





Abstract
The Database of Interacting Proteins ( http://dip.doe‐mbi.ucla.edu ) aims to integrate the diverse body of experimental evidence on protein–protein interactions into a single, easily accessible online database. Because the reliability of experimental evidence varies widely, methods of quality assessment have been developed and utilized to identify the most reliable subset of the interactions. This CORE set can be used as a reference when evaluating the reliability of high‐throughput protein–protein interaction data sets, for development of prediction methods, as well as in the studies of the properties of protein interaction networks.
Received September 18, 2003; Revised and Accepted October 6, 2003
INTRODUCTION
The Database of Interacting Proteins (DIP) was initially developed ( 1 ) to store and organize information on binary protein–protein interactions that was retrieved from individual research articles. Over the course of the last 4 years the progress in genome‐scale experimental methods has resulted in rapid identification of binary protein–protein interactions ( 2 , 3 ) and multi‐protein complexes ( 4 , 5 ). On one hand, it prompted enhancements to the database schema that allow the capture, with increased level of detail, of information on the molecular interactions. On the other hand, questions about the reliability of the experiments conducted on a genome‐wide scale stimulated development of data quality assessment methods ( 6 ).
STRUCTURE OF THE DATABASE
The DIP database is implemented as a relational database using an open source PostgreSQL database management system ( http://www.postgresql.org ). The simplified version of the current database schema is shown in Figure 1 . The key tables—PROTEIN, SOURCE and EVIDENCE—store, respectively, information on individual proteins, sources of experimental information and information on individual experiments. The information on protein–protein interactions is stored in two tables—INTERACTION and INT_PRT. Such arrangement of the tables enables description of binary interactions (two entries in the INT_PRT table for each INTERACTION entry) but also of multi‐protein complexes (more than two entries in INT_PRT for each INTERACTION entry). The METHOD table provides a list of controlled vocabulary terms, together with references to the corresponding PSI ontology entries ( 7 ), which are used to annotate the experiments.
When available, information on the details of the topology of a molecular complex that was inferred from each experiment is stored in the TOPOLOGY and LOCATION tables. The LOCATION table describes regions of proteins participating in interactions whereas the TOPOLOGY table pairs them into records that describe observed binary interactions. It also specifies the type of interaction inferred from each experiment as one of aggregate (both partners shown to be present in the same complex but not necessarily in direct contact), contact or covalent bond.
DATABASE GROWTH
Since our previous NAR report was published ( 8 ), the number of distinct binary protein–protein interactions has nearly doubled and, as of September 2003, exceeds 18 500. Even more importantly, the number of research articles referenced in DIP has grown to more than 2500, providing a broad perspective on experimental approaches used to determine protein–protein interactions. It makes DIP an ideal starting point when comparing and assessing the reliability of different experimental methodologies, including high‐throughput interaction screens.
In addition to the information extracted from the research literature, the database has been recently enriched with information obtained by analyzing the structures of protein complexes deposited in the Protein Data Bank ( 9 ). As of September 2003 analysis of protein hetero‐complexes in the PDB database resulted in the identification of ∼2000 structures describing protein–protein interactions at the atomic level. We are in the process of entering this information into the database.
QUALITY ASSESSMENT
The recent development of high‐throughput technologies for the detection of protein–protein interactions, such as large‐scale yeast two‐hybrid screens ( 2 , 3 ), protein microarrays ( 10 ) and mass spectrometric analysis of affinity purified multi‐protein complexes ( 4 , 5 ), has resulted in a rapid accumulation of protein–protein interaction data. However, small overlaps between the high‐throughput data sets and, often, lack of agreement with small‐scale experiments ( 11 ) gave rise to questions about the reliability of high‐throughput approaches and about the compatibility of their results with those obtained using conventional methods. As a result, a number of attempts has been made to assess the quality of the high‐throughput data ( 6 , 12 , 13 ). They demonstrated large differences in quality between data sets, some of which can contain many erroneously identified interactions (false positives) ( 11 ).
In order to evaluate the reliability of individual interactions reported in DIP a number of tests are used to identify the most reliable core subset of the interactions. The tests range from a simple evaluation based on the reliability of individual experimental methods to the analysis of the patterns of interactions between analogous proteins using the PVM method ( 6 ).
Besides analysis of the data already present in the DIP database, the evaluation methods are implemented as publicly available services ( http://dip.doe‐mbi.ucla.edu/dip/Services.cgi ) that can be used to evaluate the reliability of new experimental and predicted interactions. Those services include our previously described PVM and EPR methods ( 6 ) as well as the Domain Pair Verification (DPV) method, which analyses domain–domain interaction preferences as described by Deng et al. ( 14 ).
DATA ACCESS AND EXCHANGE
All the DIP data can be accessed online in both interactive and batch modes. The interactive, Web‐based interface allows users to query the database for a specific protein based on its name, annotation or species of origin. In case the protein of interest is not yet present in the database, it is also possible to perform sequence similarity (BLAST) and motif searches in order to identify closely related proteins. The pattern of interaction of these might provide insights into the potential but not yet identified interactions of the query protein.
In the batch mode, different subsets of the DIP database can be downloaded in a variety of formats ranging from the native XML‐based XIN format to simple, tab‐delimited text files that are ready to be imported into spreadsheet applications. The DIP data are also provided in the Molecular Interaction Format (MIF) developed under the auspices of the Human Proteome Organization (HUPO) Proteomics Standards Initiative ( 7 ). MIF is a community‐developed data standard that provides a database‐independent platform for the exchange of information on protein–protein interactions. It is expected to be supported by the major providers of protein interaction data, including DIP, BIND ( 15 ) and Mint ( 16 ) databases.
FUTURE DIRECTIONS
The progress in the development of high‐throughput interaction detection methods will soon result in a rapid accumulation of large amounts of protein interaction data. Organizing these data and assessing its reliability will pose significant challenges to the database providers. We foresee further development of quality assessment measures, most likely based on integration of the experimental interaction data with other sources of information, such as expression and functional data. Integration of the data will also play a key role when analyzing the topology and dynamics of protein interaction networks. It would ultimately lead to the construction of comprehensive models of protein–protein interactions amenable to computational analysis and simulation ( 17 ).
ACKNOWLEDGEMENTS
We thank the NIH and DOE for support of DIP.
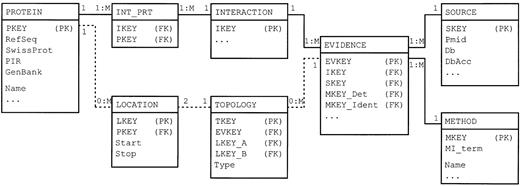
Figure 1. A simplified entity‐relationship diagram showing the key tables (rectangles) and relations (lines) of the DIP database. Dashed lines represent relationships that are used to describe the topology of protein complexes. PK, primary keys; FK, foreign keys. The full specification of the database is available at http://dip.doe‐mbi.ucla.edu/Guide.cgi .
References
Xenarios,I., Rice,D.W., Salwinski,L., Baron,M.K., Marcotte,E.M. and Eisenberg,D. (
Ito,T., Chiba,T., Ozawa,R., Yoshida,M., Hattori,M. and Sakaki,Y. (
Uetz,P., Giot,L., Cagney,G., Mansfield,T.A., Judson,R.S., Knight,J.R., Lockshon,D., Narayan,V., Srinivasan,M., Pochart,P. et al. (
Gavin,A.C., Bosche,M., Krause,R., Grandi,P., Marzioch,M., Bauer,A., Schultz,J., Rick,J.M., Michon,A.M., Cruciat,C.M. et al. (
Ho,Y., Gruhler,A., Heilbut,A., Bader,G.D., Moore,L., Adams,S.L., Millar,A., Taylor,P., Bennett,K., Boutilier,K. et al. (
Deane,C.M., Salwinski,L., Xenarios,I. and Eisenberg,D. (
Hermjakob,H., Montecchi‐Palazzi,L., Bader,G., Wojcik,J., Salwinski,L., Moore,S., Orchard,S., Sarkans,U., von Mering,C., Roechert,B. et al . (
Xenarios,I., Salwinski,L., Duan,X.Q.J., Higney,P., Kim,S.M. and Eisenberg,D. (
Westbrook,J., Feng,Z., Jain,S., Bhat,T.N., Thanki,N., Ravichandran,V., Gilliland,G.L., Bluhm,W., Weissig,H., Greer,D.S. et al. (
Zhu,H., Bilgin,M., Bangham,R., Hall,D., Casamayor,A., Bertone,P., Lan,N., Jansen,R., Bidlingmaier,S., Houfek,T. et al. (
Salwinski,L. and Eisenberg,D. (
Mrowka,R., Patzak,A. and Herzel,H. (
von Mering,C., Krause,R., Snel,B., Cornell,M., Oliver,S.G., Fields,S. and Bork,P. (
Deng,M., Mehta,S., Sun,F. and Chen,T. (
Bader,G.D., Betel,D. and Hogue,C.W. (
Zanzoni,A., Montecchi‐Palazzi,L., Quondam,M., Ausiello,G., Helmer‐Citterich,M. and Cesareni,G. (

{kind=link}
Comments