




Abstract
Background: Epithelial ovarian cancer often develops resistance to standard treatments, which is a major reason for the high mortality associated with the disease. We examined the efficacy of a treatment regimen that combines immunotherapy to block the activity of epidermal growth factor receptor (EGFR), overexpression of which is associated with the development of resistant ovarian cancer, and photodynamic therapy (PDT), a mechanistically distinct photochemistry-based modality that is effective against chemo- and radioresistant ovarian tumors. Methods: We tested a combination regimen consisting of C225, a monoclonal antibody that inhibits the receptor tyrosine kinase activity of EGFR, and benzoporphyrin derivative monoacid A (BPD)-based PDT in a mouse model of human ovarian cancer. Therapeutic efficacy was evaluated in acute treatment response and survival studies that used 9–19 mice per group. Analysis of variance and Wilcoxon statistics were used to analyze the data. All statistical tests were two-sided. Results: Mice treated with PDT + C225 had the lowest mean tumor burden compared with that in the no-treatment control mice (mean percent tumor burden = 9.8%, 95% confidence interval [CI] = 2.3% to 17.3%, P<.001). Mean percent tumor burden for mice treated with C225 only or PDT only was 66.6% (95% CI = 58.7% to 74.4%, P<.001) and 38.2% (95% CI = 29.3% to 47.0%, P<.001), respectively. When compared with PDT only or C225 only, PDT + C225 produced synergistic reductions in mean tumor burden (P<.001, analysis of variance) and improvements in survival (P = .0269, Wilcoxon test). Median survival was approximately threefold greater for mice in the PDT + C225 group than for mice in the no-treatment control group (80 days versus 28 days), and more mice in the PDT + C225 group were alive at 180 days (3/9; 33% [95% CI = 7% to 70%]) than mice in the C225-only (0/12; 0% [95% CI = 0% to 22%]) or PDT-only (1/10; 10% [95% CI = 0.2% to 44%]) groups. Conclusion: A mechanistically nonoverlapping combination modality consisting of receptor tyrosine kinase inhibition with C225 and BPD-PDT is well tolerated, effective, and synergistic in mice.
Ovarian cancer is responsible for more deaths among American women than any other cancer of the female reproductive tract ( 1 , 2 ) . Seventy percent of women diagnosed with ovarian cancer present with advanced disease and undergo the standard treatment regimen of debulking surgery followed by a combination of paclitaxel and platinum-based chemotherapy. Of these patients, 55–75% will have recurrent disease, which is rarely curable ( 3 ) ; the 5-year survival for these patients remains a dismal 31% ( 4 ) . New therapeutic approaches are therefore necessary for the management of advanced and recurrent epithelial ovarian cancer. However, because of the complex nature of this disease, it is becoming increasingly evident that combination therapies directed at nonoverlapping molecular targets are the most likely to succeed ( 5 , 6 ) . Such an approach might allow for reducing the toxicity from each treatment while also enhancing the effectiveness of each modality. In this in vivo study in a mouse model system, we examined the efficacy of combining photodynamic therapy (PDT), which destroys target tissue via light-based generation of reactive species ( 7 ) , with an antibody-based biologic treatment that induces cell cycle arrest by blocking activation of the epidermal growth factor receptor (EGFR) ( 8 , 9 ) .
PDT is a binary treatment modality that uses low-intensity illumination and photoactivatable chemicals, called photosensitizers, to produce tumoricidal effects through the generation of reactive species ( 10 , 11 ) . A variety of cellular and vascular structures can be disrupted by PDT, depending on variables such as the nature of the photosensitizer used, the duration of illumination, and PDT dosimetry ( 12 ) . PDT is approved by the U.S. Food and Drug Administration (FDA) as a first-line therapy for age-related macular degeneration and for the treatment of actinic keratoses as well as lung and esophageal cancers. Clinical trials involving PDT are ongoing for a wide array of neoplasms, including refractory brain tumors, locally recurrent prostate adenocarcinoma, chronic lymphocytic leukemia, acquired immunodeficiency syndrome-related Kaposi's sarcoma, and intraperitoneal carcinomatosis and sarcomatosis ( 13 ) .
Phase I and II clinical trials have produced encouraging results using PDT delivered at the time of surgical debulking to treat a variety of disseminated intraperitoneal tumors, even though many used porfimer sodium (Photofrin), a first-generation photosensitizer that has a poor toxicity profile due to its low tumor selectivity, particularly for intraperitoneal tumors ( 14 – 20 ) . For example, Hendren et al. ( 21 ) obtained promising results in a phase II trial of PDT for patients with advanced stage sarcomas, mesotheliomas, and ovarian and gastrointestinal carcinomas. Among patients with aggressive tumors, the median actuarial survival—21 months—exceeded expectations for this population ( 21 ) . Of the patients with disseminated solid tumors, ovarian cancer patients appeared to benefit most from the combination of debulking surgery and PDT, with more than 60% of these patients alive after 27 months ( 21 ) .
Bauer et al. ( 18 ) reported that patients with recurrent sarcomatosis, a disease for which there is no effective treatment, who were treated with Photofrin-PDT had improved disease-free interval and time to recurrence compared with historic controls. At last follow-up (1.7–17.3 months after PDT), 45% of the treated patients were alive and had no evidence of disease. Of the 55% of patients with disease recurrence (1.0–8.0 months after PDT), 66% were still alive at the last follow-up ( 18 ) . Most of the treatment-related toxicities reported in this phase II PDT study were manageable and reversible ( 18 ) . Capillary leak syndrome, the most frequent perioperative toxicity, required intensive volume resuscitation, but it typically resolved within 1 week after PDT. Other, less common toxicities included anemia, bowel obstructions, and transient liver function test abnormalities ( 18 , 19 , 21 ) .
These trials have identified dose-limiting toxicities that need to be addressed, particularly improving tumor selectivity (i.e., reducing toxicity in normal tissue) ( 22 , 23 ) . Our group has previously shown that, compared with Photofrin, the second-generation photosensitizer benzoporphyrin derivative monoacid ring A (BPD; verteporfin) has improved photophysical properties ( 24 ) and greater accumulation in tumor as compared to non-tumored tissue in a mouse model of intraperitoneally disseminated ovarian cancer ( 25 , 43 ) . Furthermore, BPD lacks the prolonged cutaneous phototoxicity of Photofrin ( 26 ) and is FDA approved for the treatment of age-related macular degeneration.
The molecular target for the second modality in the combination regimen is the EGFR, which is a receptor tyrosine kinase that regulates a number of critical cellular functions, including proliferation, differentiation, motility, and survival, via complex signaling cascades. Increased EGFR activity promotes cell cycle progression from G 1 to S phase, causing disproportionate cell proliferation. Increased expression of EGFR is associated with a poor prognosis in patients with a variety of cancers. EGFR overexpression is seen in 20–30% of patients who present with advanced stage ovarian cancer, and is associated with a more aggressive and invasive phenotype ( 8 , 27 – 32 ) . Ovarian cancer tissue takes up more 125 I-EGF, a radiolabeled form of the native ligand for the EGFR, than nonmalignant ovarian epithelium ( 33 ) . Therapies directed at preventing or blocking EGFR function could, therefore, be of benefit.
The human-mouse chimeric monoclonal antibody C225 (Erbitux, cetuximab) specifically recognizes EGFR and competes with the native ligand EGF for binding to EGFR, interrupting normal cell proliferation pathways and inducing G 1 arrest ( 34 ) . C225 is FDA approved for the treatment of metastatic colorectal cancer and is being explored for the treatment of other cancers, including advanced ovarian cancer. However, dose-limiting toxicities ( 35 , 36 ) and frequency of relapse associated with the use of C225 have stimulated the development of novel combination treatments that could both improve overall efficacy and minimize toxicity.
Combinations of C225 with either chemotherapy agents or radiotherapy are currently being tested in clinical trials, which are showing encouraging results for a variety of solid tumors ( 8 , 37 ) . We have previously shown that ex vivo photoimmunotherapy of ovarian cancer in combination with cisplatin had synergistic effects, with the greatest enhancement seen in chemoresistant cells ( 38 ) .
In this study, we tested a novel approach for the treatment of ovarian cancer that combines the biologic effect of C225-based immunotherapy with the photochemical cytotoxicity of BPD-PDT. We reasoned that combining BPD-PDT, whose primary site of action is the mitochondria ( 7 ) , with immunotherapy using EGFR-targeted blockade leading to cell cycle arrest ( 8 , 9 ) could enhance the effectiveness of each modality. We examined the efficacy of C225-based inhibition of EGFR in combination with BPD-PDT in a mouse model of human ovarian cancer.
M ATERIALS AND M ETHODS
Cell Line and Culture Conditions
Human epithelial ovarian cancer NIH:OVCAR-5 cells were obtained from Thomas Hamilton (Fox Chase Cancer Institute, Philadelphia, PA). The cells were grown in RPMI-1640 medium (Mediatech Inc., Herndon, VA) supplemented with 10% heat-inactivated fetal calf serum (GIBCO Life Technologies, Grand Island, NY), 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were maintained in an incubator at 37 °C in an atmosphere of 5% CO 2 .
Light Source
For illumination, we used a solid-state BWF 690-1 diode laser (B&W TEK, Inc., Newark, DE) to deliver monochromatic light at a wavelength (690 ± 5 nm) that closely overlapped the absorption maxima of BPD (690 nm) at a maximum power of 1 W from the diode, using a cylindrically diffusing-tip fiber (8.0 mm × 0.4 mm).
Animal Model
We used a xenograft mouse model for human ovarian carcinomatosis that was previously established in our laboratory ( 41 ) . Athymic Swiss female Nu/Nu mice (20–25 grams and 6–8 weeks old; Cox Breeding Laboratories, Cambridge, MA) were anesthetized with an intraperitoneal injection of a ketamine/xylazine mixture (ketamine, 105 mg/kg; xylazine, 15 mg/kg), weighed, and injected intraperitoneally with 31.5 × 10 6 NIH:OVCAR-5 cells suspended in 2 mL phosphate-buffered saline (PBS).
The mice had continual access to food and water and were housed in laminar flow racks under specific pathogen-free conditions. Mice were killed by CO 2 inhalation. All animal experiments were conducted according to guidelines established by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Intraperitoneal PDT
We performed intraperitoneal PDT in the nude mice as previously described ( 25 ) . Briefly, on days 10 and 20 after tumor cell inoculation, each mouse (9–16 animals per group) was injected intraperitoneally with 0.25 mg/kg body weight of liposomal BPD (QLT, Inc., Vancouver, British Columbia, Canada ) in 1 mL sterile PBS. BPD solutions were prepared immediately before use, and all work involving BPD was performed in subdued lighting. Mice that were to be illuminated were anesthetized by sequential intraperitoneal injection with a ketamine/xylazine mixture (ketamine, 105 mg/kg; xylazine, 15 mg/kg) and 2 mL of a 0.1% intralipid solution (Intralipid soybean oil emulsion for intravenous use; Baxter Healthcare Corporation, Deerfield, IL) before illumination to enhance light scattering. Mice were illuminated 90 minutes after BPD injection as follows. The diffusing-tip fiber, which was connected to the diode laser, was introduced into the peritoneal cavity of a supine anesthetized animal via a centrally placed 22-gauge catheter that traversed the abdominal cavity. A total of 20 J/cm 2 of 690-nm light was delivered via the diffusing-tip fiber at a fluence rate of 150 mW/cm 2 . One-fourth of the total light energy was delivered to each intraperitoneal quadrant over equivalent periods. This method of light delivery was chosen on the basis of previous studies of tissue dosimetry in mice, which showed that the quadrant approach was the least invasive among the geometries studied and was as effective as more complicated and intrusive methods ( 42 ) . At the completion of the treatment, the mice were placed in an animal warmer until they resumed normal activity.
C225 Treatment
On day 11 after tumor cell injection, mice (9–19 animals per group) received the first dose of C225 (0.5 mg in a volume of 0.25 mL of a preservative-free solution containing 8.48 mg/mL sodium chloride, 1.88 mg/mL sodium phosphate dibasic heptahydrate, 0.42 mg/mL sodium phosphate monobasic monohydrate, and water by intraperitoneal injection; Imclone Systems, Inc., New York City, NY). Additional doses were given on days 14, 17, and 19 after tumor cell injection, for a total dose of 2.0 mg C225 per mouse.
Study Endpoints
Therapeutic efficacy was evaluated in acute treatment response and survival studies. Four measures were used to assess treatment response: tumor burden (analyzed as mean tumor burden [mg] and as mean percent tumor burden [%]), anatomic tumor distribution, weight loss, and pathologic findings. The survival endpoint was defined as death due to disease.
Acute Treatment Response Studies
Mice were randomly assigned to the following eight experimental groups to evaluate acute treatment response: no treatment (n = 12), Light only (n = 17), BPD only (n = 12), C225 only (n = 12), C225 + light (n = 15), BPD + C225 (n = 19), PDT (n = 16), and PDT + C225 (n = 14).
Tumor burden, anatomic tumor distribution, and weight loss.
On day 21 after tumor cell injection, all mice were weighed, killed, and necropsied to assess tumor burden following treatment as previously described ( 25 ) . At necropsy, tissue from the following areas was systematically resected and weighed: subgastric omentum, mesentery, pelvis, diaphragm, pelvic omentum, and bladder. The mice were also examined carefully for the presence of distant extra-abdominal metastases. The wet weights of the tissues from the individual anatomical sites were added to determine the total tumor burden per animal.
Mean tumor burden was calculated by averaging the total tumor burden from every animal per treatment group. In the presence of tumor involvement, it can be difficult to distinguish noninvolved tissue from areas completely encased by metastatic disease. Consequently, we subtracted the mean weight of organs from healthy mice that had not been injected with tumor cells (711 mg).
Mean percent tumor burden was calculated by dividing the mean tumor burden of each treated group by the mean tumor burden of the no-treatment controls.
Pathology.
Resected tissue was fixed in 10% phosphate-buffered formalin and embedded in paraffin. Sections cut 4 μm thick were stained with hematoxylin-eosin for microscopic assessment. A gynecologic pathologist (EO), who was blinded to treatment group, evaluated tumor sections from the subgastric omentum, mesentery, pelvis, diaphragm, and pelvic omentum.
Formalin-fixed, paraffin-embedded tissue sections were immunostained for antiproliferating cell nuclear antigen using a mouse monoclonal antibody (antiproliferating cell nuclear antigen, clone 10; Dako, Inc., Carpenteria, CA). The sections were first deparaffinized for 10 minutes in xylene and rehydrated to 95% alcohol, then immersed in 3% hydrogen peroxide in methanol for 20 minutes at room temperature to quench endogenous peroxidase activity and washed in PBS. Heat-induced epitope retrieval was performed by warming the sections in a heated water bath for 30 minutes at 99 °C and then cooling the slides slowly to room temperature under cold water and washing three times with PBS. The sections were then incubated for 20 minutes with 1.5% normal horse serum (Vector Laboratories) to block nonspecific binding and washed with PBS. The sections were incubated with the primary antibody (1:500 dilution) overnight in a moist chamber at 4 °C, washed, incubated with biotinylated horse anti-mouse immunoglobulin G (1:200 dilution; Vector Laboratories), and washed again. The sections were then incubated with an avidin-peroxidase conjugate (Vector Laboratories), washed three times with PBS, incubated with 0.1% 3,3′-diaminobenzidine tetrahydrochloride (Sigma) in PBS containing 0.006% hydrogen peroxide for 2–6 minutes with frequent microscopic checking until the background staining became just barely visible, and washed three times in water. Sections were counterstained with hematoxylin (Richard-Allen Scientific, Kalamazoo, MI). We used lymphatic nodal tissue from mice in the no-treatment group as a positive control for staining. For negative controls, sections were processed as above except that the primary antibody was omitted from the overnight incubation.
Survival Studies
Mice in the following experimental groups were used to evaluate survival: no treatment (n = 10), C225 only (n = 9), PDT (n = 10), and PDT + C225 (n = 9). Mice that experienced rapid tumor progression that lead to uncontrollable ascites or systemic illness were killed if they had lost more than 15% or gained more than 20% of their original body weight, were in severe pain that did not appear to be relieved by analgesics, displayed self-mutilating behavior, or did not eat or drink. All mice that were still alive 180 days after tumor cell inoculation were weighed, killed, and necropsied. Mice that lived to 180 days and were grossly free of tumor at necropsy were considered “cured.” At the time of necropsy, tissue samples were collected and weighed as described above.
Statistical Analysis
Analysis of variance was used to examine the effects of PDT and C225 on mean tumor burden and to examine the effects of PDT + C225. For assessment of treatment effects on tumor burden, all statistical tests were done in the log scale to establish the normality assumption, which is required for analysis of variance. For the analysis of anatomic tumor distribution, mean percent tumor burden for each of the treated groups at specific sites was calculated based on the site-specific mean tumor burden of the no-treatment group. We used paired t tests to compare the mean percent tumor burdens at various sites. In addition, we evaluated synergy of interaction between C225 and PDT using analysis of variance. The product-limit method with Kaplan-Meier curves was used to summarize the distribution of survival times. We used the Wilcoxon test to compare the survival curves among groups and to test for synergism. We used Fisher's exact test to compare the proportions of mice in the different treatment groups that were still alive at day 60 and at day 180. All statistical tests were two-sided. Statistical tests were performed with the use of SAS software (version 8.2; SAS Institute, Cary, NC). P values less than .05 were considered statistically significant.
R ESULTS
Acute Treatment Response Studies
Overall tumor burden.
Mice in each of the groups that received treatment showed some reduction in mean tumor burden compared with mice in the no-treatment control group ( Figure 1 ). The most dramatic reduction in tumor burden relative to the no-treatment controls was in mice treated with PDT + C225 (mean percent tumor burden = 9.8%, 95% confidence interval [CI] = 2.3% to 17.3%, P <.001). Mean percent tumor burdens for animals treated with C225 only or PDT only were 66.6% (95% CI = 58.7% to 74.4%, P <.001) and 38.2% (95% CI = 29.3% to 47.0%, P <.001), respectively. When compared with the individual monotherapies, PDT only or C225 only, the combination treatment group, PDT + C225, produced a synergistic reduction in mean tumor burden ( P <.001, analysis of variance).
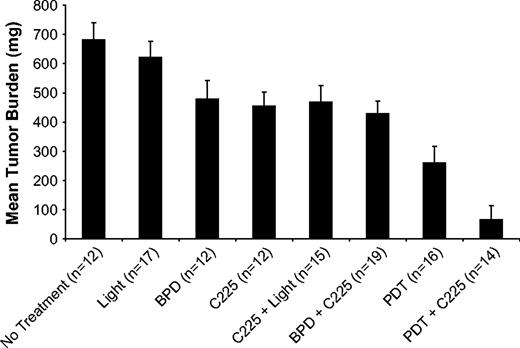
Tumor burden for control and treated groups in acute treatment response studies. Error bars represent 95% confidence intervals. BPD = benzoporphyrin derivative.
A modest tumoricidal response was also observed in mice treated with BPD only, i.e., without light activation (mean percent tumor burden = 70.1%, 95% CI = 59.8% to 80.4%, P <.001).
Anatomic tumor distribution.
Figure 2 shows the site-specific mean percent tumor burden for the following groups: no treatment, C225, PDT, and PDT + C225. For mice treated with C225 only, mean percent tumor burden was highest in the subgastric omentum (90.9%, 95% CI = 79.5% to 102.2%, P = .10) and lowest in the pelvis (73.6%, 95% CI = 63.3% to 84.0%, P <.001). The calculated difference in the mean percent tumor burden between these two anatomical sites was not statistically significant (17.3%, 95% CI = 19.5% to 34.7%; P = .053). For the mice treated with PDT only, the mean percent tumor burden was lowest in the pelvis (65.8%, 95% CI = 57.3% to 74.3%, P <.001) and highest in the diaphragm (78.4%, 95% CI = 70.9% to 85.8%, P <.001). The calculated difference in the mean percent tumor burden between these two anatomical sites was statistically significant (12.6%, 95% CI = 12.5% to 21.6%; P = .01). For the group treated with PDT + C225, substantial tumoricidal effects were seen at four anatomical sites: pelvic omentum (mean percent tumor burden = 47.7%, 95% CI = 35.2% to 60.2%, P <.001), pelvis (mean percent tumor burden = 49.4%, 95% CI = 38.3% to 60.5%, P <.001), subgastric omentum (mean percent tumor burden = 49.8%, 95% CI = 41.6% to 58.0%, P <.001), and mesentery (mean percent tumor burden = 52.1%, 95% CI = 42.4% to 61.7%, P <.001). A more limited treatment response was observed in the diaphragm (mean percent tumor burden = 66.4%, 95% CI = 57.1% to 75.7%, P <.001). The calculated difference in the mean percent tumor burden between each of the four anatomical sites where a substantial treatment effect was observed and the diaphragm was statistically significant (for pelvic omentum versus diaphragm,18.7%, 95% CI = 13.2% to 29.2%, P = .002; for pelvis versus diaphragm, 17.0%, 95% CI = 16.4% to 30.1%, P = .015; for subgastric omentum versus diaphragm, 16.6%, 95% CI = 16.2% to 29.5%, P = .016; for mesentery versus diaphragm,14.3%, 95% CI = 11.7% to 23.7%, P = .006).
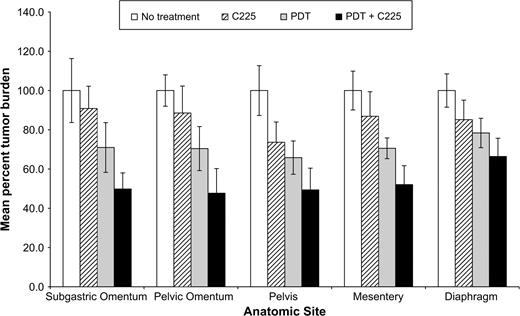
Site-specific comparison of tumor distribution. Mean percent tumor burden was evaluated for mice in the following groups: No treatment, C225 only, PDT only, and PDT + C225. Site-specific values are given for disease resected from the subgastric omentum, pelvic omentum, pelvis, mesentery, and diaphragm. Error bars represent 95% confidence intervals.
Weight loss.
All mice lost weight during the 3 weeks after tumor cell injection ( Table 1 ). Mice in the no-treatment group lost the most weight (2.66 g, 95% CI = 2.00 g to 3.32 g), and mice treated with C225 + light lost the least weight (mean weight loss [C225 + light] = 1.75 g, 95% CI = 1.37 g to 2.14 g; difference in mean weight loss [controls versus C225 + light] = 0.91 g, 95% CI = 0.17 g to 1.65 g, P = .018).
Weight loss at 3 weeks after tumor cell injection *
| Group | N | Weight loss, g Mean (95% CI) | P† |
|---|---|---|---|
| No treatment | 11 | 2.66 (2.1 to 3.2) | (referent) |
| Light | 17 | 2.63 (2.3 to 2.9) | .9388 |
| BPD | 13 | 2.01 (1.5 to 2.5) | .0989 |
| C225 | 14 | 2.11 (1.5 to 2.7) | .1545 |
| C225 + light | 15 | 1.75 (1.4 to 2.1) | .0175 |
| BPD + C225 | 25 | 1.96 (1.5 to 2.4) | .0443 |
| PDT | 15 | 2.14 (1.7 to 2.6) | .1736 |
| PDT + C225 | 16 | 2.09 (1.6 to 2.5) | .1288 |
| Group | N | Weight loss, g Mean (95% CI) | P† |
|---|---|---|---|
| No treatment | 11 | 2.66 (2.1 to 3.2) | (referent) |
| Light | 17 | 2.63 (2.3 to 2.9) | .9388 |
| BPD | 13 | 2.01 (1.5 to 2.5) | .0989 |
| C225 | 14 | 2.11 (1.5 to 2.7) | .1545 |
| C225 + light | 15 | 1.75 (1.4 to 2.1) | .0175 |
| BPD + C225 | 25 | 1.96 (1.5 to 2.4) | .0443 |
| PDT | 15 | 2.14 (1.7 to 2.6) | .1736 |
| PDT + C225 | 16 | 2.09 (1.6 to 2.5) | .1288 |
CI = confidence interval; BPD = benzoporphyrin derivative; PDT = photodynamic therapy.
Two-sided (analysis of variance).
Weight loss at 3 weeks after tumor cell injection *
| Group | N | Weight loss, g Mean (95% CI) | P† |
|---|---|---|---|
| No treatment | 11 | 2.66 (2.1 to 3.2) | (referent) |
| Light | 17 | 2.63 (2.3 to 2.9) | .9388 |
| BPD | 13 | 2.01 (1.5 to 2.5) | .0989 |
| C225 | 14 | 2.11 (1.5 to 2.7) | .1545 |
| C225 + light | 15 | 1.75 (1.4 to 2.1) | .0175 |
| BPD + C225 | 25 | 1.96 (1.5 to 2.4) | .0443 |
| PDT | 15 | 2.14 (1.7 to 2.6) | .1736 |
| PDT + C225 | 16 | 2.09 (1.6 to 2.5) | .1288 |
| Group | N | Weight loss, g Mean (95% CI) | P† |
|---|---|---|---|
| No treatment | 11 | 2.66 (2.1 to 3.2) | (referent) |
| Light | 17 | 2.63 (2.3 to 2.9) | .9388 |
| BPD | 13 | 2.01 (1.5 to 2.5) | .0989 |
| C225 | 14 | 2.11 (1.5 to 2.7) | .1545 |
| C225 + light | 15 | 1.75 (1.4 to 2.1) | .0175 |
| BPD + C225 | 25 | 1.96 (1.5 to 2.4) | .0443 |
| PDT | 15 | 2.14 (1.7 to 2.6) | .1736 |
| PDT + C225 | 16 | 2.09 (1.6 to 2.5) | .1288 |
CI = confidence interval; BPD = benzoporphyrin derivative; PDT = photodynamic therapy.
Two-sided (analysis of variance).
The only other experimental group to lose a statistically significant amount of weight compared with the no-treatment controls was the mice treated with BPD + C225 (mean weight loss [BPD + C225] = 1.96 g, 95% CI = 1.49 g to 2.43 g; difference in mean weight loss [controls versus BPD + C225] = 0.70 g, 95% CI = 0.02g to 1.37 g, P = .044).
Pathology findings.
Expression of proliferating cell nuclear antigen, a nuclear protein expressed by cells undergoing division that is commonly used as a histologic marker for cellular proliferation, was analyzed in tumor tissues by immunohistochemistry. Tumor tissues from mice in the no-treatment group stained uniformly with a monoclonal antibody against proliferating cell nuclear antigen, whereas tumor tissue from mice treated with PDT + C225 typically demonstrated patchy and weaker staining ( Fig. 3 ).
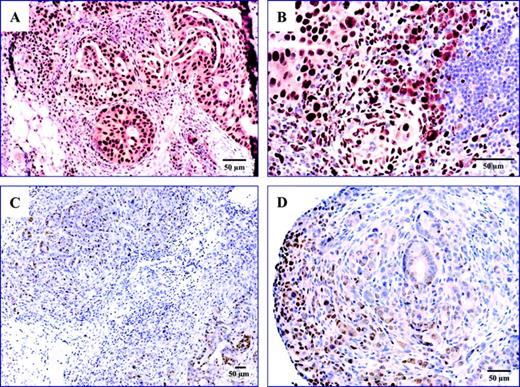
Immunohistochemical staining for proliferating cell nuclear antigen (PCNA). ( A ) Tumor from the omentum of a no-treatment mouse (i.e., a mouse that was injected with tumor cells but not treated); ( B ) lymph nodes from a no-treatment mouse; tumors from the pelvis ( C ) and mesentery ( D ) of a mouse treated with PDT + C225 resected 72 hours after the second PDT treatment. The blue color indicates nuclear staining and the brown color represents staining for PCNA. Magnification bars represent 50 μm.
At necropsy, we noted that mice treated with C225 or with C225 and light had more adhesions than the PDT-treated mice. These adhesions were typically located between the pelvic omentum and the anterior abdominal wall, among loops of the small and large bowel, and between the uterus and the cul-de-sac.
Survival
Kaplan-Meier analyses for survival.
We evaluated the survival of mice that received no treatment, C225 only, PDT only, and PDT + C225. The Kaplan-Meier curves for these four experimental groups are depicted in Fig. 4 . By day 40 after tumor cell injection, none of the untreated control mice were alive (0%; 95% CI = 0% to 31%). At 180 days after tumor cell injection, three of the nine mice treated with the PDT + C225 combination regimen were still alive (33%; 95% CI = 7% to 70%) and were considered “cured,” whereas 0 out of 12 mice treated with C225 only (0%; 95% CI = 0% to 22%) and 1 of 10 mice treated with PDT only was still alive (10%; 95% CI = 0.2% to 44%). Median survival was 28 days (interquartile range [IQR] = 21–31 days) for the no-treatment group, 36 days (IQR = 32–90 days) for the PDT-only group, 26 days (IQR = 25–37) for the C225-only group, and 80 days (IQR = 47 days – upper range not calculable secondary to censored data) for the PDT + C225 group. A comparison of the survival curves revealed no statistically significant difference between the untreated mice and the mice treated with C225 only ( P = .46) as analyzed by the Wilcoxon test. By contrast, mice treated with PDT or PDT + C225 showed statistically significant improvements in survival compared with the untreated control mice ( P = .01 and P <.001, respectively). The combination treatment, PDT + C225, resulted in a synergistic enhancement of survival compared with each of the individual monotherapies, as analyzed by the Wilcoxon test ( P = .027).
![Kaplan–Meier curves for survival. Median survival was 28 days (interquartile range [IQR] = 21–31 days) for untreated mice, 36 days (IQR = 32–90 days) for mice treated with photodynamic therapy only (PDT), 26 days (IQR = 25–37 days) for mice treated with C225 only, and 80 days (IQR = 47 days – upper range not calculable secondary to censored data) for mice treated with PDT + C225. The combination treatment, PDT + C225, resulted in a synergistic enhancement of survival compared to the individual monotherapies, as analyzed by the Wilcoxon test ( P = .027).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jnci/97/20/10.1093_jnci_dji314/2/m_jncidji314f04_lw.jpeg?Expires=1716444487&Signature=p5aFctO~RgrCTAYC1qNncVAccEh2kLhyu3BPDzWHrv3tHnuuP27Hc5sVhPaKPjclIDACC398RUvngLrnTBeUnX4mCXMxAmtNX-51--gmrByPRplY6W5o4HHoHffqGk12mo6wAIQVed7n9VXc7mn7KlZWLqhIJaa12jIhoCmLvyuVXH6cHJniw18fDaWPom7XeKm9Tar~A1gmpMAdrkJFDu-6zqlHrt1UnJNM9AxjJy~W3a-oRNws2uUsOQa1q2U1t~kOy4KhhCNG2Ie~ZUZnIwfShGOa3TinpIizxl8nVMrN5mWNB63DVH8c7BqztpYRL5LJMQnp0PjKSqICtRNLeQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Kaplan–Meier curves for survival. Median survival was 28 days (interquartile range [IQR] = 21–31 days) for untreated mice, 36 days (IQR = 32–90 days) for mice treated with photodynamic therapy only (PDT), 26 days (IQR = 25–37 days) for mice treated with C225 only, and 80 days (IQR = 47 days – upper range not calculable secondary to censored data) for mice treated with PDT + C225. The combination treatment, PDT + C225, resulted in a synergistic enhancement of survival compared to the individual monotherapies, as analyzed by the Wilcoxon test ( P = .027).
Pathology findings.
All mice that were alive at day 180 after tumor cell injection ( n = 4) were killed and examined for the presence of cancer. Three of the mice had been treated with PDT + C225, and one mouse had received PDT only. No evidence of gross tumor burden was noted in the three mice treated with the combination regimen, and they were considered “cured.” By contrast, the mouse that received PDT only and lived to 180 days showed evidence of residual carcinomatosis at the time of necropsy. This finding suggests that, in this particular mouse, PDT alone was effective in reducing the primary tumor burden and that, as a consequence, the development of lethal tumor burden was delayed for a longer time compared with the other mice in that treatment group.
D ISCUSSION
In this study, we found that C225-based EGFR blockade in combination with BPD-PDT decreased tumor burden and increased survival more than either modality alone in a xenograft mouse model that closely resembles human metastatic ovarian cancer. The synergistic response observed by using the combination treatment may reflect the fact that the individual monotherapies target and affect nonoverlapping molecular pathways. This differential molecular targeting should allow for maximal treatment efficacy with minimal toxicity and provides a possible mechanism for the synergistic interaction of C225 and PDT ( 6 , 9) .
Because recurrent epithelial ovarian cancer is rarely curable, the combination regimen used in the current study could be clinically useful in the treatment of women with ovarian cancer for three reasons: 1) C225 and BPD-PDT each employ mechanisms of action that are distinct from those employed by conventional chemotherapeutic agents; 2) the combination regimen, in part, targets tumor cells that overexpress EGFR, which is an indicator of aggressive and chemoresistant disease; and 3) PDT has been shown be effective against epithelial ovarian cancer that is refractory to chemotherapy and radiation therapy ( 17 , 18 , 44 – 46 ) .
C225-based immunotherapy is directed against the EGFR, overexpression of which is associated with a more aggressive form of ovarian cancer. Under normal conditions, EGFR expression can be critical for maintaining cellular homeostasis via receptor tyrosine kinase signaling pathways. One such pathway is the RTK-Grb2-Sos-Ras cascade, which activates both the mitogen-activated protein kinase and the phosphatidylinositol 3-kinase pathways. Increased mitogen-activated protein kinase and phosphatidylinositol 3-kinase signaling facilitates unregulated cell proliferation by shifting the normal balance of proteins to those that favor progression through the cell cycle ( 47 – 49 ) . Cancer cells that overexpress EGFR exhibit elevated levels of Cyclin-D and Myc, enhanced phosphorylation of retinoblastoma protein, increased eukaryotic translation initiation factor 4E activity ( 32 , 50 ) , and decreased levels of p27 (because of rapid degradation). This differential expression of regulatory proteins allows cancer cells to ignore checkpoint signals and to make an inappropriate transition from G 1 to S phase ( 51 ) . C225 blockade of EGFR activity prevents cancer cells that overexpress EGFR from aberrantly entering S phase, thereby inhibiting unregulated progression through the cell cycle ( 8 , 9 ) .
Cancer cells that are highly dependent on these proliferative pathways become particularly vulnerable to additional therapeutic insult and are more effectively killed via a secondary modality, such as PDT. BPD-PDT leads to tumor destruction by inducing apoptosis via mitochondrially targeted release of cytochrome c . Photochemical release of cytochrome c triggers activation of caspases 3, 6, 7, and 8, which in turn leads to cleavage of either Poly (ADP-ribose) Polymerase or Bap31, ultimately resulting in apoptosis ( 39 , 52 ) .
An alternative explanation for the observed synergistic activity of C225 and PDT is based on recent findings that PDT can enhance EGFR phosphorylation as well as increase the activity of downstream signaling members, such as ERK 1 and ERK 2 ( 53 , 54 ) . Treatment of NIH:OVCAR-5 cells in vitro with C225-based immunoconjugates was shown to mitigate this activation of EGFR and suppress downstream survival signals, thereby enhancing the efficacy of PDT ( 53 ) .
PDT has a unique translational applicability for the treatment of ovarian cancer because this disease, even in its advanced stages, is generally limited to the peritoneal cavity, and thus can be treated via local therapies. PDT with fiber optics could therefore be used to treat small-volume residual disease following initial debulking and cytoreduction. PDT is also a useful alternative to conventional regimens, either as a consolidation treatment or as an effective salvage therapy. The use of a photosensitizer such as BPD, which has better pharmacokinetics than Photofrin and none of its prolonged cutaneous phototoxicity effects ( 7 , 25 , 26 ) , reduces the potential for treatment-related side effects. Combining BPD-PDT with a second modality that functions via a different molecular pathway allows for the use of lower doses of both drugs and light. In the case of PDT, reduced doses of photosensitizer and light often translate into improved selectivity because cytotoxic effects due to PDT are not observed below a threshold level of interaction between drug and light ( 55 ) .
Several clinical trials have investigated the use of PDT for the treatment of disseminated intraperitoneal malignancies. Results from phase I/II clinical trials of unoptimized PDT treatment conditions in intraperitoneal sarcomatosis and carcinomatosis suggest that PDT may have promise for treatment of patients with minimal residual disease after cytoreductive surgery or small-volume chemoresistant disease ( 16 , 21 , 22 ) . Results from a phase II clinical trial of patients with disseminated carcinomatosis treated with PDT indicate a survival advantage over historic controls, even though the nonspecific photosensitizer Photofrin was used ( 21 ) . Among all the intraperitoneal solid tumors studied in that trial, ovarian cancer appeared to be one of the most responsive to PDT ( 21 ) . Documented treatment-associated toxicities were acute but reversible ( 21 ) .
The combination treatment of C225 + PDT investigated in the current study represents a substantial improvement in efficacy from previous in vivo studies and further optimization of PDT-mediated therapeutic regimens. BPD has enhanced photobiologic activity, accumulates more rapidly in tumors, and clears more quickly from the body than the dihematoporphyrin ethers in Photofrin ( 24 – 26 , 40 ) . Molpus et al. ( 25 ) investigated the efficacy of BPD-PDT in vivo using a xenograft mouse model for disseminated ovarian cancer and found that three to five PDT treatments were necessary to achieve therapeutic benefit in reduction of mean tumor burden and survival. By contrast, we found that BPD-PDT in combination with anti-EGFR therapy required fewer treatments to achieve improved efficacy compared with that reported by Molpus et al. with minimal toxicity, less weight loss, and no treatment-related morbidity ( 25 ) .
We qualitatively assessed adhesion formation following treatment, and found that adhesions increased with the use of C225 only but decreased with the use of BPD-PDT. We speculate that BPD-PDT results in less adhesion formation because of the favorable wound healing effects of PDT with certain photosensitizers, as have been noted in other clinical and preclinical studies ( 61 – 63 ) ; these effects would tend to counteract the adhesion formation potential of C225. The ability of BPD to prevent adhesion is further intriguing because other photosensitizers, such as Photofrin and chlorin e6, have instead been associated with adhesion formation ( 64 ) .
Among the mice treated with BPD-PDT or the combination therapy, a less substantial reduction in tumor burden was observed in the diaphragm relative to the other anatomical sites, presumably reflecting the poor accessibility of this site to light. Consistent with this notion, Molpus et al. ( 25 ) also found that the mesentery, pelvic omentum, pelvis, and subgastric omentum were more effectively treated than the diaphragm.
The limitations of the current investigation are that this is a proof-of-concept study without any optimization of the treatment parameters (i.e., doses of light, photosensitizer, and antibody, timing of treatments, and sequence of treatments). Further optimization of PDT parameters would include the use of more selective photosensitizers and monitoring of intratumoral hypoxia ( 22 , 23 , 25 , 57 ) . In addition, the low-level tumoricidal effect observed with photosensitizers alone (in the absence of light activation) needs to be better understood and exploited ( 58 – 60 ) .
In summary, our results suggest that the rational use of a combination of biologic and photochemical therapies may be effective for the treatment of advanced and recurrent epithelial ovarian cancer and merits consideration for clinical development. EGFR overexpression is associated with advanced and resistant disease, whereas PDT in experimental models is effective against chemo- and radioresistant ovarian cancer cells. The combination of EGFR immunotherapy and PDT addresses some of the fundamental problems in the treatment of ovarian cancer with each of the monotherapies. On the PDT side, the major limitation to its use has been the nonspecific (acute and prolonged) phototoxicity with Photofrin and excessive adhesion formation. These problems are minimized with BPD-PDT. With C225, the primarily cytostatic, as opposed to cytotoxic, effect necessitates prolonged treatment, resulting in frequent relapse and antibody-associated toxicities. The combination, BPD-PDT + C225, was well tolerated and demonstrated statistically significant tumoricidal response, acceptable toxicity, and improved survival in a murine ovarian cancer model for disseminated disease. The marked reduction noted in total tumor volume with fewer PDT treatments than previously described ( 25 ) could be useful in the application of PDT in the treatment of microscopic epithelial ovarian cancer at the time of initial diagnosis and debulking or in the setting of small-volume recurrent disease. Our results invite investigation of other PDT-immunotherapy combination regimens such as the application of immunoconjugates in conjunction with other treatment modalities.
This work was supported by Public Health Service grant R01AR40352 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Department of Health and Human Services. Marcela G. del Carmen was supported by a Department of Energy Massachusetts General Hospital Laser Center Fellowship in Gynecologic Oncology from Vincent Memorial Obstetrics and Gynecology Service.
We thank QLT, Inc. (Vancouver, British Columbia, Canada), for the BPD and Imclone Systems, Inc. (New York, NY), for the monoclonal antibody C225.
References
Ozols RF. Treatment of ovarian cancer: current status.
Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000.
Bolis G, Villa A, Guarnerio P, Ferraris C, Gavoni N, Giardina G, et al. Survival of women with advanced ovarian cancer and complete pathologic response at second-look laparotomy.
American Cancer Society. Cancer facts and figures 2004. Atlanta (GA): American Cancer Society;
Chabner B, Boral A, Multani P. Translational research: walking the bridge between idea and cure.
Chabner B. Cytotoxic agents in the era of molecular targets and genomics.
Runnels JM, Chen N, Ortel B, Kato D, Hasan T. BPD-MA-mediated photosensitization in vitro and in vivo: cellular adhesion and beta1 integrin expression in ovarian cancer cells.
Ciardiello F, Bianco R, Damiano V, De Lorenzo S, Pepe S, De Placido S, et al. Antitumor activity of sequential treatment with topotecan and anti-epidermal growth factor receptor monoclonal antibody C225.
Mendelsohn J. Epidermal growth factor receptor inhibition by a monoclonal antibody as anticancer therapy.
Hasan T, Moor ACE, Ortel B. Photodynamic therapy of cancer. In: Holland JF, Frei EI, Bast RCJ, Kufe DW, Morton DL, Weichselbaum RR, editors. Cancer medicine. 5th ed. London (United Kingdom): B.C. Decker, Inc.;
Spikes J. Photobiology of phorphyrins. In: Dorian DR, Gomer CL, editors. Porphyrin localization and treatment of tumors. New York (NY): Arlan R. Liss;
Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, et al. Photodynamic therapy.
NLM-NIH. ClinicalTrials.gov: National Institutes of Health-National Library of Medicine. Available at: http://www.clinicaltrials.gov/ . [Last accessed: December 20,
Sindelar WF, DeLaney TF, Tochner Z, Thomas GF, Dachoswki LJ, Smith PD, et al. Technique of photodynamic therapy for disseminated intraperitoneal malignant neoplasms. Phase I study.
Tochner Z, Mitchell JB, Hoekstra HJ, Smith P, DeLuca AM, Barnes M, et al. Photodynamic therapy of the canine peritoneum: normal tissue response to intraperitoneal and intravenous photofrin followed by 630 nm light.
Wierrani F, Fiedler D, Grin W, Henry M, Dienes E, Gharehbaghi K, et al. Clinical effect of meso-tetrahydroxyphenylchlorine based photodynamic therapy in recurrent carcinoma of the ovary: preliminary results.
DeLaney TF, Sindelar WF, Tochner Z, Smith PD, Friauf WS, Thomas G, et al. Phase I study of debulking surgery and photodynamic therapy for disseminated intraperitoneal tumors.
Bauer TW, Hahn SM, Spitz FR, Kachur A, Glatstein E, Fraker DL. Preliminary report of photodynamic therapy for intraperitoneal sarcomatosis.
Canter RJ, Mick R, Kesmodel SB, Raz DJ, Spitz FR, Metz JM, et al. Intraperitoneal photodynamic therapy causes a capillary-leak syndrome.
Wilson JJ, Jones H, Burock M, Smith D, Fraker DL, Metz J, et al. Patterns of recurrence in patients treated with photodynamic therapy for intraperitoneal carcinomatosis and sarcomatosis.
Hendren SK, Hahn SM, Spitz FR, Bauer TW, Rubin SC, Zhu T, et al. Phase II trial of debulking surgery and photodynamic therapy for disseminated intraperitoneal tumors.
Busch TM, Hahn SM, Wileyto EP, Koch CJ, Fraker DL, Zhang P, Putt M, Gleason, K, Shin DB, Emanuele MJ, Jenkins K, Glatstein E, Evans SM. Hypoxia and photofrin uptake in the intraperitoneal carcinomatosis and sarcomatosis of photodynamic therapy patients.
Busch TM, Wileyto EP, Emanuele MJ, Del Piero F, Marconato L, Glatstein E, et al. Photodynamic therapy creates fluence rate-dependent gradients in the intratumoral spatial distribution of oxygen.
Aveline B, Hasan T, Redmond RW. Photophysical and photosensitizing properties of benzoporphyrin derivative monoacid ring A (BPD-MA).
Molpus KL, Kato D, Hamblin MR, Lilge L, Bamberg M, Hasan T. Intraperitoneal photodynamic therapy of human epithelial ovarian carcinomatosis in a xenograft murine model.
Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies.
Gullick WJ. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers.
Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model.
Divgi CR, Welt S, Kris M, Real FX, Yeh SD, Gralla R, et al. Phase I and imaging trial of indium 111-labeled anti-epidermal growth factor receptor monoclonal antibody 225 in patients with squamous cell lung carcinoma [see comments].
Schwartz GK. Invasion and metastases in gastric cancer: in vitro and in vivo models with clinical correlations.
Kohler M, Janz I, Wintzer HO, Wagner E, Bauknecht T. The expression of EGF receptors, EGF-like factors and c-myc in ovarian and cervical carcinomas and their potential clinical significance.
Bauknecht T, Kiechle M, Bauer G, Siebers JW. Characterization of growth factors in human ovarian carcinomas.
Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer.
Tonini G, Vincenzi B, Santini D, Olzi D, Lambiase A, Bonini S. Ocular toxicity related to cetuximab monotherapy in an advanced colorectal cancer patient.
Bonner JA, Raisch KP, Trummell HQ, Robert F, Meredith RF, Spencer SA, et al. Enhanced apoptosis with combination C225/radiation treatment serves as the impetus for clinical investigation in head and neck cancers.
Duska L, Hamblin M, Miller J, Hasan T. Combination photoimmunotherapy and cisplatin: effects on human ovarian cancer ex vivo.
Granville DJ, Carthy CM, Jiang H, Shore GC, McManus BM, Hunt DW. Rapid cytochrome c release, activation of caspases 3, 6, 7 and 8 followed by Bap31 cleavage in HeLa cells treated with photodynamic therapy.
Ratkay LG, Chowdhary RK, Iamaroon A, Richter AM, Neyndorff HC, Keystone EC, et al. Amelioration of antigen-induced arthritis in rabbits by induction of apoptosis of inflammatory cells with local application of transdermal photodynamic therapy.
Molpus KL, Koelliker D, Atkins L, Kato DT, Buczek-Thomas J, Fuller AF Jr, Hasan T. Characterization of a xenograft model of human ovarian carcinoma which produces intraperitoneal carcinomatosis and metastases in mice.
Lilge L, Belcuig M, Singh G, Grenier J, Li Y, Chopp M. Light dosimetry for intraperitoneal photodynamic therapy in a murine xenograft model of human epithelial ovarian carcinoma.
Peterson CM, Reed R, Jolles CJ, Jones KP, Straight RC, Poulson AM. Photodynamic therapy of human ovarian epithelial carcinoma, OVCAR-3, heterotransplanted in the nude mouse.
Kulapaditharom B, Boonkitticharoen V. Photodynamic therapy in management of head and neck cancers and precancerous lesions.
Lustig R, Vogl T, Fromm D, Cuenca R, Alex HR, D'Cruz A, et al. A multicenter phase I safety study of intratumoral photoactivation of talaporfin sodium in patients with refractory solid tumors.
Manyak M, Ogan K. Photodynamic therapy for refractory superficial bladder cancer: long-term clinical outcomes of single treatment using intravesical diffusion medium.
Gschwind A, Fischer O, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy.
Bjorge J, Chan T, Antczak M, Kung H, Fujita D. Activated type I phosphatidylinositol kinase is associated with the epidermal growth factor (EGF) receptor following EGF stimulation.
Rafferty M, Fenton J, Jones A. An overview of the role and inter-relationship of epidermal growth factor receptor, cyclin D and retinoblastoma protein on the carcinogenesis of squamous cell carcinoma of the larynx.
Bill H, Knudsen B, Moores S, Muthuswamy S, Rao V, Brugge J, et al. Epidermal growth factor receptor-dependent regulation of integrin-mediated signaling and cell cycle entry in epithelial cells.
Granville DJ, Jiang H, An MT, Levy JG, McManus BM, Hunt DW. Overexpression of Bcl-X(L) prevents caspase-3-mediated activation of DNA fragmentation factor (DFF) produced by treatment with the photochemotherapeutic agent BPD-MA.
Moor A, Rizvi I, Savellano MD, Yu W, Del Carmen MG, Hasan T. Photoimmunotargeting of the epidermal growth factor receptor for the treatment of ovarian cancer. San Francisco (CA): 13th International Congress on Photobiology and 28th Annual Meeting ASP;
Tong Z, Singh G, Rainbow AJ. Sustained activation of the extracellular signal-regulated kinase pathway protects cells from photofrin-mediated photodynamic therapy.
Patterson M, Wilson B, Graff R. In vivo tests of the concept of photodynamic threshold dose in normal rat liver photosensitized by aluminum chlorosulphonated phthalocyanine.
Griffin GM, Zhu T, Solonenko M, Del Piero F, Kapakin A, Busch TM, et al. Preclinical evaluation of motexafin lutetium-mediated intraperitoneal photodynamic therapy in a canine model.
Busch TM, Hahn SM, Evans SM, Koch CJ. Depletion of tumor oxygenation during photodynamic therapy: detection by the hypoxia marker EF3 [2-(2-nitroimidazol-1[H]-yl)-N-(3,3,3-trifluoropropyl)acetamide].
Dennis MV, Judy MM, Matthews JL, Sogandares-Bernal F. Protection of NIH 3T3 cells from infection by trypomastigotes and sphaeromastigotes of Trypanosoma cruzi, Telahuen strain, by porphyrins in the presence and absence of light (630 and 690 nm).
Runnels JM, Chen N, Ortel B, Kato D, Hasan T. BPD-MA-mediated photosensitization in vitro and in vivo: cellular adhesion and beta1 integrin expression in ovarian cancer cells.
Morgan J, Potter WR, Oseroff AR. Comparison of photodynamic targets in a carcinoma cell line and its mitochondrial DNA-deficient derivative.
Bown S, Rogowska A. New photosensitizers for photodynamic therapy in gastroenterology.
Jenkins M, Buonaccorsi G, Raphael M, Nyamekye I, McEwan J, Bown S, et al. Clinical study of adjuvant photodynamic therapy to reduce restenosis following femoral angioplasty.
Sullivan L, Hasan T, Wright M, Mankin H, Towle C. Photodynamic treatment has chondroprotective effects on articular cartilage.


{kind=link}
{kind=link}
{kind=link}
{kind=link}