





Abstract
Zeocin is a member of the bleomycin/phleomycin family of antibiotics, known to bind and cleave DNA. We established human SK-OV-3 cells that stably express the Zeocin resistance gene (Sh ble) using an ecdysone-inducible mammalian expression system. Surprisingly, our results demonstrated that Zeocin, added in the culture medium to maintain the expression of the ecdysone receptor, was responsible for the formation of DNA strand breaks in the recombinant cells. This suggests that the Zeocin is not completely detoxified and is still able to cleave DNA, despite the stable expression of the Sh ble gene in the recombinant clones. Our study indicates that one needs to be very cautious in the interpretation of data involving stable cell lines selected with Zeocin.
Introduction
Mammalian cells have evolved a number of repair pathways to deal with the various types of DNA damage and to maintain genomic integrity (1). The double strand break (DSB) is generally regarded as the most toxic of all DNA lesions. DSBs arise spontaneously during normal DNA processing, such as DNA replication, meiosis and immunoglobulin gene rearrangement (2) and can be induced by a variety of DNA-damaging agents, including ionizing radiations (IR) (3) and radiomimetic drugs, such as bleomycin (4). DSBs trigger the activation of specific checkpoint signaling pathways that transduce the appropriate biological responses, including cell cycle arrest, DNA repair and apoptosis (5). The two main mechanisms of DSB repair are non-homologous end joining (NHEJ) (6) and homologous recombination (HR) (7). Generally, HR is considered error-free and most active in the late S and G2 phases (8). It appears to involve a large number of proteins, including Rad51, Rad52, Rad54, BRCA1, BRCA2, the Rad51 paralogues and the Mre11/Rad50/Nbs1 (MRN) complex (9). In addition, the involvement of the tumor suppressor p53 in HR has been demonstrated by a number of studies (10–12). As a matter of fact, cells containing the mutant forms of p53 or missing p53 often show increased rates of HR (13,14). In order to extend these studies, we decided to examine the involvement of p53 in the repair of DNA strand breaks in isogenic cell lines, by establishing a system that can inducibly express wild-type p53 in p53-null human ovarian cancer SK-OV-3 cells (15). For that purpose, we used the pIND and pVgRXR vectors, which are components of an ecdysone-inducible mammalian expression system, to select for stable transfectants (16). Selection of transfectants is permitted by the neomycin resistance gene in pIND, and the Zeocin resistance gene in pVgRXR. We thus established recombinant SK-OV-3 ZeoR G418R cells that stably express the ecdysone receptor and the wild-type p53 gene cloned into pIND in an ecdysone-inducible system. Using this isogenic system, we analyzed the formation and repair of strand breaks induced by ionizing radiations at the level of the ribosomal gene (rDNA). We detected total single strand breaks (SSB) as alkaline sensitive sites in this fragment, using alkaline agarose gel electrophoresis followed by Southern blot analysis, as described previously (17,18). DSBs were detected in the rDNA region using neutral gel electrophoresis and Southern blot analysis. Surprisingly, we found that the initial yield of total strand breaks and DSBs was abnormally elevated in all the recombinant cell lines. Our results demonstrated that the antibiotic Zeocin, a bleomycin analogue added in the culture medium to maintain the expression of the ecdysone receptor, was mainly responsible for the formation of these strand breaks. Zeocin causes cell death by directly intercalating into the DNA and cleaving it predominantly through reactive radicals. Resistance to Zeocin is conferred by the Sh ble gene, which encodes a protein that binds to the antibiotic and prevents it from binding DNA (19). Thus, our data suggest that Zeocin is not completely detoxified and is still able to cleave DNA, despite the stable expression of the Zeocin resistance gene in recombinant ZeoR SK-OV-3 clones, and are of importance in studies using this antibiotic.
Materials and methods
Cell line
Human ovarian cancer SK-OV-3 cells were obtained from the American Type Culture Collection (ATTC) and were cultured in RPMI 1640 medium supplemented with l-glutamine (2 mM), streptomycin (100 µg/ml), penicillin (100 U/ml), insulin (0.2 U/ml) and 10% fetal bovine serum (all from Life Technologies, Inc.). Cells were maintained at 37°C in a 5% CO2 humidified atmosphere and were in exponential phase at the time of irradiation.
Establishment of an inducible system and stable recombinant cell lines
Before transfection, we determined the optimal Zeocin concentration for selection of SK-OV-3 transfectants using a kill curve. After plating SK-OV-3 cells, Zeocin was added at concentrations ranging from 100 to 800 µg/ml for up to 3 weeks. Fresh medium and antibiotic were added every 3 days. The dose of 500 µg/ml Zeocin killed all cells by day 15. No spontaneously resistant colonies appeared at this concentration after 3 weeks. All transfections were achieved using 40 µg/ml Polybrene (Sigma) as described previously (20). SK-OV-3 cells were first transfected with the ecdysone-inducible system regulatory vector, pVgRXR (Invitrogen) and selected with 500 µg/ml Zeocin (Cayla) to establish recombinant SK-OV-3 ZeoR cells that stably express the ecdysone receptor. Clones were isolated and were next transfected with a pIND vector (Invitrogen) that either contained or did not contain a 1.2-kb fragment of the cDNA encoding wild-type p53 (p53wt). Stably transfected cells were selected with 500 µg/ml G418 (Sigma). Individual clones were then screened for the inducible expression of wild-type p53 after treatment with 10 µM ponasterone (an analogue of ecdysone, Invitrogen) for 48 h by western analysis. The integration of the pVgRXR and the pINDwtp53 (or the pIND as control) was confirmed by Southern blot in recombinant ZeoR G418R clones. The established recombinant cell lines (SK-OV-3/pVgRXR/pINDp53wt and control SK-OV-3/pVgRXR/pIND) were subsequently grown in complete RPMI 1640 medium supplemented with 250 µg/ml Zeocin and 400 µg/ml G418 to maintain the selection pressure.
γ Irradiation and DNA extraction
Cells were irradiated on ice with a 137Ce source for blood products (IBL 437C, Cis-Bio International) at graded doses (dose rate 6.7 Gy/min) and were then lysed immediatly to measure the amount of DNA strand breaks or allowed to repair for different times in culture before lysis. Genomic DNA was isolated and purified by the salt lysis method (21).
Detection of DNA strand breaks by Southern analysis
Genomic DNA was restricted with HindIII and treated with (for detection of total SSB) or without (for detection of DSB) 100 mM NaOH for 30 min at 37°C. Equal samples were electrophoresed in 0.6% alkaline (for total SSB) or neutral (for DSB detection) agarose gels at 30 V for 16 h and then transferred to nylon membranes, as described previously (17,18). The membranes were hybridized with a randomly 32P-labeled probe pABB (Ready-to-go kit, Pharmacia) for the rDNA, which detects a 22 kb-fragment containing the 28S region of human rDNA (Figure 1). Intensity of the DNA bands was quantified using a PhosphorImager™ and ImageQuant software (Molecular Dynamics Inc.). The average frequency of strand breaks in the gene of interest was calculated from the amount of DNA present in the irradiated sample and compared with the unirradiated control. We applied the Poisson expression [−ln (irradiated/unirradiated)] to calculate the number of breaks/10-kb fragment. Data were generated from three biological experiments and two gels/experiment.
Map of the region of the human ribosomal genes. Filled boxes indicate the template positions of 18S and 28S rRNAs. Vertical lines indicate the position of the 22 kb-HindIII fragment used in this study.
Results and discussion
In order to assess the involvement of p53 in the repair of DNA strand breaks in an isogenic system, we have stably transfected human ovarian SK-OV-3 cells, which harbor a deletion of the p53 gene (15), with vectors that allow the inducible expression of wild-type p53. We used the pIND and pVgRXR vectors, components of an ecdysone-inducible mammalian expression system (16). SK-OV-3 cells were first transfected with the ecdysone-inducible system regulatory vector, pVgRXR, and selected with Zeocin to establish recombinant SK-OV-3 ZeoR cells that stably express the ecdysone receptor. We next cloned the cDNA encoding human p53wt into the vector pIND, to obtain pINDp53wt. This construct was transfected into the SK-OV-3 cells, which already contained pVgRXR. After transfection and double selection using G418 (selection for p53) and Zeocin (selection for pVgRXR) for 2 weeks, 18 transfectant cell lines were established, which were subsequently grown in the presence of both antibiotics for several weeks. Of these, four cell lines could be induced to generate p53 at various expression levels after treatment with 10 µM ponasterone for 24 h. We selected one of the transfectants, SK-OV-3/pVgRXR/pINDp53wt for further study, since it displayed the highest induction of p53 in the presence of ponasterone and no background expression without ponasterone. In parallel, we have also established a control transfectant cell line SK-OV-3/pVgRXR/pIND using pIND without cloning p53. Both recombinant cell lines SK-OV-3/pVgRXR/pINDp53wt and control SK-OV-3/pVgRXR/pIND did not show obvious morphological changes and their doubling time was only slightly reduced when compared with the untransfected SK-OV-3 cells.
Using the control SK-OV-3/pVgRXR/pIND cell line that did not carry the p53 gene, we began by performing a dose–response experiment to determine the amount of DSBs produced by γ irradiation. DNA strand breaks can be quantitated at the level of individual genes (17,18), using a methodology similar to that previously described for the detection of UV dimers, which utilizes agarose gel electrophoresis, followed by a quantitative Southern analysis (22). Using this assay, presence of strand breaks in the DNA fragment of interest appears as a less intense full-length band on the Southern blot. We decided to measure the number of γ irradiation-induced DSBs in a 22 kb-HindIII rDNA fragment (Figure 1), using neutral agarose gel electrophoresis. Since data from the literature have shown that doses <500 Gy do not allow the detection of strand breaks in a small target such as the rDNA fragment (18), we first tried to detect DNA strand breaks following IR doses ranging from 500 to 800 Gy. Surprisingly, the frequency of DSBs introduced after those irradiations was so high that we were able to detect only very faint bands corresponding to the genomic region of interest. The detection of high yields of DSBs detected in ZeoR SK-OV-3 cells following 600 Gy is illustrated in Figure 2A. We then performed an assay to detect DSBs following much lower irradiation doses than induced previously. As shown in Figure 2A, we were able to determine a significant level of DSBs or breaks on opposite strands in close proximity, which were produced in the rDNA fragment following γ irradiation with 20 and 50 Gy. The dose of 50 Gy introduced ∼0.2 DSB/10 kb-fragment (Figure 2B), an amount that is 60 to 70 times higher than the yield of DSBs usually measured in mammalian cells (3). Since it is difficult to conceive of such a high yield of DSBs produced after 50 Gy, we hypothesized that the antibiotic Zeocin, a bleomycin analogue that we added in the culture medium to maintain the expression of the ecdysone receptor, was not completely detoxified and still able to cleave DNA. To support this hypothesis, we determined the level of strand breaks induced by γ irradiation in the control recombinant cells grown for several days in a medium without Zeocin. As shown in Figure 3A, we were not able to detect DSBs up to a dose of 600 Gy. Total SSBs introduced in the rDNA fragment were also measured following the same IR dose–response in cells grown without Zeocin, using alkaline gel electrophoresis followed by Southern blot analysis. This assay reveals SSBs as a whole, including both SSBs and DSBs, as well as abasic sites and sites of incomplete repair. The dose of 600 Gy introduced ∼0.35 SSB/10 kb-fragment (Figure 3A), a measurement that is consistent with previous studies quantifying SSB formation by Southern blots (18,23) and with studies measuring SSB production by other techniques (24,25). This strongly suggested that the high rate of DSBs, which we previously detected in the ZeoR recombinant cells, was due to DNA cleavage by Zeocin in addition to exposure to γ irradiation.
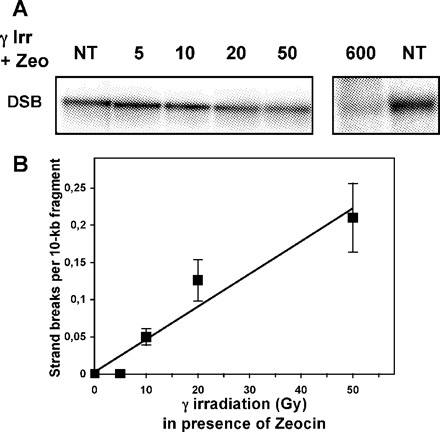
Detection of γ irradiation-induced DSB in the rDNA gene of recombinant SK-OV-3 cells in the presence of Zeocin. (A) Representative autoradiogram of Southern blot of HindIII-genomic fragments, isolated from SK-OV-3/pVgRXR/pIND cell line exposed to increasing doses of γ irradiation in the presence of Zeocin and hybridized with 32P-labeled probe for the rDNA region. The DSBs were assayed by neutral agarose gel electrophoresis. DSB, double strand breaks; NT, non-irradiated DNA. (B) γ Irradiation-induced DSB dose–response at the level of the rDNA gene of recombinant SK-OV-3 cells in the presence of Zeocin. Data were generated from three biological experiments with two gels/assay. The average frequency of strand breaks in the gene of interest was calculated from the amount of DNA present in the irradiated sample compared with the unirradiated control using a PhosphorImager™ and ImageQuant software (Molecular Dynamics Inc.).

Detection of γ irradiation-induced strand breaks in the rDNA gene of recombinant SK-OV-3 cells in the presence or absence of Zeocin. (A) Representative autoradiograms of Southern blot of HindIII-genomic fragments, isolated from SK-OV-3/pVgRXR/pIND cell line exposed to increasing doses of γ irradiation in the absence of Zeocin and hybridized with 32P-labeled probe for the rDNA region. The total strand breaks were assayed by alkaline agarose gel electrophoresis (lower panel) and the DSB by neutral agarose gel electrophoresis (upper panel). (B) Representative autoradiograms of Southern blot of HindIII-genomic fragments, isolated from unirradiated SK-OV-3/pVgRXR/pIND cells grown in the presence (+) or absence (−) of Zeocin and hybridized with 32P-labeled probe for the rDNA region. The total strand breaks were assayed by alkaline agarose gel electrophoresis and the DSB by neutral agarose gel electrophoresis. SSB, single strand breaks.
Finally, to confirm that the presence of Zeocin in the medium was responsible for a significant production of strand breaks, we measured the yield of SSBs at the level of the rDNA gene from unirradiated ZeoR G418R SK-OV-3 cells growing in the presence or in the absence of Zeocin. As expected, alkaline gel electrophoresis revealed a high generation of SSBs in this genomic region in cells grown in medium supplemented with 250 µg/ml Zeocin, compared with cells grown in the absence of the antibiotic (Figure 3B). We estimated that >60% of the DNA fragments analyzed contained at least 1 SSB when the cells were grown in medium supplemented with Zeocin. This significant number of SSBs caused by the chronic cell exposure to Zeocin is probably the result of a balance between the process of strand break formation and repair. It should be noted that we were not able to detect DSBs at the level of the rDNA gene using neutral gel electrophoresis when unirradiated cells were grown in the presence of Zeocin (Figure 3B). These results indicate that the Zeocin treatment induced a significant amount of SSBs, but not DSBs, in the DNA fragment of interest in ZeoR SK-OV-3 cells. The formation of DSBs was only visualized in these cells following γ irradiation in the presence of Zeocin, as shown in Figure 2A. Therefore, we found greater DSBs formation following the combination treatment (Zeocin and γ irradiation) than expected from the individual frequency induced by each treatment alone. This was observed previously following bleomycin–radiation combination, which had an enhancing effect on the formation of DNA strand breaks in treated CHO cells (26).
Our data clearly demonstrate that Zeocin is still able to cleave DNA despite the expression of the Sh ble resistance gene in recombinant ZeoR SK-OV-3/pVgRXR/pIND cells. Since SK-OV-3 cells, similar to many p53-null ovarian cancer cell lines, are known to be radioresistant (27), we examined the consequences of p53 deficiency on the Zeocin-induced SSB formation in our recombinant cells. Using the ZeoR SK-OV-3/pVgRXR/pINDp53wt clone that displayed the highest induction of p53 following treatment with ponasterone, we measured total strand breaks at the level of the rDNA gene. As previously observed with the control SK-OV-3/pVgRXR/pIND cell line (Figure 3B), we found a significant amount of SSBs at the level of the DNA fragment of interest, when SK-OV-3/pVgRXR/pINDp53wt cells were grown in the presence of Zeocin and in the absence of ponasterone (data not shown). In addition, restoration of the p53 function by ponasterone induction did not markedly affect the total amount of DNA strand breaks produced in ZeoR SK-OV-3/pVgRXR/pINDp53wt cells grown in a medium supplemented with Zeocin (data not shown), indicating that the absence of p53 expression in ZeoR SK-OV-3 cells could not account for the significant amount of strand breaks detected in the presence of Zeocin.
Thus, our findings imply that Zeocin is not fully detoxified by the Sh ble protein expressed in ZeoR recombinant SK-OV-3 cells. This suggests that these cells exposed to chronic doses of a radiomimetic drug during clonal selection and subsequent proliferation may have undergone cumulative damages for a large number of divisions. Biological consequences of such a chronic exposure may include mutagenesis and/or adaptive response (28,29), indicating that genomic integrity and metabolism of the recombinant cells may have been altered.
In conclusion, our study indicates that one needs to be very cautious in the interpretation of data involving stable cell lines selected with Zeocin.
Present address: LBCMCP, UMR 5088 CNRS, Bat 4R3B1, 118 Route de Narbonne, 31062 Toulouse, France
This work was supported by a grant from Electricité de France (EDF).
References
Friedberg,E.C., Walker,G.C. and Siede,W. (
Haber,J.E. (
Ward,J.F. (
Valerie,K. and Povirk,L.F. (
Barnes,D.E. (
van den Bosch,M., Lohman,P.H. and Pastink,A. (
Johnson,R.D. and Jasin,M. (
Thompson,L.H. and Schild,D. (
Sturzbecher,H.W., Donzelmann,B., Henning,W., Knippschild,S. and Buchhop,B. (
Yang,Q., Zhang,R., Wang,X.W., Spillare,E.A., Linke,S.P., Subramanian,D., Griffith,J.D., Li,J.L., Hickson,I.D., Loeb,L.A., Mazur,S.J., Appella,E., Brosh,R.M.Jr, Karmakar,P., Bohr,V.A. and Harris,C.C. (
Mekeel,K.L., Tang,W., Kachnic,L.A., Luo,C.M., DeFrank,J.S. and Powell,S.N. (
Saintigny,Y. and Lopez,B.S. (
Linke,S.P., Sengupta,S., Khabie,N., Jeffries,B.A., Buchhop,S., Miska,S., Henning,W., Pedeux,R., Wang,X.W., Hofseth,L.J., Yang,Q., Garfield,S.H., Sturzbecher,H.W. and Harris,C.C. (
Yaginuma,Y. and Westphal,H. (
No,D., Yao,T.P. and Evans,R.M. (
Fritz,L.K., Suquet,C. and Smerdon,M.J. (
May,A. and Bohr,V.A. (
Bennett,R.P., Cox,C.A. and Hoeffler,J.P. (
Abe,A., Miyanohara,A. and Friedmann,T. (
Miller,S.A., Dykes,D.D. and Polesky,H.F. (
Bohr,V.A. and Okumoto,D.S. (
Alrefai,R.H., Beecham,E.J., Bohr,V.A. and Gearhart,P.J. (
Elkind,M.M. and Redpath,J.L. (
van Loon,A.A., Raadsheer,F.C., Timmerman,A.J., Haanen,C., Wessels,J., van der Schans,G.P., Lohmans,P.H. and Baan,R.A. (
Wu,D.Z., Zhang,Y.Q., Keng,P., Sutherland,R.M. and Lasagna,L. (
Concin,N., Zeillinger,C., Stimpfel,M., Schiebel,I., Tong,D., Wolff,U., Reiner,A., Leodolter,S. and Zeillinger,R. (
Kosinska,W., Pelle,E., von Pressentin,M.M., Chen,M. and Guttenplan,J.B. (

{kind=link}
{kind=link}
{kind=link}