstrated so far.
Type 2 Diabetes mellitus is a growing problem to public health, affecting more than 5% of western populations. Progressive, late onset insulin resistance and inadequate compensation through insulin secretion from pancreatic β-cells leading to fasting hyperglycemia characterizes the course of the disease. Efforts of the pharmaceutical industry focus on the development of small molecules that increase the sensitivity of insulin receptor signaling (9). Here, animal models of insulin resistance are essential tools for understanding the molecular basis of the disease. Furthermore, drug target validation and evaluation of test compounds in appropriate disease model mice support the drug discovery process. The use of current genetic mouse models of type 2 diabetes is not optimal for this purpose as they do not allow modulating the onset and the degree of systemic insulin resistance, and the genetic modifications are not reversible (10,11). The first aspect is particularly important for developing therapeutic interventions during the early phase of the disease, whereas the latter would allow dissecting acute effects of insulin resistance from late diabetic symptoms such as neuropathy and arteriosclerosis. As the current technologies for gene manipulation do not facilitate these aspects, we established a new approach for temporal modulation of gene expression in vivo.
In cultured cell lines, inducible expression of antisense or shRNAs has been achieved by using engineered, RNA polymerase III-dependent promoters containing operator sequences (tetO) of the E. coli tetracycline resistance operon (12–15). Binding of the tetracycline resistance operon repressor (tetR) to tetO blocks transcription, whereas de-repression is achievable by adding the inductor doxycycline (dox), causing release of TetR. Here we show that a codon optimized tet repressor, but not the wt coding region as employed in previous attempts, facilitates tight control of RNAi in different cell types and throughout development of transgenic mice. We found efficient, doxycycline-inducible silencing of both, a luciferase reporter and the insulin receptor gene, reaching 80–90% repression in most tissues. By introducing single-vector-constructs into the ES cell genome via recombinase mediated cassette exchange (RMCE) and applying tetraploid blastocyst complementation we demonstrate the rapid generation of a reversible mouse model of type 2 Diabetes mellitus within 3 months.
MATERIALS AND METHODS
Rosa targeting and exchange vectors
Rosa26 Rluc vector: The Renilla luciferase gene (Promega) was inserted into the rosa26 targeting vector (5) 3′ of the splice acceptor site. ShRNA cassettes were inserted 3′ of the Renilla luciferase gene.
ShRNA cassettes: The U6- and the H1-promoter fragments were amplified from human genomic DNA using a 3′ primer containing the tet-operator sequences (bold: TATA-box, underlined: tet-operator):
H1-tet: gaaatgtctttggatttgggaatcttataagtccctatcagtgatagagattccc
U6-tet: gaaagtatttcgatttcttggctttatatatctccctatcagtgatagagaaag
Both fragments were cloned 5′ of the Fluc-specific shRNA (16), followed by five thymidines as termination signal. Inducible shRNA expression cassettes were flanked by two loxP-sites (lox) to allow cre-mediated deletion.
Reporter configuration (Figure 1B): The firefly luciferase gene (Promega) was inserted into the rosa26-targeting vector (17) followed by the CAGGS-tetR cassette and a PGK-hygromycin resistance gene.
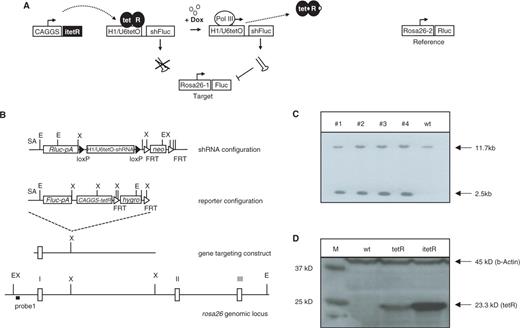
Schematic representation of the inducible system and gene targeting strategy. (A) Principle of tetR-mediated control of shRNA expression. Transcription of the RNA polymerase III-dependent promoter is blocked in cells expressing the tet repressor (tetR). Upon induction by doxycycline, tetR is removed from the tet-operator sequences (tetO) inserted into the promoter, allowing transcription of shFluc. ShRNA expression leads to RNAi-mediated knockdown of the target gene firefly luciferase. Renilla luciferase is used for reference to quantify firefly luciferase activity. (B) Scheme of the targeting strategy. ShRNA and reporter constructs were independently inserted into the rosa26 locus by homologous recombination in ES cells. Genes encoding the Renilla (Rluc) and firefly luciferases (Fluc) along with an adenovirus splice acceptor sequence and a polyadenylation signal (pA) were placed downstream of the endogenous rosa26 promoter. The Fluc-specific shRNA is expressed under the control of the U6-tet or H1-tet promoter, and terminated by five thymidines (shRNA). The loxP-sites flanking the shRNA expression cassettes were used to generate a negative control through cre-mediated recombination in ES cells. (C) Southern blot analysis of genomic DNA from transfected ES cell clones containing the shRNA- (lane #1 and #2) or the reporter-constructs (lanes #3 and #4). Homologous recombination at the rosa26 locus is detectable by using EcoRV-digested genomic DNA and probe 1, resulting in a 11.7 kb band for the wt and a 2.5 kb band for targeted allele. E: EcoRV; X: XbaI; neo: FRT-flanked neomycin resistance gene; hyg: FRT-flanked hygromycin resistance gene. (D) Western blot analysis from protein extracts of ES cells expressing either the wt tetR or the itetR using tetR- or β-Actin-specific antiserum. Control: protein extracts from wt ES cells.
Reporter configuration carrying itetR: Similar configuration as the reporter configuration described above, except that the tetR coding region is replaced by the codon optimized tet-repressor gene (itetR (18)).
ShRNA test vectors: The following shRNA sequences against the Insr mRNA (NM_010568) together with five thymidines for transcript termination were cloned 3′ of pH1tetO:
IR1: agtccgcatcgagaagaatattcaagagatattcttctcgatgcggact
IR2: atcgagaagaataatgagctttcaagagaagctcattattcttctcgat
IR3: actacattgtactgaacaattcaagagattgttcagtacaatgtagt
IR4: agggcaagaccaactgtcctttcaagagaaggacagttggtcttgccct
IR5: agaccagacccgaagatttcttcaagagagaaatcttcgggtctggtct
IR6: agcctggctgccaccaatacttcaagagagtattggtggcagccaggct
The resulting vectors are called pIR1-pIR6·pIR5-tet exchange vector (Figure 3A). The vector contains the F3 site and the FRT site in a similar configuration as described before (5). pIR5-tet was generated by insertion of the H1-tet-IR5 fragment from pIR5 into MCS of pINV-2 containing the following elements in 5′ to 3′ direction: synthetic polyA signal, F3-site, neomycin-resistance gene lacking the start ATG, hgH polyadenylation signal, MCS, CAGGS promoter (19), the codon optimized tet-repressor gene, synthetic polyA signal, FRT-site.
pFluc-tet exchange vector (Figure 2A): Similar configuration as the pIR5-tet exchange vector, except a Fluc-specific shRNA (16) instead of IR5.
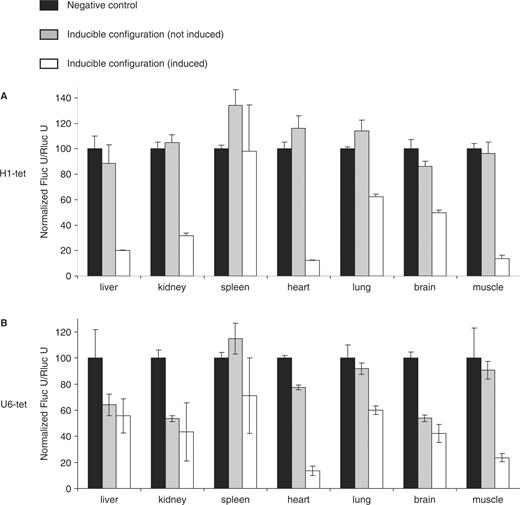
Efficiency of H1/U6-shRNA-mediated firefly luciferase (Fluc) knockdown in mice expressing the codon-optimized tetR. Each configuration (control, H1-tet shRNA, U6-tet shRNA) was analyzed using two to four mice at the age of 8–10 weeks, respectively. Percentages of shRNA-mediated repression of firefly luciferase activity with standard error of the mean are shown for untreated controls (gray bars) and in animals upon 10 days feeding with 2 mg/ml doxycycline in the drinking water (white bars). In negative control animals (black bars), the shRNA expression cassettes are removed through cre-mediated recombination. Relative values of relative firefly luciferase expression level in different organs were calculated by using Renilla luciferases activities for reference. (A) H1-tet promoter-driven shRNA expression. (B) shRNA expression through the U6-tet promoter.
Cell culture
ES cell culture was carried out as described before (20). Transfection of Art4.12 ES cells carrying the FRT/F3 configuration at rosa26 was performed as described (5).
Doxycycline treatment of ES cells
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS), 4500 mg/l glucose, 1× non-essential amino acids and 1 µg/ml doxycycline (Sigma D-9891) for 48 h. Medium containing doxycycline was prepared and changed every day.
Transient transfection of muscle cells
C2C12 myoblasts were grown at 37°C/5% CO2 in DMEM containing 10% FCS, 4500 mg/l glucose and 1× non-essential amino acids (GIBCO BRL). Transfection studies were carried out with 1.35 × 105 cells plated on a 6-well plate. Cells were transfected with 2.5 µg DNA (1.25 µg GFP-vector + 1.25 µg of either shRNA expression vector pIR1 to 6). DNA was mixed with 10 µl Lipofectamin (Invitrogen, #18324-111) and 200 µl Optimem (Gibco BRL, #51985-026) and incubated for 45 min at RT. For transfection, cells were washed with 1× PBS and incubated for 5 h in 2 ml starving medium containing the Optimen-DNA-Solution. After 5 h, medium DMEM with 20% FCS was added to the cells. Twenty four hours after transfection, cells were washed with 1 × PBS and fixed with methanol for 3 min, washed with 1× PBS and dried. Cells were stained with DAPI in Vectashield (Vector). Cells were analyzed for GFP expression and transfection efficiency.
Mice
All animal studies were approved according to the German Animal Welfare Act. Mice were kept in the animal facility at Artemis Pharmaceuticals GmbH in microisolator cages (Tecniplast Sealsave). B6D2F1 mice for the generation of tetraploid blastocysts were obtained from Harlan, NL.
Doxycycline treatment
Solutions containing 2 mg/ml doxycycline (Sigma D-9891), 10% sucrose or 20 µg/ml doxycycline, 1% sucrose or 2 µg/ml doxycycline, 0.1% sucrose in drinking water were prepared every second day and kept dark.
Luciferase detection
Organs were homogenized at 4°C in lysis buffer (0.1 M KH2PO4, 1 mM DTT, 0.1% Triton X-100) using a tissue grinder and centrifuged for 5 min at 2000 × g (4°C). Supernatant was assayed for Fluc activities using the Dual Luciferase Assay (Promega, Inc.) according to the manufacturer's protocol. The luminescence was detected using a Lumat LB 9507 (EG&G Berthold). Relative values of firefly luciferase activity in different organs are given as indicated. All values of Fluc activity were normalized by using the Rluc activity for reference.
Protein isolation
Cells were lysed in protein extraction buffer containing 1% Triton X-100, 0.1% SDS, 10 mM Tris-HCl pH 7.4, 1.25 mM Tris Base, 10 mM EDTA, 50 mM NaCl, 50 mM NaF, 50 µg Aprotinin. Protein concentration was calculated according to Warburg–Christian.
Western blot
Proteins were fractionated on a 10% SDS-PAGE gel and semi-dry blotted for 30 min at 200 mA. Primary antibodies: αINSR (Rβ (C-19) #sc-711, Santa Cruz Biotechnology) was diluted 1:200, αAKT (AKT/Proteinkinase B: AKT #9272, Cell Signaling Technology) 1:1000, tet02 (Mobitec, #tet02) 1:500 and β-Actin 1:500 (Santa Cruz Biotechnology #sc-47778) in TBS containing 2% milk powder. Goat anti-rabbit IgG (whole molecule)-peroxidase (Sigma, #A6154-1ML) was used as secondary antibody to detect INSR and AKT. For detection of tetR and β-Actin, goat anti-mouse IgG (Mobitec #A1-900) was diluted 1:1000 in 2% MP/TBS and detected using ECL reagents (Amersham, #RPN 2105).
RNA isolation
Total RNA was isolated with peqGOLD TriFast (peqLab, #30-2020) using 2.5 ml for a confluent grown 10-cm plate. Cells were centrifuged for 15 min at 13 000 r.p.m. and 4°C. Supernatant was transferred in a siliconized 2 ml Eppendorf tube and 0.3× volume Chloroform was added to the supernatant. The solution was mixed and centrifuged for 15 min at 13 000 r.p.m. and 4°C. The supernatant was transferred into a new siliconized 1.5 ml tube and protein was precipitated by adding the same volume of isopropanol. RNA was subsequently dissolved in DEPC-H2O.
Northern blot
A 30 µg RNA was fractionated on a 15% denaturating polyacrylamide gel and blotted on a nylon membrane at 3.3 mA/cm2 for 35 min. RNA was cross-linked to the membrane using UV-light and incubated at 80°C for 30 min. The membrane was subsequently incubated for 2 h in 10 ml pre-hybridization solution and labeled using radioactive, oligo probes against antisense strand of shRNA-IR5 (gaccagacccgaagatttct) and against 5srRNA (tcctgcaattcacattaattctcgcagctagc). A 10 U T4-Polynucleotide-kinase (NEB) and 10 µCi γ- (32P)-ATP (10 U µCi/µl) were used for probe labeling.
Reverse transcription
cDNA was synthesized using the Reverse Transcription Core Kit (Eurogentec). A real-time reaction of 20 µl contained the following components: 1 µl cDNA, 1 µl Probe (αInsr, Mm 00439693-m1), 8 µl DEPC-H2O and 10 µl qPCR Mastermix Plus (Eurogentec). Real-time PCR reactions were performed using an iCycler Thermal cycler instrument (Bio-Rad).
Measurement of insulin levels in plasma
Plasma mouse insulin levels were determined by using the Insulin Mouse Ultrasensitive ELISA kit (DRG Diagnostics) according to the manufacturer's protocol.
RESULTS
Establishing a configuration for inducible shRNA expression in mice
We inserted tetO sequences 3′ of the TATA box of the human H1- and U6-promoter (H1-tet, U6-tet), similar to the constructs described in two recent publications (12,13). Both promoter constructs were tested using a dual reporter system consisting of firefly luciferase (Fluc) as a test substrate and Renilla reniformis luciferase (Rluc) as a reference (Figure 1A). A firefly-luciferase-specific shRNA sequence (16) under the control of either H1-tet or U6-tet along with the luciferase reporter constructs and a gene encoding a codon-optimized version of tetR (itetR (18)) were introduced into the Rosa26 locus through homologous recombination in embryonic stem (ES)-cells (Figure 1B and C).
We have chosen rosa26 for this purpose as this locus mediates ubiquitous transgene expression in vivo using RNA polymerase II- as well as RNA polymerase III-dependent promoters (5,17). In contrast to random transgenesis that often results in variable transgene expression due to copy number, configuration and integration site, we expected this strategy to facilitate an optimal shRNA expression.
Recombinant ES cells were injected into blastocysts and chimeric mice were obtained upon transfer of blastocysts into pseudopregnant females using standard protocols (20). The activity of firefly luciferase was determined in different organs of the resulting mice. While the U6-tet promoter showed residual activity even in the absence of inductor (Figure 2B), regulation of H1-tet appeared tight in all organs (Figure 2A). The activity of H1-tet was undetectable before induction, but mediated a similar pattern of RNAi in the presence of doxycycline as the wild-type (constitutive) human H1 promoter in all tissues, except brain (Figure 2A, (5)). In brain of doxycycline-treated animals, 50% knockdown was reached (Figure 2A) in comparison to 80% silencing achieved by employing the constitutive H1 promoter (5). The limited activity of the inducible shRNA construct in brain may reflect a lower local concentration of doxycycline (21). A weak activity of the shRNA construct was also seen in spleen and thymus (Figure 2A and data not shown), which is in accordance with similar results using the constitutive H1 promoter in the same chromosomal context (5). The reason for this phenomenon is unclear. In all other tissues, induction with doxycycline resulted in efficient repression of firefly luciferase activity, ranging between 50 and 90% gene silencing (Figure 2A, white bars).
In previous experiments, we also tested the activity of U6-tet and H1-tet using the wt tetR in a similar configuration as described above (Figure 1B). Here we detected a high degree background shRNA activity in the absence of doxycycline, particularly in kidney and brain (Figure S1). In other organs such as liver, muscle and heart, leakiness seemed less pronounced, indicating that limited expression of tetR might be the reason for the incomplete block of RNAi in some tissues. Comparing the expression level of wt and codon optimized tetR in transfected ES cells by western blotting supported this speculation (Figure 1D).
Generation of a reversible diabetes model
We next applied our H1-tet configuration for generating an inducible mouse model of insulin resistance and type 2 Diabetes mellitus. Six different shRNA sequences directed against the insulin receptor (Insr) mRNA were tested in the INSR expressing muscle cell line C2C12 (22). shRNA coding regions were cloned into the H1-tet expression vector pH1tetO (pIR1 to pIR6) and transiently transfected into C2C12 cells. Western blot analysis of protein extracts derived from transfected cells revealed a strong RNAi activity using constructs pIR5 and pIR6, leading to >80% reduction of INSR expression (Figure S2). The RMCE strategy (5) was subsequently adapted for targeted insertion shRNA sequence #IR5 under the control of the H1-tet promoter along with a constitutive expression cassette encoding the codon optimized itetR (Figure 3A). Recombinase mediated integration resulted in >90% correctly targeted ES cell clones upon transfection of the exchange vector (Figure S3A). Northern blot analysis of isolated ES cell clones showed a high level of doxycycline-dependent shRNA expression (Figure 3B), resulting in a ∼70% knockdown of Insr mRNA as quantified by real-time PCR analysis (Figure S3B).
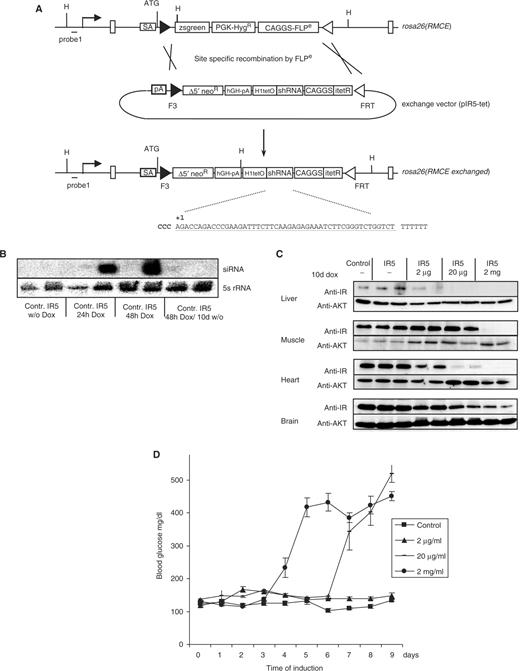
Single vector configuration for INSR gene knockdown in vivo. (A) RMCE through Flpe-mediated recombination using the exchange vector generates the rosa26(RMCE exchanged) allele. The exchange vector carries the shRNA coding region under the control of the H1-tet promoter, the codon-optimized tetRi gene under the control of the CAGGS promoter and a truncated neoR gene for positive selection of clones upon successful RMCE. The insr-specific shRNA sequence IR5 is shown in the figure. (B) Detection of processed IR5-shRNA. shRNA-transgenic ES cell clone #1 (IR5) and control cells transfected with an RMCE exchange vector lacking the shRNA-expression cassette (control) were cultured in the absence and/or presence of 1 µg/ml doxycycline as indicated. RNA extracts were analyzed by northern blot using IR5-shRNA and 5S-rRNA-specific oligonucleotide probes. (C) Doxycycline dose response of INSR knockdown in mice. IR5 transgenic animals were treated in the absence or presence of 2 μg/ml, 20 μg/ml or 2 mg/ml doxycycline for 10 days, as indicated. Protein extracts were prepared from various tissues and subjected to western blot analysis using INSR- or AKT-specific antiserum. Control: protein extracts from animals carrying a firefly luciferase-specific shRNA. (D) Serum glucose levels in shRNA-transgenic mice treated with 2 μg/ml (triangles), 20 μg/ml (bars) or 2 mg/ml doxycycline in the drinking water (circles) for the indicated number of days. Control animals (rectangles) carry a firefly luciferase-specific shRNA construct. Serum glucose levels ± SEM are shown. All assays were performed using groups of six mice at the age of two months.
Recombinant ES cell clones were injected into tetraploid blastocysts (23) and 35 eight-week-old ES mice were derived within three months. Transgenic ES mice harboring expression cassettes for inducible shRNA expression were fed with increasing concentrations of doxycycline in the drinking water for 10 days and the degree of INSR knockdown was analyzed in liver, muscle, heart and brain. Western blot analysis revealed a near-complete removal of INSR in animals treated with 2 mg/ml doxycycline, whereas the expression of INSR in untreated controls was unaltered (Figure 3C). In liver and heart, 20 µg/ml doxycycline was sufficient for inducing the INSR knockdown. In contrast, 2 µg/ml doxycycline in the drinking water had no effect on INSR expression (Figure 3C).
As a consequence of INSR knockdown, mice displayed signs of insulin resistance such as severe hyperglycemia. Blood glucose levels reached a maximum of ∼500 mg/dl at day 9 when treated with 20 µg/ml and at Day 5 when treated with 2 mg/ml doxycycline in the drinking water (Figure 3D), indicating a dosage-dependent progression of the disease. In contrast, treatment with 2 µg/ml doxycycline did not result in any change of blood glucose levels (Figure 3D). Control animals expressing an unrelated shRNA against firefly luciferase showed no alteration of glucose homeostasis, even when treated with high concentrations of doxycycline (Figure 3D). When crossed to wild-type C57BL/6 animals, INSR knockdown mice transmitted the shRNA transgene at mendelian ratios. Upon induction with 20 µg/ml doxycycline, shRNA-transgenic offspring displayed the same phenotype as parental mice (data not shown), indicating that the transgenes are not silenced following germ-line transmission.
Upon removal of doxycycline from the drinking water, glucose levels declined within seven days and serum insulin returned to normal levels after 14 days, demonstrating the reversibility of the inducible promoter (Figure 4A and B). In these animals, glucose homeostasis was fully restored as confirmed by glucose tolerance testing (Figure 4C). Accordingly, discontinuation of doxycycline treatment resulted in the reappearance of INSR protein in the liver within 21 days (Figure 4D).
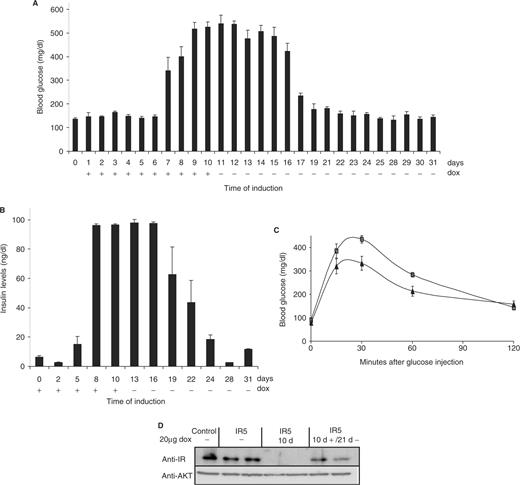
Reversible induction of hyperglycemia in mice. A group of six 2-month old, shIR5-transgenic mice were fed for 10 days with 20 µg/ml doxycycline in the drinking water and subsequently kept in the absence of doxycycline for the following 21 days. (A) Blood glucose levels were determined in venous blood samples at the indicated day of treatment. (B) Serum insulin concentrations were measured as indicated. Each bar represents the mean value ± SEM. (C) Glucose tolerance test were performed using IR5-transgenic mice before (triangles) and 21 days after dox treatment (rectangles). Results are expressed as mean blood glucose concentration ± SEM from at least six animals of each group. (D) Reversible knockdown of the insulin receptor using 20 µg/ml doxycycline for 10 days and 21 days after removal of dox in two independent mice. Protein extracts prepared from liver were subjected to western blot analysis using an INSR-specific antiserum and an AKT-specific antiserum.
DISCUSSION
In this work, we describe a straightforward method for the generation of drug-inducible knockdown mouse models. Several key issues for the state-of-the-art development of transgenic mice are addressed: (i) Reproducible transgene expression is warranted by the well-characterized rosa26 locus. (ii) High targeting efficiency of transgene constructs into the rosa26 locus is achieved by exploiting recombinase-mediated cassette exchange. (iii) Minimization of elaborated genetic modifications is allowed by a one-, rather than a multi-vector expression cassette. (iv) Tight and reversible regulation of shRNA expression is obtained by the usage of a codon-optimized tet-repressor. (v) Rapid generation of mice without further breeding is achieved by tetraploid embryo complementation. The type 2 diabetes mouse model described in this article illustrates the capability of this technology.
The model for insulin resistance presented herein, exhibits several advantages over existing ones (10,11). First of all, induction of insulin resistance allows to circumvent potential confounding developmental effects or compensatory mechanisms altering the phenotype. Moreover, the temporal control of insulin receptor knockdown can provide further insights into the impact of environmental factors prior to the onset of the disease. Second, reversibility of hyperglycemia provides a unique model to mimic the effect of an ideal pharmacological intervention. Along this line, it has so far not been established to what extent perfect control of metabolism after onset of disease is successful to prevent occurrence and/or progression of diabetic complications or lead even to reversion of them. Therefore, this model will be of central importance to gain better insights into not only diabetic retinopathy, nephropathy, polyneuropathy, but also macrovascular complications such as myocardial infection and stroke. Third, the obvious doxycycline-dependent reduction of insulin repressor expression can be extended to carefully determine the critical threshold of insulin action necessary to (i) induce a compensatory increase in pancreatic β-cell secretion and (ii) the occurrence of hyperglycemia. Thereby, these experiments will help to define the therapeutic efficiency required for reversal of clinically relevant insulin resistance. Finally, the single-allele configuration of the transgene circumvents time- and cost-consuming crossing experiments and makes it an ideal model for genetic screens for insulin resistance modulations, i.e. in a sensitized ENU-screen.
Previous examples of doxycycline-inducible shRNA expression were restricted to in vitro applications or gene knockdown in tumor cells transplanted into nude mice (15,24–27). The activity of such systems in selected cell clones are not predictive for in vivo applications in which a tight control of RNAi needs to be achieved in different cell types and throughout development. In addition, these approaches included random transgenesis or lentiviral infection (28), each resulting in clones with unique, irreproducible shRNA expression patterns. Screening of several cell lines was required, which is laborious and time consuming. Therefore, these techniques, albeit being useful for the particular experiment presented, are not generally applicable for different biological questions.
Here, we show that a codon optimized rather than the wild-type tetR mediates tight control of the H1-tet promoter when integrated at the rosa26 locus. ShRNA-mediated gene knockdown is achieved within 10 days of doxycycline treatment. The system appeared to be reversible within 14 days after withdrawal of the inductor. This is in accordance with previous observations that saturation of a system with doxycycline is an intrinsically faster process than depletion (21,29). The kinetics of reversible gene modulation may be further improvable by applying other tetracycline derivates with a shorter half-life in vivo.
The targeted insertion of all elements required for inducible shRNA expression into rosa26 using RMCE, and the subsequent ES cell injection using tetraploid blastocysts accelerate the generation of conditional knockdown mice significantly: 2 weeks for cloning of the RMCE exchange vector, 3 weeks for generating targeted ES cell clones, 4 weeks for the generation of newborn mice. Therefore, our system facilitates a rapid performance of in vivo gene function studies, providing a generally applicable tool for reverse mouse genetics research. Moreover, it permits the investigation of the effect of gene knockdown after the onset of a chronic or acute disease. This aspect is of particular interest for pharmaceutical drug target validation, as inducible gene silencing in mouse models of human disease can provide an ideal surrogate for the treatment of patients with antagonistic drugs. Even multiple dose regimes can be tested in preclinical studies by applying several rounds of induction. Furthermore, reversible knockdown of drug targets can help to distinguish target dependent from target independent effects of drug action.
Taken together, the technology described provides a novel, rapidly applicable approach for the inducible and reversible analysis of gene function in mice, overcoming the limitations of recombinase-based conditional gene targeting.
ACKNOWLEDGEMENTS
We wish to thank Heidrun Kern, Maria da Silva, Marco Schneider (Artemis Pharmaceuticals GmbH, Cologne) and Sabine Jordan (Institute for Genetics, Cologne) for technical advice as well as Holger Kissel (Artemis Pharmaceuticals GmbH, Cologne) for critical reading of the manuscript. We also thank Rolf Sprengel (Max-Planck Institute for Medical Research, Heidelberg) for kindly providing the codon-optimized tet-repressor. This work was supported by Artemis Pharmaceuticals GmbH and the German Ministry for Education and Science (BMBF) grant no. 0313066H. Funding to pay the Open Access publication charge was provided by Artemis Pharmaceuticals GmbH, Cologne/Germany.
Conflict of interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments