





Abstract
Multidrug resistance protein-1 (MRP1) belongs to subfamily C of the ATP-binding cassette transporters, and exports leukotriene C4 and organic anions including the fluorescent calcium indicator indo-1. The observation that leukocytes from patients with an autoimmune disease exported indo-1 at a higher rate than controls prompted the hypothesis that MRP1 contributes to the function of activated cells. To test this, we defined the expression of MRP1 on resting and activated human T cells, and determined whether T cell activation is dependent upon MRP1 function. MRP1 is expressed on resting memory but not on naive CD4 and CD8 T cells. After activation through the TCR, cord blood CD4 T cells express high levels of MRP1. Blockade of MRP1 with the specific inhibitor MK-571 abrogated superantigen-induced expression of IFN-γ, tumor necrosis factor-α, IL-10, IL-2, IL-4 and CD69 by T cells without affecting their viability, and was reversible upon removal of MK-571 from the culture media. Electrophoretic mobility shift assays demonstrate that MRP1 blockade with MK-571 induces activation of the transcriptional repressor peroxisome proliferator-activated receptor-γ in CD4 T cells, thus providing insight into the potential mechanism by which their responses are abrogated.
Introduction
Multidrug resistance protein-1 (MRP1) is one of a family of ATP-binding cassette transporters that protects transformed cells from chemotherapeutic agents and healthy cells from xenobiotics. MRP1 also serves physiological functions, such as transport of glutathione (GSH), GSH conjugates, oxidized GSH and the cysteinyl-leukotriene (cys-LT) leukotriene C4 (LTC4) (1–3). GSH maintains intracellular oxidative homeostasis within cells; severe GSH depletion decreases T cell proliferation (4, 5), IL-12 production by antigen-presenting cells and type 1 responses (6–8). LTC4 is primarily a product of monocyte/macrophages, eosinophils and mast cells (9), and after export and subsequent conversion to LTD4, is a ligand for cys-LTRs CysLTR1 and CysLTR2. Cys-LT causes bronchoconstriction and eosinophil migration, and its role in asthma is supported by proven benefit of pharmacological antagonists of the cys-LT synthesis pathway or CysLTR1. Thus, redox balance and leukotrienes provide two potential nexus points between MRP1 and regulation of inflammation (10, 11).
A role for MRP1 in immune function was reported in murine models in which the gene was deleted. These mice have decreased local inflammatory responses to irritant stimuli (12), and early disease is after challenge with Mycobacterium tuberculosis is slightly more severe (13). Both these findings may be attributed to the LTC4-dependent defect in dendritic cell migration to CCR7 ligands observed in mrp1−/− mice by Robbiani et al. (14). However, mrp1−/− mice are more susceptible to experimentally induced colitis (15). Furthermore, murine T cells with memory or activation markers more actively export the fluorescent probe fluo-3, and inflammatory cytokines such as IL-2 increase fluorescent export activity (16). Blockade with the MRP1 inhibitor MK-571 decreases IFN-γ and IL-4 secretion by IL-2-stimulated Th1 and Th2 cell lines, respectively (17). Thus, MRP1 affects T cell function and potentially impacts the murine response beyond straightforward impairment of CCR7-mediated chemotaxis.
Reports of a role for MRP1 in human immune responses are few. Human MRP1 is expressed on CD4 and CD8 T cells, B cells, NK cells (18), monocytes and monocyte-derived dendritic cells (19). It has been reported that MRP1 blockade with the small-molecule inhibitor MK-571 enhances LPS and IL-1-induced secretion of IL-6 by peripheral blood monocytes (PBMs) (20).
Our interest in MRP1 was prompted by an observation that leukocytes from subjects with the auto-inflammatory disease Wegener's granulomatosis retained less of the anionic fluorescent calcium indicator indo-1 when compared with normal controls (R. L. Rabin, unpublished observation). We considered the possibility that higher export activity may be due to higher expression and function of MRP1 in activated cells, and in turn, that MRP1 might regulate immune function. Since T cell-derived inflammatory cytokines are implicated in the pathogenesis of Wegener's granulomatosis (21), we specifically asked whether MRP1 might be implicated in inflammatory T cell responses.
In this report, we demonstrate that MRP1 is differentially expressed in naive versus memory T cells, is up-regulated with activation, that MRP1 blockade with MK-571 reversibly abrogates cytokine secretion by superantigen-stimulated human T cells and that the anti-inflammatory effect of MRP1 blockade is associated with activation of the transcriptional repressor peroxisome proliferator-activated receptor-γ (PPARγ). These data suggest a novel pathway of modulation of T cell responses and a potential source of novel therapeutic targets for treatment of auto-inflammatory diseases in which T cell-derived cytokines have an integral role.
Methods
Cells and reagents
Buffy coats, leukopaks and elutriated monocytes and lymphocytes were obtained from the National Institutes of Health (NIH) Department of Transfusion Medicine, Bethesda, MD, USA. Cord blood was obtained from Shady Grove Adventist Hospital, Gaithersburg, MD, USA. The Institutional Review Boards of the Food and Drug Administration, NIH and Shady Grove Adventist Hospital approved these studies.
RPMI, PBS and HBSS were obtained from Biofluids (Gaithersburg, MD, USA), Ficoll-Hypaque from Sigma (St Louis, MO, USA) and FBS from Hyclone (Logan, UT, USA). The hybridomas OKT3 (CD3), OKT4 (CD4), OKT8 (CD8), UCHL1 (CD45RO) and DREG-56 (CD62L) were obtained from American Type Culture Collection (Manassas, VA, USA); mAbs were purified from ascites fluid or culture supernatants with a Protein G column (Amersham Biosciences, Piscataway, NJ, USA), and when used for flow cytometry, conjugated to fluorochromes according to established protocols (22). Anti-CD28 (mAb 9.3) was a kind gift of Carl June, University of Pennsylvania. Human rIL-4, rIL-12 and antibodies against IFN-γ (4S.B3), IL-4 (MP4-25D2), IL-12 (C11.5 and C8.6), tumor necrosis factor (TNF)-α (Mab1 and Mab11), IL-6 (MQ2-13A5 and MQ2-39C3), CD45RA (L48) and CD69 (L78) for neutralization, staining and/or ELISA were obtained from BD Biosciences Pharmingen (San Diego, CA, USA). Human rIL-2 was obtained from Hoffman-La Roche (Nutley, NJ, USA) and human rIL-7 from Peprotech (Rocky Hill, NJ, USA). Toxic shock syndrome toxin-1 (TSST-1) was purchased from Toxin Technology, Inc. (Sarasota, FL, USA); Staphylococcus enterotoxins A and B (SEA, SEB), Escherichia coli O111:B4 LPS, phorbol myristate acetate (PMA) and ionomycin were purchased from Sigma and Pansorbin® Staphylococcus aureus Cowan (SAC) from Calbiochem (San Diego, CA, USA). Indo-1, phalloidin, FITC–phalloidin, ethidium bromide and acridine orange were purchased from Molecular Probes (Eugene, OR, USA). MK-571 was obtained from Biomol (Plymouth Meeting, PA, USA). Nucleic acid primers and probes were synthesized in the CBER Facility for Biological Reagents, or by Keystone Laboratories, Menlo Park, CA, USA.
Cell responses to stimulation
PBMCs were isolated by Ficoll-Hypaque density centrifugation and stimulated at a concentration of 5 × 106 cells ml−1 with TSST-1, SEA or SEB (10, 100 or 1000 ng ml−1, respectively, determined by titration). Unless otherwise indicated, MK-571 was added to the cells 30 min prior to superantigen, and the cells were incubated overnight. Supernatants were harvested, and after cellular debris was removed by centrifugation, were stored at −70°C. For intracellular cytokine staining, monensin (2 μM, Calbiochem) was added to the cell suspension.
Elutriated PBMs were suspended in RPMI, 10% FBS and 10% human serum (Sigma) and stimulated with E. coli O111:B4 LPS, 10 ng ml−1, or with SAC diluted 1:10 000. After an overnight incubation, supernatants were harvested for measurement of IL-6 and TNF-α by ELISA (BD Biosciences).
Cytokine secretion into supernatants by PBMC was analyzed with a fluorescent bead assay (CBA, BD Pharmingen) after staining according to the manufacturer's instructions. Cytokine secretion into supernatants by monocytes was analyzed by ELISA with Immulon #4 ELISA plates. Cytokine was revealed with HRP-avidin (BD Biosciences) and 3,3′,5,5′-tetramethylbenzidine (KPL, Gaithersburg, MD, USA) and after acidification, measured on a 96-well spectrophotometer at 450 nm (Dynex Technologies).
Flow cytometry and cell sorting
Freshly isolated PBMCs were sorted into B cells (CD19+), NK cells (CD16 or CD56+), monocytes (CD14+) and memory and naive CD4 and CD8 T cells based on expression of CD45 isoforms and CD62L (23, 24). Cord blood mononuclear cells that had been stimulated with mAb against CD3 and CD28 were stained with PECy5-conjugated anti-CD4 and FITC-conjugated mAb against CD8, CD16, CD19 and CD14 and sorted on a FACSDiva (BD Biosciences Immunocytometry Systems, San Jose, CA, USA), or isolated by positive selection with magnetic beads (Miltenyi Biotec, Auburn, CA, USA). Cells were stained for surface phenotype in HBSS with Ca++ and Mg++, 10 mM HEPES and 1% FBS for 15 min on ice (25), and for intracellular cytokine as previously described (26).
The cells and beads were analyzed on an LSR or LSR II flow cytometer (BD Biosciences) equipped with lasers at 325, 488 and 633 nm, and in the LSR II, 407 nm. The data files were analyzed using Flowjo software (Treestar, Ashland, OR, USA).
Semi-quantitative reverse transcription–PCR
RNA was isolated (RNeasy, Qiagen, Valencia, CA, USA) and subjected to reverse transcription (RT) using random hexamer or oligo-dT primers (Superscript, Invitrogen, Carlsbad, CA, USA). Sequences for the primers and probes are shown in Table 1. Thirty-five-cycle PCR was performed with Taq DNA polymerase (Invitrogen) according to the manufacturer's instructions. PCR products obtained from expression of genes of MRP family members and actin were run on a 1.5% agarose gel. PCR for Taqman analysis was done with Amplitaq Gold (ABI, Foster City, CA, USA) using the ABI Prism Sequence Detection System. Relative expression was determined with Sequence Detection System Software (ABI, version 1.9.1) and shown as a ratio of amplified cDNA to reverse transcribed 18S RNA or Glyseraldehyde-3-phosphate dehydrogenase (GAPDH) using commercially obtained primers and probes (Biosource, Camarillo, CA, USA). Cytokine and transcription factor primers and probes were designed with Primer Express Software (ABI) or taken from the literature (27, 28).
Oligonucleotide sequences used for experiments in this report
Thirty-five-cycle RT–PCR (Fig. 1) | |||
| Gene | Forward primer | Reverse primer | |
| MRP1 | CATGAAGGCCATCGGACTCT | CAGGTCCACGTGCAGACA | |
| MRP3 | GGCACTGCTGATTGAAGACA | AATGGCTGCTTTCTCCTCCT | |
| MRP5 | CATGCTGATCCTGAAAGCCAT | GAATCAGGACCCTGGAGACA | |
| Actin | ATCTGGCACCACACCTTCTA CAATGAGCTGCG | CGTCATACTCCTGCTTGCTGA TCCCACATCTGC | |
| Quantitative RT–PCR (Taqman) | |||
| Gene | Forward primer | Probe | Reverse primer |
| IFN-γ | CTAATTATTCGGTAACTGACTTGA | TCCAACGCAAAGCAATACATGAAC | ACAGTTCAGCCATCACTTGGA |
| IL-4 | AACAGCCTCACAGAGCAGAAGAC | TGCTGCCTCCAAGAACACAACTGA | GCCCTGCAGAAGGTTTCCTT |
| IL-13 | TGAGGAGCTGGTCAACATCA | AGGCTCCGCTCTGCAATGGC | CAGGTTGATGCTCCATACCAT |
| GATA-3 | AACCGGCCCCTCATTAAGC | TCCTGTGCGAACTGTCAGACCACCAC | GGCATTCCTCCTCCAGAGTGT |
| T-bet | AGGCTGAGTTTCGAGCAGTCA | CTGCATTCTTGCCCTCTGCCCCT | TGGCCTCGGTAGTAGGACATG |
| MRP1 | TCTACCTCCTGTGGCTGAATCTG | ACCTGATACGTCTTGGTCTTCATCGCCAT | CCGATTGTCTTTGCTCTTCATGT |
| Promoter site | Electrophoretic mobility shift assay | ||
| Consensus PPRE | TGAAACTAGGGTAAA GTTCA | ||
| Mutant PPRE | TGAAACTAGCCTAAA GAACA |
Thirty-five-cycle RT–PCR (Fig. 1) | |||
| Gene | Forward primer | Reverse primer | |
| MRP1 | CATGAAGGCCATCGGACTCT | CAGGTCCACGTGCAGACA | |
| MRP3 | GGCACTGCTGATTGAAGACA | AATGGCTGCTTTCTCCTCCT | |
| MRP5 | CATGCTGATCCTGAAAGCCAT | GAATCAGGACCCTGGAGACA | |
| Actin | ATCTGGCACCACACCTTCTA CAATGAGCTGCG | CGTCATACTCCTGCTTGCTGA TCCCACATCTGC | |
| Quantitative RT–PCR (Taqman) | |||
| Gene | Forward primer | Probe | Reverse primer |
| IFN-γ | CTAATTATTCGGTAACTGACTTGA | TCCAACGCAAAGCAATACATGAAC | ACAGTTCAGCCATCACTTGGA |
| IL-4 | AACAGCCTCACAGAGCAGAAGAC | TGCTGCCTCCAAGAACACAACTGA | GCCCTGCAGAAGGTTTCCTT |
| IL-13 | TGAGGAGCTGGTCAACATCA | AGGCTCCGCTCTGCAATGGC | CAGGTTGATGCTCCATACCAT |
| GATA-3 | AACCGGCCCCTCATTAAGC | TCCTGTGCGAACTGTCAGACCACCAC | GGCATTCCTCCTCCAGAGTGT |
| T-bet | AGGCTGAGTTTCGAGCAGTCA | CTGCATTCTTGCCCTCTGCCCCT | TGGCCTCGGTAGTAGGACATG |
| MRP1 | TCTACCTCCTGTGGCTGAATCTG | ACCTGATACGTCTTGGTCTTCATCGCCAT | CCGATTGTCTTTGCTCTTCATGT |
| Promoter site | Electrophoretic mobility shift assay | ||
| Consensus PPRE | TGAAACTAGGGTAAA GTTCA | ||
| Mutant PPRE | TGAAACTAGCCTAAA GAACA |
Oligonucleotide sequences used for experiments in this report
Thirty-five-cycle RT–PCR (Fig. 1) | |||
| Gene | Forward primer | Reverse primer | |
| MRP1 | CATGAAGGCCATCGGACTCT | CAGGTCCACGTGCAGACA | |
| MRP3 | GGCACTGCTGATTGAAGACA | AATGGCTGCTTTCTCCTCCT | |
| MRP5 | CATGCTGATCCTGAAAGCCAT | GAATCAGGACCCTGGAGACA | |
| Actin | ATCTGGCACCACACCTTCTA CAATGAGCTGCG | CGTCATACTCCTGCTTGCTGA TCCCACATCTGC | |
| Quantitative RT–PCR (Taqman) | |||
| Gene | Forward primer | Probe | Reverse primer |
| IFN-γ | CTAATTATTCGGTAACTGACTTGA | TCCAACGCAAAGCAATACATGAAC | ACAGTTCAGCCATCACTTGGA |
| IL-4 | AACAGCCTCACAGAGCAGAAGAC | TGCTGCCTCCAAGAACACAACTGA | GCCCTGCAGAAGGTTTCCTT |
| IL-13 | TGAGGAGCTGGTCAACATCA | AGGCTCCGCTCTGCAATGGC | CAGGTTGATGCTCCATACCAT |
| GATA-3 | AACCGGCCCCTCATTAAGC | TCCTGTGCGAACTGTCAGACCACCAC | GGCATTCCTCCTCCAGAGTGT |
| T-bet | AGGCTGAGTTTCGAGCAGTCA | CTGCATTCTTGCCCTCTGCCCCT | TGGCCTCGGTAGTAGGACATG |
| MRP1 | TCTACCTCCTGTGGCTGAATCTG | ACCTGATACGTCTTGGTCTTCATCGCCAT | CCGATTGTCTTTGCTCTTCATGT |
| Promoter site | Electrophoretic mobility shift assay | ||
| Consensus PPRE | TGAAACTAGGGTAAA GTTCA | ||
| Mutant PPRE | TGAAACTAGCCTAAA GAACA |
Thirty-five-cycle RT–PCR (Fig. 1) | |||
| Gene | Forward primer | Reverse primer | |
| MRP1 | CATGAAGGCCATCGGACTCT | CAGGTCCACGTGCAGACA | |
| MRP3 | GGCACTGCTGATTGAAGACA | AATGGCTGCTTTCTCCTCCT | |
| MRP5 | CATGCTGATCCTGAAAGCCAT | GAATCAGGACCCTGGAGACA | |
| Actin | ATCTGGCACCACACCTTCTA CAATGAGCTGCG | CGTCATACTCCTGCTTGCTGA TCCCACATCTGC | |
| Quantitative RT–PCR (Taqman) | |||
| Gene | Forward primer | Probe | Reverse primer |
| IFN-γ | CTAATTATTCGGTAACTGACTTGA | TCCAACGCAAAGCAATACATGAAC | ACAGTTCAGCCATCACTTGGA |
| IL-4 | AACAGCCTCACAGAGCAGAAGAC | TGCTGCCTCCAAGAACACAACTGA | GCCCTGCAGAAGGTTTCCTT |
| IL-13 | TGAGGAGCTGGTCAACATCA | AGGCTCCGCTCTGCAATGGC | CAGGTTGATGCTCCATACCAT |
| GATA-3 | AACCGGCCCCTCATTAAGC | TCCTGTGCGAACTGTCAGACCACCAC | GGCATTCCTCCTCCAGAGTGT |
| T-bet | AGGCTGAGTTTCGAGCAGTCA | CTGCATTCTTGCCCTCTGCCCCT | TGGCCTCGGTAGTAGGACATG |
| MRP1 | TCTACCTCCTGTGGCTGAATCTG | ACCTGATACGTCTTGGTCTTCATCGCCAT | CCGATTGTCTTTGCTCTTCATGT |
| Promoter site | Electrophoretic mobility shift assay | ||
| Consensus PPRE | TGAAACTAGGGTAAA GTTCA | ||
| Mutant PPRE | TGAAACTAGCCTAAA GAACA |
Nuclear extract isolation and electrophoretic mobility shift assay
Nuclear proteins were extracted with Nuclear Extract Kit from Active Motif (Carlsbad, CA, USA) and stored at −70°C. Protein concentration was determined with the BCA (bicinchoninic acid) Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA). Double-stranded oligonucleotide containing the consensus peroxisome proliferator response elements (PPRE) site was end-labeled by the T4 polynucleotide kinase (New England Biolabs, Beverly, MA, USA) in the presence of [γ-32P]ATP. Samples of 10 μg of each nuclear extract were incubated with 0.2 ng of double-stranded radiolabeled oligonucleotides in 20 μl containing 10 mM Tris–HCl, pH 7.9, 50 mM NaCl, 5% glycerol, 0.5 mM dithiothreitol plus 0.5 μg of poly[d(I-C)]. Incubation was performed for 20 min at room temperature. The samples were analyzed by electrophoresis in 6% polyacrylamide gels containing 0.5× TBE for 70 min at 100 V, transferred to Whatman 3M paper, dried under vacuum at 80°C and exposed to a phosphor screen. The image was visualized by a Typhoon image system (GE Healthcare, Piscataway, NJ, USA) and the densitometry was measured with Imagequant software (GE Healthcare). To determine the specificity of the DNA–protein complex, competition assay was performed using excess of the corresponding unlabeled, double-stranded consensus or mutant oligonucleotide.
Statistics
Differences between experimental groups were analyzed using t test for paired samples or repeated measures analysis of variance. Differences were considered statistically significant if P < 0.05. Microsoft Excel (Microsoft, Redmond, WA, USA) or JMP (SAS, Cary, NC, USA) were used for statistical analyses.
Linear regression was carried out using Microsoft Excel. In addition, the goodness-of-fit parameter, Q, as defined by Press et al. (29), was obtained using functions available in Excel. In brief, if Q < 0.05, the points are not considered to be well fit to a line.
Results
Expression and function of MRP1 in resting and activated T cells
We first sought to define expression of MRP1 in primary human PBMCs and compare it with other members of the MRP family. PBMCs were isolated from buffy coats, stained for flow cytometry and sorted into naive and memory (CD45RO−, CD62L+ and CD45RO+, respectively) subsets of CD4 and CD8 T cells (Fig. 1A, lanes 1–4), B cells (CD19+, CD14−, CD16/CD56−, lane 5), NK cells (CD16/CD56+, CD19−, CD14−, lane 6) and monocytes (CD14+, CD16/CD56−, CD19−, lane 7). Semi-quantitative RT–PCR analysis for gene expression revealed that in addition to B cells, MRP1 RNA is highly expressed by memory CD4 and CD8 T cells, and poorly expressed by their naive counterparts. By comparison, two other MRP family members, MRP3 and MRP5, are expressed only by monocytes, and at similar levels by all PBMC subsets, respectively.
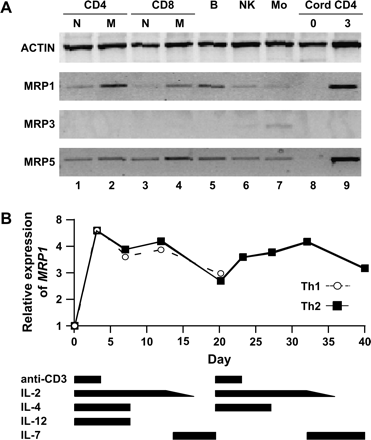
MRP1 is expressed in memory T cells and induced by activation. (A) Thirty-five-cycle RT–PCR of mRNA from FACS sorted subsets of human PBMC, and in umbilical cord CD4 T cells upon isolation and after 3 days of stimulation (lanes 8 and 9, respectively) with anti-CD3 and anti-CD28. Lanes 1–4: naive and memory CD4 and CD8 T cells; lanes 5–7, B cells, NK cells and monocytes. (B) MRP1 expression increases and decreases with activation and rest, respectively. Cord CD4 T cells were stimulated with anti-CD3 and anti-CD28 in the presence of IL-2 and polarizing cytokines. The bars at the bottom indicate the duration of cytokines and stimulation. PBMCs were exposed to IL-4 or IL-12 for 7 days, and IL-2 was tapered in the presence of IL-7. Figures are representative of two experiments each.
Since MRP1 was expressed at low levels in the naive T cells (lanes 1 and 3), and since there may be memory cell contamination within CD45RO−, CD62L+ T cells (30), we purified CD4 T cells from umbilical cord blood and stimulated them with anti-CD3 + anti-CD28. MRP1 was not detectably expressed on day 0 (lane 8), but was highly expressed after 3 days of activation (lane 9). Expression was seen as early as 2 h after stimulation and reached high levels by 24 h. Stimulation in the presence of monensin (a transport inhibitor of proteins that blocks their exit from the Golgi apparatus) did not diminish MRP1 expression at 2, 4 or 8 h (data not shown). In addition, cord blood CD4 T cells expressed low levels of MRP1 after 3 days in media in the absence of stimulation, which was not enhanced by the addition of IL-2 (data not shown). Taken together, these data suggest that the expression of MRP1 mRNA was a consequence of signaling through the TCR, and not due to cytokines that are associated with T cell stimulation such as IL-2.
We then tested whether activation-induced expression of MRP1 is biased in the context of polarization, by stimulating cord CD4 T cells in type 1 (IL-12 + anti-IL-4) versus type 2 (IL-4 + anti-IL-12) polarizing conditions, followed by rest with IL-7 (31). Quantitative RT–PCR (Taqman, ABI) shows that while MRP1 expression increased with activation and fell during rest, Th1 and Th2 cell lines expressed MRP1 equally (Fig. 1B).
MRP1 blockade inhibits activation-induced cytokine secretion
When stimulated with superantigen such as TSST-1, PBMC cultures acquire an ‘activated’ morphology that consists of large aggregates of cells, and express CD69 and cytokines such as IFN-γ. To determine whether these responses are dependent upon MRP1, we inhibited it with MK-571 and observed that morphologic changes and expression of CD69 and IFN-γ were suppressed in CD4 T cells (Fig. 2A and B) as well as CD8 T cells (data not shown).
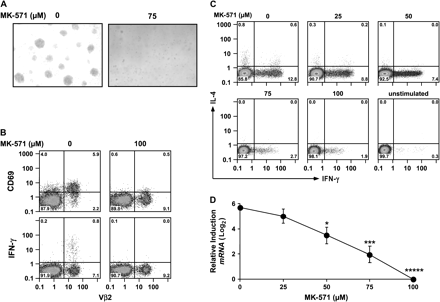
MK-571 inhibits MRP1 function and blocks T cell activation by superantigen. (A) MK-571 blocks superantigen-induced morphologic changes. PBMCs were stimulated overnight with TSST-1 (10 ng ml−1) in the absence or presence of MK-571 (75 μM). (B–D) MK-571 blocks superantigen-induced CD69, IFN-γ and IL-4 expression by CD4 T cells. (B) PBMCs were stimulated with TSST-1 prior to harvesting, fixing and staining for CD4, Vβ2 (the primary Vβ group to which TSST-1 binds), CD69 and IFN-γ. Distribution of CD4+ lymphocytes is shown. (C) IL-4 and IFN-γ are suppressed in a dose-dependent manner. PBMCs were exposed to increasing concentrations of MK-571 and stimulated with the combination of superantigens—SEA, SEB and TSST-1—for 4–6 h after which they were harvested, fixed and stained. Distribution of CD4 T cells is shown. (D) IFN-γ gene transcription is inhibited by MK-571. RT–PCR (Taqman) analysis of IFN-γ gene expression. Cumulative data are from six experiments; error bars display SD. *P ≤ 0.05, ***P ≤ 0.005, *****P < 0.0001 compared with no MK-571.
To evaluate whether IL-4 is also suppressed by MK-571, we increased the number of IL-4+ cells by combining the superantigens SEA, SEB and TSST-1. As seen in Fig. 2(C), MK-571 suppresses both IFN-γ and IL-4 in a dose-dependent manner. Suppression is at the level of transcription for both IFN-γ (Fig. 2D) and IL-4 (data not shown).
Analysis of PBMC culture supernatants shows that similar to IFN-γ, TNF-α and IL-10 are also suppressed (Fig. 3). While IL-2 secretion was also suppressed by MRP1 blockade, the pattern was somewhat different: IL-2 inhibition exhibited a linear versus semi-logarithmic relationship for IFN-γ, IL-10 and TNF-α (see insets), and a dramatic drop in IL-2 secretion at the highest dose of MK-571 (open circle). Calculation of the goodness of fit for the linear fits (29) supports the exclusion of this last point from the present analysis and suggests that IL-2 responds somewhat differently to MRP1 blockade than the other cytokines shown here.
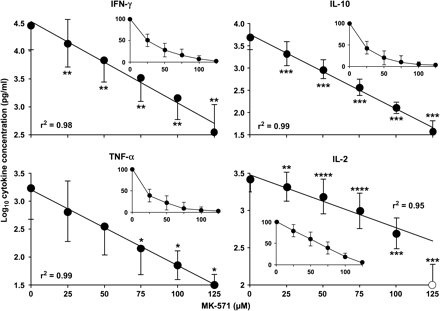
MK-571 blocks secretion of multiple cytokines in a dose-dependent manner. PBMCs were stimulated overnight with TSST-1 in the presence of MK-571. Supernatants were tested with CBA fluorescent bead ELISA kit (BD Biosciences). Data from eight experiments were log transformed and plotted as mean ± SD. Note that the y-axis varies between graphs. Inset: means and SD of responses normalized to 100%. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.005, ****P < 0.001, *****P < 0.0001 compared with no MK-571.
We then tested whether cytokine suppression by MRP1 blockade is limited to T cells by stimulating freshly isolated purified human PBMs with either LPS from E. coli O111:B4 or SAC. Figure 4 shows that MRP1 blockade by treating PBMs with MK-571 decreased secretion of TNF-α induced by LPS in a dose-dependent manner (Fig. 4A). However, TNF-α secretion induced by SAC and IL-6 secretion induced by LPS or SAC were unaffected by MK-571. In addition to demonstrating that MRP1 blockade may inhibit cytokine responses by PBM, these data demonstrate specificity in that the consequences of MRP1 blockade are limited to specific inflammatory stimuli, and affect the secretion of specific cytokines.
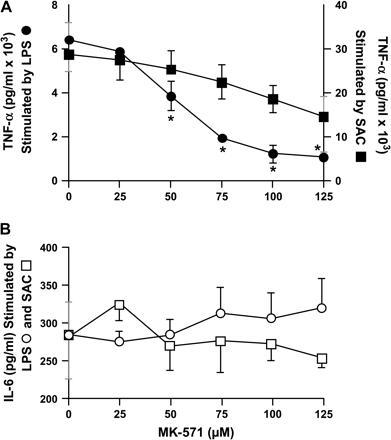
MRP1 blockade suppresses TNF-α but not IL-6 in response to LPS and SAC. Elutriated human PBMs were cultured overnight with LPS (10 ng ml−1) or SAC (1:104) in RPMI with 10% FCS and 10% human serum. Cumulative data from three experiments are shown. *P < 0.05.
MRP1 blockade does not diminish cell viability, and suppression of IFN-γ secretion is reversible and rapid in onset
To insure that inhibition of cytokine secretion by MRP1 blockade is not due to loss of cell viability, we cultured PBMCs, either at rest or with TSST-1, with increasing doses of MK-571 overnight and assessed viability with the fluorescent dyes acridine orange and ethidium bromide. Figure 5(A) shows that MK-571 at doses of ≤75 μM have no effect on viability of stimulated T cells. Resting cells were also unaffected (data not shown).
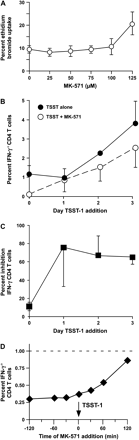
MK-571 is not toxic to superantigen-stimulated PBMCs, and blockade of T cell responses is reversible with rapid onset. (A) Effect of MK-571 on viability. PBMCs were stimulated overnight with SEB and TSST-1 and increasing doses of MK-571. Viability was assessed with acridine orange/ethidium bromide staining, in which all nucleated cells stain with acridine orange, and ethidium bromide permeates only dead cells. (B and C) Suppression by MK-571 is reversible. MK-571 (75 μM) was added to PBMCs on day 0. The PBMCs were then washed and returned to culture in aliquots to be stimulated overnight with TSST-1 immediately (day 0), or 1, 2 or 3 days (day 1–3) after washing out MK-571. For comparison, cells that were not exposed to MK-571 were maintained in culture and stimulated with TSST-1. After overnight stimulation, the cells were harvested and stained for IFN-γ and CD4. Data from four experiments are shown as percentage of CD4 T cells expressing IFN-γ (B) and as percentage inhibition by MK-571 (C). (D) Rapid onset of cytokine blockade by MK-571. MK-571 was added at various time points relative to the addition of TSST-1. After an overnight stimulation, the cells were harvested and stained for IFN-γ and CD4. The dashed line indicates the percentage of cells expressing IFN-γ after stimulation in the absence of MK-571. Figure is representative of two experiments.
We then tested reversibility of MRP1 blockade by incubating PBMCs overnight with MK-571 (75 μM), after which they were washed and re-suspended in fresh media. After the overnight incubation with MK-571, aliquots of PBMC were stimulated with TSST-1 immediately, 1 day and 2 days afterward. The PBMCs were harvested 1 day after stimulation and stained for IFN-γ and CD4. Figure 5(B and C) shows that the inhibitory effect of overnight culture with MK-571 was substantially reversed after 1 or 2 days of culture in media free of MK-571.
To gain insight into the mechanism by which MRP1 blockade may inhibit T cell function, we added MK-571 (75 μM) at different time points relative to stimulation of PBMC with TSST-1. Figure 5(D) shows that MRP1 blockade is equally effective whether the MK-571 is added to cells 120 min prior to addition of TSST-1 or simultaneously with it. This suggests that MRP1 blockade does not depend on transcription of inhibitory factors, but instead, either activates repressors or blocks signaling pathways that lead to activation. Since we found no differences in CD3 cross-linking induced calcium flux between untreated and MK-571-exposed cells (data not shown), we focused on repressors.
PPARγ is a transcription factor that may repress gene expression either indirectly by blocking activation of inflammatory transcription factors such as nuclear factor-κB (NF-κB) or directly by binding to DNA sequences referred to as PPRE. PPARγ agonists include ciglitazone and similar pharmacological agents, and eiconasoids such as 15-deoxy-prostaglandin J2 (15-PGJ2) (32). The role of MRP1 as the exporter of LTC4 suggested the possibility that MRP1 blockade resulted in retention of an endogenous agonist of PPARγ. We thus compared binding to a consensus PPRE DNA sequence of nuclear extracts from CD4 T cells after culture alone or with MK-571 (50 μM), versus 20 μM of ciglitazone, trosiglitazone or rosiglitazone. Compared with untreated CD4 T cells (Fig. 6, lane 2), the known PPARγ agonists (lanes 3–6) and MK-571 (lanes 7–11) increased binding of PPARγ to the consensus PPRE sequence. While there was donor variability in binding of PPARγ from untreated resting CD4 T cells to the consensus PPRE, treatment with ciglitazone, its analogs and MK-571 always showed a significant increase in retarded migration of the labeled PPRE oligonucleotide.
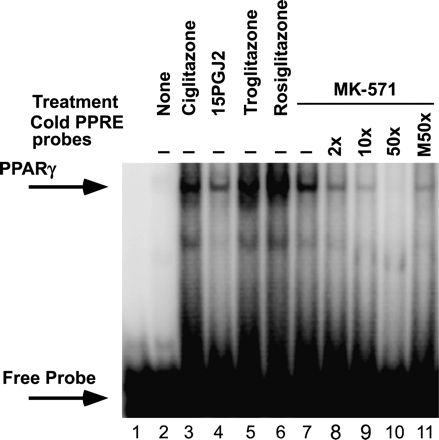
Treatment of PBMCs with MK-571 increases binding of cellular PPARγ to the consensus PPRE element. Nuclear extracts from purified CD4 T cells were exposed to 32P-labeled oligonucleotide coding the consensus PPRE sequence and analyzed by electrophoretic mobility shift assay. CD4 T cells were either not treated (lane 2) or treated for 2 h with synthetic PPARγ ligands (20 μM each, lanes 3, 5 and 6), a natural ligand 15-PGJ2 (5 μM, lane 4) or MK-571 (50 μM) in the absence or presence of unlabeled PPRE oligonucleotide (lanes 7–10), or an unlabeled mutant PPRE oligonucleotide (lane 11). Figure is representative of three experiments.
In contrast to the effects on unstimulated CD4 T cells of PPARγ ligands and MK-571, adding them 1 h after stimulation with TSST showed no increase, and possibly a decrease in intensity of the PPRE activation (data not shown). This time dependence of PPARγ activation by MK-571 is consistent with Fig. 5(D), showing similar time dependence for inhibition of cytokine responses by MK-571.
Discussion
In this study, we asked whether MRP1 is selectively expressed by T cell subsets, and whether it regulates human T cell function. Expression of MRP1 by human leukocytes has been previously reported (19). Using the fluorescent extrusion assay, Laupeze et al. (18) found similar levels of MRP1 activity among all subsets of primary human leukocytes. Oselin et al. (33) reported a hierarchy of MRP1 gene expression by human PBMC as CD4 T cells > CD8 T cells > B cells = NK cells. We extend their observations by demonstrating that among T cells, MRP1 expression is restricted to the memory subsets, is up-regulated during T cell activation and, contrary to reports using murine cells (16), is not preferentially expressed by human Th1 versus Th2 cell lines (Fig. 1). Since fluorescent extrusion assays may not accurately reflect cellular levels of MRP1 (34), and staining T cells with anti-MRP mAb yields unimodal histograms (HH and RLR, unpublished observations) inconsistent with expression that is limited to the memory subset, analysis of gene expression may be the most accurate tool currently available for determining levels of MRP1 among human leukocyte subsets.
To our knowledge, this is the first study that comprehensively explores the consequences of MRP1 blockade on human T cell function. We report that T cell activation and cytokine secretion are inhibited by MK-571, and IFN-γ and IL-4, and most likely TNF-α, IL-2 and IL-10, are suppressed at the level of gene transcription. Since intracellular staining for cytokines requires blockade of secretion with monensin or Brefeldin A, and since addition of MK-571 simultaneous to stimulation with superantigen is effective, it is unlikely that the effects seen here are due to cytokine-induced expression of PPARγ, as has been reported for IL-4 (35, 36), or that IL-10 and IFN-γ induced or repressed each other's expression.
It is possible that the effects of MK-571 we are observing are direct, and not relevant to its function as an MRP1 inhibitor. Other MRP1 inhibitors such as probenecid, and small interfering RNA (siRNA) knockdown of MRP1 expression did not inhibit T cell activation (data not shown). MK-571, however, binds to MRP1 with high affinity, and inhibits MRP1 function almost completely with an IC50 60-fold lower than probenecid (37). In addition, siRNA decreased expression of MRP1 only 75–80% (data not shown), which may be insufficient for inhibiting T cell activation through MRP1 blockade. Taken together, the data in this report suggest that the effects of MK-571 are due to its activity as a potent inhibitor of MRP1.
A putative role for the MRP family as exporters of toxins and xenobiotics raises suspicion that MRP1 blockade may inhibit the function of T cells by killing them. However, concentrations that clearly inhibited IFN-γ gene and protein expression did not kill cells, and ‘wash-out’ studies were largely reversible (Fig. 5B and C). Cellular toxicity is thus not responsible for the attenuation of functional T cell responses, and other possible mechanisms were explored.
The rapid onset of action of MRP1 blockade (Fig. 5D) suggests that signaling pathways are blocked either by post-translational modification of constitutively present proteins or by small molecules that are rapidly synthesized and normally exported by MRP1. Since cytokine expression by T cells in response to PMA and ionomycin is suppressed (data not shown), calcium flux in response to TCR ligation is unaffected (data not shown), and both GATA-3 and T-bet expressions are unaffected (data not shown), the process by which MK-571 blocks T cell activation is independent of protein kinase C, calcium flux and the master regulators of inflammatory cytokine gene transcription, GATA-3 and T-bet.
Among the possibilities for mechanisms of repression of cytokine secretion is the role of MRP1 as an exporter of GSH and GSH conjugates. In this context, the cytoplasmic oxidation–reduction environment may become inhospitable for signal transduction critical for T cell responses (4). MRP1 also exports GSH conjugates of prostaglandins A1 and A2 (PGA) (38). The electrophilic carbon in the cyclopentenone ring of these prostaglandins reacts with free sulfhydryls of cysteine residues in GSH or cytosolic proteins. GSH-conjugated PGA is exported, but GSH-conjugated cytosolic proteins may be inactivated (39). If abrogating the export of GSH-conjugated PGA by MRP1 blockade shifts covalent modification from GSH to vulnerable cytosolic proteins such as IκB kinase (40, 41), then NF-κB-mediated responses will be shut down.
Since resting T cells do not express CysLTR1 (42) and are not acknowledged sources of LTC4 (11), it would appear to have no role in the effects of MRP1 blockade that we have shown. However, Cifone et al. (43) demonstrated 5-lipoxygenase activity in primary T cells and recovered cys-LTs from supernatants after stimulation. Cytoplasmic LTC4 may serve a regulatory function by binding to an internal receptor similar or identical to the one recently characterized in eosinophils (44).
We hypothesize that blockade of MRP1 in CD4 T cells results in retention of an agonist for the transcriptional repressor PPARγ. PPARγ has been demonstrated to suppress transcription of genes for the cytokines TNF-α, IL-1β, IL-6 and IL-4 in T cells and macrophages (45–48). While eiconasoids such as PGA1, 15-PGJ2, 9- and 13-hydroxyoctadecadienoic acid and 12- and 15-hydroxyeicosatetaenoic acid are ligands for PPARγ (49, 50), to our knowledge LTC4 itself has never been described as a PPARγ agonist. The data presented here raise the possibility of a novel eiconasoid substrate of MRP1 that when retained (i.e. not pumped out because of MRP1 blockade) activates PPARγ and blocks activation of T cells and monocytes.
In conclusion, we have demonstrated that MRP1 is expressed by resting memory T cells and activated naive T cells, and that blockade of MRP1 suppresses T cell activation. The mechanism by which MRP1 regulates immune function may be due to indirect activation of the transcriptional repressor PPARγ. Deciphering the precise mechanism by which MRP1 regulates and MRP1 blockade suppresses T cell function may reveal novel therapeutic targets.
Disclaimer
The views in this article are those of the authors and do not reflect the official policy or position of the Food and Drug Administration or the US Government.
Transmitting editor: W. Strober
We thank Richard Pastor for help with statistical analysis and Karen Elkins for careful review of the manuscript, and the nurses ad Shady Grove Adventist Hospital for collecting cord blood. This work was supported by intramural funds from the Center for Biologics Evaluation and Research, US Food and Drug Administration. J.Z. was supported by Oak Ridge Associated Universities, Oak Ridge, TN, USA.
References
Leier, I., Jedlitschky, G., Buchholz, U., Cole, S. P., Deeley, R. G. and Keppler, D.
Borst, P., Evers, R., Kool, M. and Wijnholds, J.
Borst, P., Evers, R., Kool, M. and Wijnholds, J.
Iwata, S., Hori, T., Sato, N. et al.
Hehner, S. P., Breitkreutz, R., Shubinsky, G. et al.
Kuppner, M. C., Scharner, A., Milani, V. et al.
Dobashi, K., Aihara, M., Araki, T. et al.
Peterson, J. D., Herzenberg, L. A., Vasquez, K. and Waltenbaugh, C.
Soberman, R. J. and Christmas, P.
Staal, F. J. T., Roederer, M., Herzenberg, L. A. and Herzenberg, L. A.
Funk, C. D.
Wijnholds, J., Evers, R., van Leusden, M. R. et al.
Verbon, A., Leemans, J. C., Weijer, S., Florquin, S. and van Der Poll, T.
Robbiani, D. F., Finch, R. A., Jager, D., Muller, W. A., Sartorelli, A. C. and Randolph, G. J.
ten Hove, T., Drillenburg, P., Wijnholds, J., Te Velde, A. A. and van Deventer, S. J.
Lohoff, M., Prechtl, S., Sommer, F. et al.
Prechtl, S., Roellinghoff, M., Scheper, R., Cole, S. P., Deeley, R. G. and Lohoff, M.
Laupeze, B., Amiot, L., Payen, L. et al.
Laupeze, B., Amiot, L., Bertho, N. et al.
Vellenga, E., Tuyt, L., Wierenga, B. J., Muller, M. and Dokter, W.
Sanders, J. S., Stegeman, C. A. and Kallenberg, C. G.
Roederer, M.
Roederer, M., Dubs, J. G., Anderson, M. T., Raju, P. A. and Herzenberg, L. A.
Rabin, R. L., Roederer, M., Maldonado, Y., Petru, A., Herzenberg, L. A. and Herzenberg, L. A.
Rabin, R. L., Park, M. K., Liao, F., Swofford, R., Stephany, D. and Farber, J. M.
Prussin, C. and Metcalfe, D. D.
Hartel, C., Bein, G., Kirchner, H. and Kluter, H.
Stordeur, P., Poulin, L. F., Craciun, L. et al.
Press, W. H., Teukolsky, S. A., Vettering, W. T. and Flannery, B. P.
De Rosa, S. C., Herzenberg, L. A. and Roederer, M.
Rabin, R. L., Alston, M. A., Sircus, J. C. et al.
Daynes, R. A. and Jones, D. C.
Oselin, K., Mrozikiewicz, P. M., Pahkla, R. and Roots, I.
Meaden, E. R., Hoggard, P. G., Khoo, S. H. and Back, D. J.
Huang, J. T., Welch, J. S., Ricote, M. et al.
Cunard, R., Ricote, M., DiCampli, D. et al.
Lania-Pietrzak, B., Michalak, K., Hendrich, A. B. et al.
Evers, R., Cnubben, N. H., Wijnholds, J., van Deemter, L., van Bladeren, P. J. and Borst, P.
van Iersel, M. L., Cnubben, N. H., Smink, N., Koeman, J. H. and van Bladeren, P. J.
Straus, D. S., Pascual, G., Li, M. et al.
Rossi, A., Kapahi, P., Natoli, G. et al.
Figueroa, D. J., Breyer, R. M., Defoe, S. K. et al.
Cifone, M. G., Cironi, L., Santoni, A. and Testi, R.
Bandeira-Melo, C., Woods, L. J., Phoofolo, M. and Weller, P. F.
Ricote, M., Li, A. C., Willson, T. M., Kelly, C. J. and Glass, C. K.
Jiang, C., Ting, A. T. and Seed, B.
Welch, J. S., Ricote, M., Akiyama, T. E., Gonzalez, F. J. and Glass, C. K.
Chung, S. W., Kang, B. Y. and Kim, T. S.
Zhang, X. and Young, H. A.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}