






Abstract
Zika virus (ZIKV) is an emerging pathogen that can cause microcephaly and other neurological defects in developing fetuses. The cellular response to ZIKV in the fetal brain is not well understood. Here, we show that ZIKV infection of human fetal astrocytes (HFAs), the most abundant cell type in the brain, results in elevated expression and secretion of fibroblast growth factor 2 (FGF2). This cytokine was shown to enhance replication and spread of ZIKV in HFAs and human fetal brain explants. The proviral effect of FGF2 is likely mediated in part by suppression of the interferon response, which would represent a novel mechanism by which viruses antagonize host antiviral defenses. We posit that FGF2-enhanced virus replication in the fetal brain contributes to the neurodevelopmental disorders associated with in utero ZIKV infection. As such, targeting FGF2-dependent signaling should be explored further as a strategy to limit replication of ZIKV.
Zika virus (ZIKV) is an emerging pathogen within the family Flaviviridae. While most infections are asymptomatic [1], recent outbreaks revealed that ZIKV can cause microcephaly and other neurological deficits in developing fetuses [2–4]. Elucidating how the virus persists in human fetal brain tissue is key to understanding its teratogenic nature.
Degenerated or reactive astrocytes have been reported following ZIKV infection of mice [5, 6] and macaque [7] models, as well as human fetuses [8]. We showed that ZIKV-infected human fetal astrocytes (HFAs) have a dampened interferon (IFN) response and are resistant to apoptosis [9]. This may underlie the ability of the virus to persist in these cells for a least 1 month ex vivo. Other studies have shown that ZIKV infects human astrocytes derived from induced pluripotent stem cells [10, 11] and adult brain tissue [12], but infection of isolated bona fide fetal astrocytes has not been investigated in significant depth until now.
To understand more about how ZIKV establishes productive infection in the human fetal brain, we investigated how it affects the transcriptome of HFAs, the most abundant cell type in the brain and potential reservoirs for ZIKV [9]. Fibroblast growth factor 2 (FGF2) was among the most upregulated transcripts, in addition to those of innate immune genes, in ZIKV-infected HFAs. FGF2 is the prototypical member of the FGF family [13], a group of cytokines that bind to and activate receptor tyrosine kinases that signal through multiple downstream pathways to modulate cell growth and survival, angiogenesis, neurodevelopment, and wound healing [14]. Whereas elevated levels of FGF2 have been detected in sera from ZIKV-infected individuals [15] and hepatitis C virus–infected patients [16], the role of this cytokine in virus-host interactions is not known.
We found that FGF2 significantly enhances replication of ZIKV and a closely related pathogen, dengue virus (DENV). The proviral activity of FGF2 may be mediated in part by dampening the IFN response. Conversely, blocking FGF2 and downstream signaling significantly inhibited ZIKV replication in HFAs and human fetal brain explants. Together, our data provide new insights into how ZIKV establishes productive infection in the human fetal brain, as well as a potential basis for novel antiviral approaches to block flavivirus infection.
METHODS
Ethics Statement
Human fetal brain tissues were obtained from fetuses aborted at gestation weeks 15–19, with written consent from the donor parents and prior approval under protocol 1420 (University of Alberta Human Research Ethics Board).
Cells, Tissue Explants, and Viral Infections
ZIKV (MR766, PLCal, and PRVABC-59) and DENV-2 (New Guinea C) were propagated in Aedes albopictus C6/36 cells grown in minimum essential medium. HFAs [17] and brain tissue explants were prepared as described elsewhere [18]. Vero cells (ATCC) were maintained in Dulbecco’s modified Eagle’s medium. For infection, cells or tissue explants were incubated with the virus (multiplicity of infection [MOI], 0.3–3) for 2 hours or overnight at 37°C, using fresh medium supplemented with fetal bovine serum (Gibco). Culture of HFAs and cell lines, tissue explants, and use of ZIKV strains are described in more detail in the Supplementary Materials.
Quantitative Reverse Transcription–Polymerase Chain Reaction (qRT-PCR) Analysis
RNA was extracted using RNeasy mini kits (Qiagen). Real-time RT-PCR was performed in an Mx3000P thermal cycler (Agilent Technologies) by the use of ImProm-II reverse transcriptase (Promega). More details about the protocol and primers [19] are available in the Supplementary Materials.
Cytotoxicity Assays
Cytotoxicity assays were performed using CellTiter-Glo Luminescent Cell Viability and CytoTox 96 Nonradioactive Cytotoxicity kits (Promega) as described in the Supplementary Materials.
Immunostaining and Imaging
After fixation and permeabilization, cells on coverslips or 96-well plates were incubated with mouse primary antibodies antiflavivirus group antigen (clone 4G2) diluted 1:500 (Millipore) at room temperature for 1.5 hours, washed, and then incubated with DAPI and Alexa Fluor–conjugated secondary antibodies against mouse primary antibody for 1 hour at room temperature. Antibodies were diluted in blocking buffer, and phosphate-buffered saline containing 0.3% bovine serum albumin was used for wash steps. Confocal images were acquired using an Olympus 1×81 spinning disk confocal microscope or an Operetta High-Content Imaging System (Perkin Elmer). Experimental details are available in the Supplementary Materials.
Enzyme-Linked Immunosorbent Assay (ELISA)
Supernatants from mock or ZIKV-infected HFAs were collected at indicated time points (1–5 days), and secreted FGF2 was quantified using a commercial ELISA kit (RayBiotech).
FGF2 Inhibition and Stimulation Assays
HFAs in 96-well plates (Greiner) were infected with ZIKV (MOI, 0.3 or 3). For FGF2 neutralization assays, infected HFAs were treated with 6 or 12 µg/mL of mouse anti-FGF2 antibody (catalog no. 05-117; Millipore) or an isotype-matched irrelevant mouse monoclonal antibody (R&D Systems) as a negative control. To block FGF receptor signaling, infected HFAs were treated with BGJ398 (Adooq Bioscience) at 1.25–2.5 nM. To determine the effect of FGF2 on ZIKV or DENV infection, HFAs were pretreated for 24 hours and treated after infection with 6.25–12.5 ng/mL of human recombinant FGF2 (Sigma-Aldrich). Inhibition of downstream signaling of FGF2 was tested with PD98059 (MEK kinase inhibitor) at 25–50 µM, U-73122 (PLCγ inhibitor) at 5–10 µM, and LY294002 (PI3K/Akt inhibitor) at 12.5–25 µM from Adooq Bioscience. A total of 48–72 hours after infection, viral genomes in cells were quantified by qRT-PCR, and/or viral titers in cell supernatants were determined by plaque assay.
HFAs were grown in 24-well plates at 1 × 105 cells/well (Greiner) and, after adding FGF2, were infected with ZIKV (MOI, 3) to perform a 5-day analysis of viral titer kinetics. Where indicated, noninfected HFAs were treated with FGF2, harvested by trypsin detachment, and counted with a Moxi Z mini automated cell counter (Orflo) on the second and fourth days.
For FGF2 inhibition and stimulation assays conducted at lower MOIs (ie, 0.3), cells were cultured in 96-well plates (Cellstar) for immunofluorescence imaging using an Operetta High-Content Imaging System (Perkin Elmer).
Polyinosinic:Polycytidylic Acid (Poly[I:C]) Treatment
HFAs were pretreated for 24 hours with 12 µg/mL of mouse anti-FGF2 antibody (Millipore) or an isotype-matched nonspecific mouse monoclonal antibody (R&D Systems) as a negative control. In another set of experiments, cells were pretreated for 24 hours with PD98059 (50 µM) or dimethyl sulfoxide as a negative control. Next, cells were transfected with poly(I:C) (Sigma-Aldrich) at a concentration of 0.02 or 0.1 μg/well, using TransIT (0.3 µL/well; Mirus Bio). Antibodies and the drug (or vehicle) were also present during and after transfection. At 12 hours after transfection, total RNA was extracted, and transcripts levels for immune genes were quantified by qRT-PCR.
Recombinant IFN-α Treatment
HFAs were pretreated with BGJ398 (2.5 nM) or PD98059 (50 µM) for 24 hours, while control cells were treated with dimethyl sulfoxide. Next, cells were treated with 25 U/mL of human recombinant IFN-α (Sigma-Aldrich) in the presence of BGJ398 or PD98059. Total RNA was isolated from cells at 4-, 8-, and 12-hour time points, followed by qRT-PCR analysis to measure expression of IFN-stimulated genes (ISGs).
Statistical Analyses
A paired Student t test was used for pair-wise statistical comparison. The standard error of the mean is shown in all line and bar graphs. GraphPad Prism software was used in all statistical analyses.
RNA Sequencing (RNAseq)
Construction of RNAseq libraries and bioinformatics analysis are described in the Supplementary Materials.
Sequence Accession Number
The accession number for the raw data for the transcriptional response of HFAs to acute ZIKV infection reported in this article have been deposited to the Sequence Read Archive of the National Center for Biotechnology Information to be publicly available under the project accession number PRJNA356760.
RESULTS
ZIKV Infection of HFAs Upregulates FGF2
Prior to assessing how ZIKV affects the transcriptome of HFAs, 1-step viral growth assays were conducted to determine the period when peak virus replication occurs in these cells. Data in Figure 1A show that HFAs are highly permissive for ZIKV infection and that maximum virus production occurs between 48 and 72 hours. Recently, we showed that, despite supporting robust viral replication, >95% of cells in ZIKV-infected HFA cultures were viable after 48 hours [9]. Next, HFAs were infected with a contemporary ZIKV strain (PLCal; MOI, 3), and RNAseq analysis was performed using total RNA extracted from cells 48 hours after infection. HFAs from 3 different donors were used for these analyses, to minimize potentially aberrant responses from any one sample. In the aggregate data, 2403 cellular transcripts were upregulated and 238 downregulated in response to ZIKV infection (Supplementary Table 1). While there were significant differences between the basal transcriptome profiles of the 3 HFA samples, the general patterns of virus-induced gene expression were similar. Within the most downregulated group of transcripts (P < .05; Figure 1B) were those that encode proteins involved in promoting apoptosis (JADE1, RHOT1, and HIPK1) [20–22], cell proliferation (PICALM) [23], and neural development (PTK7 and TMEM237) [24, 25]. Among the most upregulated host genes in ZIKV-infected HFAs, 25 were ISGs, including OAS, CXCL, MX, RSAD2 (viperin), and IFN-β. In addition to ISGs, transcript levels for FGF2 were increased 270-fold during ZIKV infection (Figure 1B). Analyses of medium from ZIKV-infected HFAs revealed that viral infection increased secretion of this cytokine up to 4-fold (P < .05; Figure 1C).
![Zika virus (ZIKV) infection induces expression of fibroblast growth factor 2 (FGF2). Human fetal astrocytes (HFAs) were infected with ZIKV strain PLCal (multiplicity of infection [MOI], 3). A, Virus production was assessed by determining viral titers in supernatants at indicated times after infection by a plaque assay. B, Mean of fold change corrected P values of the top upregulated and downregulated transcripts in HFAs (from 3 donors) 2 days after infection. C, Secreted levels of FGF2 from mock- and ZIKV-infected HFAs (MOI, 3) were measured daily for up to 5 days by an enzyme-linked immunosorbent assay. Values are expressed as the mean of 3 independent experiments. Error bars represent standard errors of the mean. PFU, plaque-forming units. *P < .05, by the Student t test.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/220/8/10.1093_infdis_jiz073/1/m_jiz073f0001.jpeg?Expires=1716477213&Signature=bshoY034PDkCdAD7CZ5dQerGkc6QIdtyETOHrucycP~RYKxr1hCfexrgXS39g8k~F25CrkBmvcz5NH4YQQuK4wvBIQ8TS4QYUINDGzjbZzchX3SV2ntZiKcNHyKDgkKlu2vpNXknuZEzmtDxKdBxZMprHL11sU-O-Kmlr0-PZhKk0lXw8ge5XEeFfjK5VBvctU0qKT4RBW~CLUid~co3xRANZzG28GpMRVjmbVqUOP3Ml6xm~Auhl4eB1fKGBzYBziZVEBdEDNwQKC-HxAd0Stg6DlgkEdiClTtrI-ro9squB3JbmVyYhnAJLmHFstPA1ASVzn06WAUqwJTGxH~8Tg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Zika virus (ZIKV) infection induces expression of fibroblast growth factor 2 (FGF2). Human fetal astrocytes (HFAs) were infected with ZIKV strain PLCal (multiplicity of infection [MOI], 3). A, Virus production was assessed by determining viral titers in supernatants at indicated times after infection by a plaque assay. B, Mean of fold change corrected P values of the top upregulated and downregulated transcripts in HFAs (from 3 donors) 2 days after infection. C, Secreted levels of FGF2 from mock- and ZIKV-infected HFAs (MOI, 3) were measured daily for up to 5 days by an enzyme-linked immunosorbent assay. Values are expressed as the mean of 3 independent experiments. Error bars represent standard errors of the mean. PFU, plaque-forming units. *P < .05, by the Student t test.
FGF2 Enhances Replication of ZIKV in HFAs
To determine how FGF2 affects virus replication, recombinant human FGF2 was added to ZIKV-infected HFAs, and viral titers were determined over a 5-day period. FGF2 had a dose-dependent stimulatory effect on ZIKV production, with the greatest effect occurring 48 hours after infection, which coincided with the peak of virus replication (Figure 2A). At this time point, levels of viral RNA were >3-fold higher in HFAs supplemented with FGF2 (P < .01; Figure 2B). Because FGF2 can induce cellular proliferation, it was possible that the effect of this cytokine on viral replication was due to increased numbers of susceptible HFAs. However, the numbers of cells and levels of ATP in FGF2-treated HFA cultures were not significantly elevated, compared with those in mock samples (Supplementary Figure 1A). This indicates that the concentrations of FGF2 used in our experiments were below the threshold to stimulate cell proliferation.
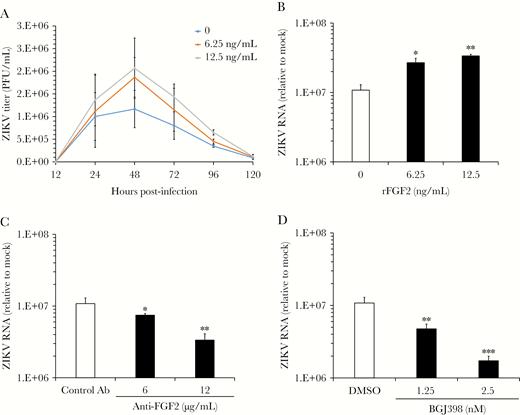
Fibroblast growth factor 2 (FGF2) enhances viral replication in human fetal astrocytes (HFAs). HFAs were pretreated for 24 hours with or without recombinant human FGF2 (6.25–12.5 ng/mL) and then infected with Zika virus (ZIKV) strain PLCal (multiplicity of infection, 3). A, Medium was collected daily from infected HFAs cultured with or without FGF2, and viral titers were determined by a plaque assay. B, Total RNA was extracted 2 days after infection from HFAs and then subjected to quantitative reverse transcription–polymerase chain reaction (qRT-PCR) analysis to determine how FGF2 affected levels of viral RNA. C and D, ZIKV-infected HFAs were cultured in the presence of a neutralizing mouse anti-FGF2 antibody (Ab) or an isotype-matched mouse immunoglobulin G (IgG; C) or the FGF receptor inhibitor BGJ398 or dimethyl sulfoxide (DMSO; D) for 2 days, after which total RNA was extracted and viral RNA quantitated by qRT-PCR. Values are expressed as the mean of 3 independent experiments. Error bars represent standard errors of the mean. PFU, plaque-forming units. *P < .05, **P < .01, and ***P < .001, by the Student t test.
Given that FGF2 promotes ZIKV replication, we expected that blocking FGF2 from binding to receptors on the cell surface would have the opposite effect. Indeed, addition of neutralizing anti-FGF2 but not an isotype-matched control antibody reduced ZIKV replication in HFAs in a dose-dependent manner by as much as 5-fold (P < .01; Figure 2C). Similarly, inhibition of FGF receptor kinase activity with the drug BGJ398 also had an inhibitory effect on ZIKV replication in HFAs (P < .001; Figure 2D). Neither anti-FGF2 nor BGJ398 at concentrations that showed antiviral activity impacted the viability of HFAs (Supplementary Figure 1A). Finally, we tested whether FGF2 enhances replication of a closely related flavivirus, DENV-2. Similar to what was observed with ZIKV, addition of FGF2 significantly increased replication of DENV-2 in HFAs (P < .05; Supplementary Figure 1B and 1C).
Strain-Dependent Effects of FGF2 on ZIKV Replication
To determine whether FGF2 production and proviral effects varied among ZIKV strains, HFAs were infected at a low MOI (ie, 0.3) with 3 different strains of ZIKV, followed by quantitation of FGF2 and release of infectious virions by ELISA and plaque assay, respectively. Interestingly, regardless of the strain, infected HFAs secreted similar amounts of FGF2, which were 50%–100% higher than levels in mock-infected cells (Figure 3A); however, the more recent virus isolates, including the pandemic ZIKV strain PRVABC-59, were significantly more responsive to FGF2 supplementation (P < .01; Figure 3A). Similarly, replication of PLCal and PRVABC-59 was more susceptible than MR766 to the effects of FGF2-neutralizing antibody (P < .05; Figure 3A) and the FGF receptor–inhibiting drug BGJ398 (P < .05; Figure 3A).
![Strain-dependent effects of fibroblast growth factor 2 (FGF2) on Zika virus (ZIKV) replication. A, Human fetal astrocytes (HFAs) were infected with MR766, PLCal, or PRVABC-59 strains of ZIKV (multiplicity of infection [MOI], 0.3). Two and 3 days after infection, levels of FGF2 in the culture medium were determined by an enzyme-linked immunosorbent assay (i). ZIKV-infected HFAs were cultured in the presence or absence of FGF2 for 2 days, after which viral titers were determined by a plaque assay (ii). ZIKV-infected HFAs were cultured in medium containing a neutralizing mouse anti-FGF2 antibody (Ab) or an isotype-matched mouse Ab. Viral titers in supernatants were determined by a plaque assay (iii). ZIKV-infected HFAs were treated with FGF receptor inhibitor (BGJ398) or dimethyl sulfoxide (DMSO) alone for 2 days, after which viral titers were determined by a plaque assay (iv). Values are expressed relative to control, which was set to 1.0. All statistically significant differences are relative between control and experimentally treated (rFGF2, anti-FGF2 Ab or BGJ398) samples. B and C, ZIKV PRVABC-59–infected HFAs (MOI, 0.3) were cultured in the presence or absence of 12.5 µg/mL recombinant human FGF2 (B) or 2.5 nm BGJ398 or DMSO (C) for 2 days, after which automated high-content immunofluorescence imaging was used to determine the numbers of cells positive for ZIKV E protein. Values are expressed as the mean of 3 independent experiments. Error bars represent standard errors of the mean. *P < .05 and **P < .01, by the Student t test.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/220/8/10.1093_infdis_jiz073/1/m_jiz073f0003.jpeg?Expires=1716477213&Signature=1kHvBhM58sT5ZiQiyFo2uvkFO2YYx8Iz6jVB3qSzeAoPdMseYg~jMabWIFLZj1NNRJOtkNK5ph0csv6W36Iioa1FlP920ZV0rolOC0HS58mK0g~CLkirVB2xHAIwh3T35Sh-HhFom6Bo8Qf7RPYwkBukLIx7bWWrE8kEp3ZON9Idj3AopDQAiBprvC4KD0PfhCOl-7K5t~vzlJb0DSa~1H93WETw9RLYAeVYYtGAyOXh1umT2CBe-Ol69~nVVAVW~kqnsEzJad6gCQJRc8ySjInOB5~FpG8Vl9FZBH3v5cW5Rc18xV~~qs7xUex1CHlqadU-WCWyprKlPygRvDHICw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Strain-dependent effects of fibroblast growth factor 2 (FGF2) on Zika virus (ZIKV) replication. A, Human fetal astrocytes (HFAs) were infected with MR766, PLCal, or PRVABC-59 strains of ZIKV (multiplicity of infection [MOI], 0.3). Two and 3 days after infection, levels of FGF2 in the culture medium were determined by an enzyme-linked immunosorbent assay (i). ZIKV-infected HFAs were cultured in the presence or absence of FGF2 for 2 days, after which viral titers were determined by a plaque assay (ii). ZIKV-infected HFAs were cultured in medium containing a neutralizing mouse anti-FGF2 antibody (Ab) or an isotype-matched mouse Ab. Viral titers in supernatants were determined by a plaque assay (iii). ZIKV-infected HFAs were treated with FGF receptor inhibitor (BGJ398) or dimethyl sulfoxide (DMSO) alone for 2 days, after which viral titers were determined by a plaque assay (iv). Values are expressed relative to control, which was set to 1.0. All statistically significant differences are relative between control and experimentally treated (rFGF2, anti-FGF2 Ab or BGJ398) samples. B and C, ZIKV PRVABC-59–infected HFAs (MOI, 0.3) were cultured in the presence or absence of 12.5 µg/mL recombinant human FGF2 (B) or 2.5 nm BGJ398 or DMSO (C) for 2 days, after which automated high-content immunofluorescence imaging was used to determine the numbers of cells positive for ZIKV E protein. Values are expressed as the mean of 3 independent experiments. Error bars represent standard errors of the mean. *P < .05 and **P < .01, by the Student t test.
FGF2 Promotes Viral Spread in HFAs
Next, automated high-content immunofluorescence imaging was used to determine whether FGF2 affected viral spread in HFA cultures. Data in Figure 3B show that the proportion of virus-infected HFAs was nearly 2-fold higher in samples treated with FGF2 (P < .05). Conversely, blocking FGF2-dependent signaling with BGJ398 reduced the number of infected HFAs >2-fold (P < .01; Figure 3C). Representative images from these experiments are shown in Supplementary Figure 2A. Together, these data indicate that FGF2 increases cell-to-cell spread of ZIKV in HFA cultures.
Mitogen-Activated Protein (MAP) Kinase Signaling Is Important for ZIKV Infection
FGF signaling can activate at least 3 downstream signal transduction pathways, including MAP kinase, phosphatidylinositol 3-kinase (PI3K)/Akt, and phospholipase C γ (PLCγ). Treatment of ZIKV-infected HFAs with subcytotoxic concentrations of LY294002 and U-73122, which inhibit PI3K/Akt and PLCγ, respectively, did not significantly affect virus replication (Supplementary Figure 2B). In contrast, when infected HFAs were treated with PD98059, a selective inhibitor of MEK (an upstream kinase that phosphorylates MAP kinases), levels of ZIKV genomic RNA and viral titers were reduced >5-fold and 100-fold, respectively (P < .001; Figure 4A and 4B), without affecting the viability of HFAs (Supplementary Figure 3A). Moreover, addition of PD98059 largely abrogated the proviral effects of recombinant FGF2 on infected HFAs (P < .001; Figure 4C). Viral spreading in HFA cultures was also reduced by inhibiting MEK activity (P < .001; Figure 4D). Together, these data suggest that the proviral effect of FGF2 is mediated in part by signaling through MAP kinase pathways.
![MEK-1 activity is important for Zika virus (ZIKV) replication and infectivity. Human fetal astrocytes (HFAs) were infected with ZIKV strain PRVABC-59 (multiplicity of infection [MOI], 0.3) in the presence of the MEK inhibitor PD98059 or dimethyl sulfoxide (DMSO). Two days after infection, total RNA extracted from cells and supernatants were subjected to quantitative reverse transcription–polymerase chain reaction analysis (A) and plaque assays (B), respectively. The effects of PD98059 (50 µM) on production of ZIKV progeny virions (C) and spread (D) were assessed in the presence and absence of recombinant human fibroblast growth factor 2 (FGF2; 12 ng/mL) 2 days after infection. D, Automated high-content immunofluorescence imaging was used to determine the numbers of cells positive for ZIKV E protein. Values are expressed as the mean of 3 independent experiments. Error bars represent standard errors of the mean. PFU, plaque-forming units. *P < .05 and ***P < .001, by the Student t test.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/220/8/10.1093_infdis_jiz073/1/m_jiz073f0004.jpeg?Expires=1716477213&Signature=wlIwLQu5kBB1cHR6Y55Dyzk7h3cF2uTQDHdIX2xEVvVu96Q8Z7mOvba8m3w~oPcrhRvstQxy6nlcGtzcIufm968Ek934i2Rpflc1s27GhMscSq7wBy4awLGY8hfBUCWggtcXjuKTPotsltxmIXdmHr5cyryiC3eCNMwJ~CG6B7HRzaHdHG1fKJHDmFH42CsECSV9tphmwjhzj1vqYboC0q-25bCLH8kW40DAe3G3SUX3R0I9GKz46aW7bXjRY44iBcpR61UVa8VFZCAVwBKRhJJWG5rnWspZeBvjGOMx5VvIcVC0ZOS0VxwRiSJFp5YXmGbR95Y2IfD1lBQJbsQ~YA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MEK-1 activity is important for Zika virus (ZIKV) replication and infectivity. Human fetal astrocytes (HFAs) were infected with ZIKV strain PRVABC-59 (multiplicity of infection [MOI], 0.3) in the presence of the MEK inhibitor PD98059 or dimethyl sulfoxide (DMSO). Two days after infection, total RNA extracted from cells and supernatants were subjected to quantitative reverse transcription–polymerase chain reaction analysis (A) and plaque assays (B), respectively. The effects of PD98059 (50 µM) on production of ZIKV progeny virions (C) and spread (D) were assessed in the presence and absence of recombinant human fibroblast growth factor 2 (FGF2; 12 ng/mL) 2 days after infection. D, Automated high-content immunofluorescence imaging was used to determine the numbers of cells positive for ZIKV E protein. Values are expressed as the mean of 3 independent experiments. Error bars represent standard errors of the mean. PFU, plaque-forming units. *P < .05 and ***P < .001, by the Student t test.
FGF2 Signaling Suppresses the IFN Response in HFAs
In addition to activating cell survival and proliferation pathways, FGF2 has recently been reported to suppress the innate immune response during viral infection of mouse cells [26] and human cancer cells [27]. To investigate whether FGF2 affected the innate immune response in HFAs, cultures were pretreated with a neutralizing antibody to FGF2 or an isotype matched control mouse antibody and then transfected with poly(I:C), a mimic of viral double-stranded RNA. Because the concentration of FGF2 in cell culture-grade fetal bovine serum is only 30–40 pg/mL (application note S0801; Thermo Fisher) and the anti-FGF2 monoclonal antibody used here neutralizes human and bovine FGF2, we assumed that the anti-FGF2 antibody was able to block the activity of all FGF2 in the culture medium.
Twelve hours after transfection, levels of messenger RNA for IFN-β and the ISGs MX2, viperin, and OAS1 were quantitated by qRT-PCR. Data in Figure 5A and Supplementary Figure 3B (P < .001) show that blocking FGF2 signaling in HFAs enhanced the innate immune response to poly(I:C). Specifically, transcript levels for IFN- β were increased up to 6-fold, whereas downstream antiviral genes, such as those encoding MX2, viperin, and OAS1 were upregulated 2–3-fold by anti-FGF2 treatment. Pharmacological inhibition of downstream FGF2 signaling with the MEK inhibitor PD98059 also resulted in 2–4.5-fold increased expression of IFN-β and ISGs in response to poly(I:C) (P < .01; Figure 5B and Supplementary Figure 3B).
![Blocking fibroblast growth factor 2 (FGF2) signaling increases expression of type I interferon (IFN) and IFN-stimulated genes (ISGs). A, Human fetal astrocytes (HFAs) were pretreated with mouse anti-FGF2 antibody (Ab; 12 µg/mL) or an isotype-matched nonspecific mouse monoclonal Ab for 24 hours, followed by transfection with polyinosinic:polycytidylic acid (poly[I:C]) (0.02–1 μg/well). Twelve hours after transfection, total RNA was extracted from cells, and quantitative reverse transcription–polymerase chain reaction (qRT-PCR) analysis was used to measure relative levels of IFN-β (i), MX2 (ii), viperin (iii) and OAS-1 (iv) transcripts. B, HFAs were pre-treated with the MEK-1 inhibitor PD98059 (50 µM) or dimethyl sulfoxide (DMSO) alone for 24 hours and then transfected with poly(I:C) (0.02 μg/well) for 12 hours after which total RNA was extracted from cells, and qRT-PCR analysis was used to measure relative levels of IFN-β, MX2, viperin, and OAS-1 transcripts. C and D, Transcripts levels of indicated ISGs were determined by qRT-PCR after pretreatment of HFAs with BGJ398 (2.5 nM; C), PD98059 (50 µM; D), or DMSO alone for 24 hours, followed by addition of human recombinant IFN-α (rIFN-α; 25 U/mL) for 4–12 hours (C) or 12 hours (D). Values are expressed as the mean of 3 independent experiments. Error bars represent standard errors of the mean. mRNA, messenger RNA. *P < .05, **P < .01, and ***P < .001, by the Student t test).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/220/8/10.1093_infdis_jiz073/1/m_jiz073f0005.jpeg?Expires=1716477213&Signature=ochYHdHMjj0mNbuvbm2PbMlMf5lGwMRoZRPXgYAjEJSx1WvzsvN-9zQXmoqoLtYsz6MwBpEKWyD-064xOdBsI5Ugw1uhZXIZ-TbLjZb2Ig~K42X2Cj8BYxaA-qPQd02p3DCghx8nAFYVDupqyokN2plcUW8y2AkZFAMGTJPb-7LPLMtd0afip81dtAER4Z~ExEBz6Y1V7WdBV3D7VSmXv6O440C-dpn2HUWbETFSkt~oylAsIyTDtAkMemBdcFz1dPeYz13qgs6hptC08YG07eNief072RnPzW0dY5bcKVloa~8LQenivN4JdbfXjOgrChS9R4YtCEDb~uHZingZ1w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Blocking fibroblast growth factor 2 (FGF2) signaling increases expression of type I interferon (IFN) and IFN-stimulated genes (ISGs). A, Human fetal astrocytes (HFAs) were pretreated with mouse anti-FGF2 antibody (Ab; 12 µg/mL) or an isotype-matched nonspecific mouse monoclonal Ab for 24 hours, followed by transfection with polyinosinic:polycytidylic acid (poly[I:C]) (0.02–1 μg/well). Twelve hours after transfection, total RNA was extracted from cells, and quantitative reverse transcription–polymerase chain reaction (qRT-PCR) analysis was used to measure relative levels of IFN-β (i), MX2 (ii), viperin (iii) and OAS-1 (iv) transcripts. B, HFAs were pre-treated with the MEK-1 inhibitor PD98059 (50 µM) or dimethyl sulfoxide (DMSO) alone for 24 hours and then transfected with poly(I:C) (0.02 μg/well) for 12 hours after which total RNA was extracted from cells, and qRT-PCR analysis was used to measure relative levels of IFN-β, MX2, viperin, and OAS-1 transcripts. C and D, Transcripts levels of indicated ISGs were determined by qRT-PCR after pretreatment of HFAs with BGJ398 (2.5 nM; C), PD98059 (50 µM; D), or DMSO alone for 24 hours, followed by addition of human recombinant IFN-α (rIFN-α; 25 U/mL) for 4–12 hours (C) or 12 hours (D). Values are expressed as the mean of 3 independent experiments. Error bars represent standard errors of the mean. mRNA, messenger RNA. *P < .05, **P < .01, and ***P < .001, by the Student t test).
To assess how blocking FGF receptors and MEK signaling affected type I IFN-induced expression of ISGs, IFN-α was added to HFAs pretreated with BGJ398 or PD98059, and levels of ISG transcripts were assessed by qRT-PCR. Inhibiting FGF receptor activity with BGJ398 increased viperin transcript levels between 4 and 12 hours (P < .05; Figure 5C and Supplementary Figure 3C), but MX2 and OAS1 transcript levels were not significantly altered. Pretreatment of HFAs with the MEK-1 inhibitor PD98059 also potentiated the effect of IFN-α on viperin expression (P < .05; Figure 5D and Supplementary Figure 3D). OAS1 and MX2 transcript levels were slightly elevated in PD98059-treated HFAs, but the changes were not statistically different from mock samples. These data suggest that secreted FGF2 suppresses induction of some but not all ISGs during viral infection of HFAs.
Blocking FGF Signaling in Human Fetal Brain Explants Inhibits ZIKV Replication
Finally, to determine whether blocking FGF signaling affected ZIKV replication in a more anatomically representative tissue model, fetal brain explants were infected with the pandemic ZIKV strain PRVABC-59 for 48 hours in the presence or absence of BGJ398. Similar to the results observed with pure HFA cultures, blocking FGF receptor signaling reduced viral titers and genomic RNA in the explant cultures approximately 10-fold (P < .001; Figure 6A and 6B). Under the conditions used here, BGJ398 treatment did not negatively impact the structure of and was not cytotoxic to brain explant tissue, as revealed by histologic and lactate dehydrogenase–release assays (Supplementary Figure 4A–D). In fact, treatment of brain explant tissue with BGJ398 had a significant cytoprotective effect against ZIKV infection (P < .05; Figure 6C), and there was also a corresponding decrease in the amount of infected and apoptotic neural cells in the explant tissue (Supplementary Figure 5A–D). Together, data from this explant infection model are consistent with an important role for FGF signaling in ZIKV infection of human fetal brain tissue.
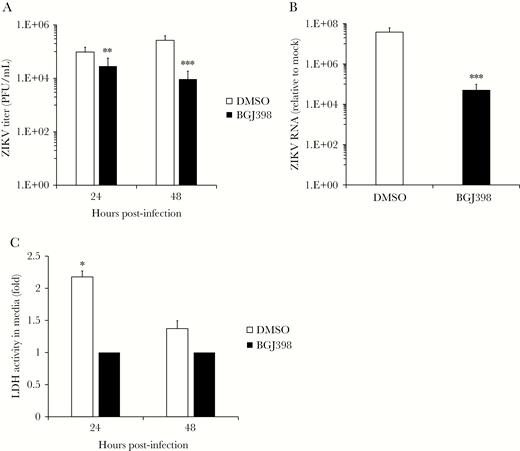
Fibroblast growth factor 2 (FGF2) signaling is important for replication of Zika virus (ZIKV) infection in human fetal brain explant cultures. A, Human fetal brain explants cultured in medium containing the FGF receptor inhibitor BGJ398 (10 nM) or dimethyl sulfoxide (DMSO) alone were infected overnight with 106 plaque-forming units (PFU)/mL of ZIKV strain PRVABC-59. Resulting viral titers were determined by a plaque assay 1 and 2 days after infection. B, Total RNA extracted from infected brain explants 2 days after infection was subjected to quantitative reverse transcription–polymerase chain reaction analysis to measure virus replication. C, The relative levels of lactate dehydrogenase (LDH) activity in the medium of infected fetal brain explant cultures with BGJ398 (10 nM) or DMSO were assessed 1 and 2 days after infection.
DISCUSSION
ZIKV infection can cause microcephaly and other neurodevelopmental defects in the developing fetus [2, 8, 28, 29] through a process that is thought to require persistence of the virus in fetal brain tissue [9]. Astrocytes have critical roles in support of neurons and brain homeostasis and are the most abundant cell type in the brain [30]. As such, infection of these cells is likely to have major consequences for fetal brain development. Here, we provide evidence that ZIKV-induced expression of FGF2 facilitates virus replication and cell-to-cell spread in HFAs. We recently reported that ZIKV infection of human Sertoli cells also upregulates FGF2, but the mechanism of its proviral activity was not elucidated nor was its effect on production of infectious virus determined [31].
The fact that levels of FGF2 are elevated in sera of patients during acute ZIKV infection [15] is yet more evidence that this cytokine is an important proviral factor. Intriguingly, in a study of ZIKV-infected pregnant women in Brazil, the highest concentrations of FGF2 were found in sera from women carrying developmentally affected fetuses [32]. FGF2 also has proviral effects on hepatitis C virus replication, and levels of this cytokine are elevated in chronically infected patients [16], but to our knowledge, the mechanisms by which this cytokine affects virus-host interactions has not been investigated until now.
It has long been known that FGF2 can block apoptosis by activating phosphatidylinositol-3-kinase [33]. This would seem to be an advantage for the virus, and indeed many flaviviruses are known to stimulate the activity of AKT, a prosurvival kinase that is downstream from phosphatidylinositol-3-kinase [34–37]. However, we did not see significant effects on ZIKV replication when HFAs were treated with an inhibitor of phosphatidylinositol-3-kinase. Thus, while levels of apoptosis in ZIKV-infected HFAs is relatively low [9], it has yet to be determined whether increased FGF2 expression plays any role in this process.
A number of recent reports are consistent with a scenario in which the proviral effect of FGF2 [26, 27] and downstream MAP kinase signaling [38–40] is mediated at least in part by IFN suppression. Indeed, we showed that blocking the kinase activity of FGF receptors or the downstream kinase MEK in uninfected cells increases expression of type I IFN and ISGs, including viperin, which has been shown to restrict ZIKV replication [41, 42]. Accordingly, secreted IFN would be expected to make bystander cells resistant to viral infection, thereby limiting ZIKV replication and spread. A concomitant decrease in ZIKV replication was observed when FGF receptor or MEK signaling was inhibited. While MAP kinase signaling appears to be critical for the proviral effect of FGF2, it is unclear how the IFN response is inhibited downstream of MEK activation.
Intriguingly, the effect of FGF2 on ZIKV replication was strain dependent even though all strains tested caused HFAs to secrete more FGF2 during infection. Specifically, replication of contemporary ZIKV strains, including the pandemic Puerto Rico strain PRVABC-59, were stimulated to a greater extent by supplemental FGF2 than by MR766. Conversely, the contemporary strains were more sensitive to anti-FGF2 and an FGF receptor inhibitor. While the evidence is only circumstantial at this point, it will be of interest to determine whether this phenomenon underlies the pathology caused when ZIKV infects immune-privileged FGF2-producing tissues, including fetal brain or testis [43]. Alternatively, it may simply be related to interstrain replication differences [12, 44]. In this regard, the African strain used in our study is derived from an infectious clone [45], while the contemporary strains are virus isolates.
Data from this study suggest that FGF signaling facilitates infection of HFAs and fetal brain explant cultures, both of which may serve as platforms for testing drugs against ZIKV. FGF2 released by HFAs may create a more conducive microenvironment by acting in a paracrine and/or autocrine fashion to facilitate viral spread within the fetal brain. As such, blocking FGF2 signaling could be a viable strategy to treat in utero ZIKV infections. Indeed, the Food and Drug Administration–approved drug BGJ398 [46] significantly reduced replication of ZIKV in human fetal brain explant cultures, without any apparent cytotoxicity. Of note, there are a number of ongoing clinical trials for cancer indications focused on blocking FGF receptor signaling with monoclonal antibodies [47] and small molecules [48]. Moreover, in a recent trial, oral BGJ398 (Infigratinib) at concentrations as high as 3 µg/mL was shown to be safe after 4 weeks of treatment in patients [48]. In addition to blocking FGF and/or FGF receptors, pharmacological inhibition of downstream signaling pathways had an even greater inhibitory effect on ZIKV production. As such, further investigation into the potential antiviral activities of these drugs should be followed up, using preclinical animal models for ZIKV. Finally, the observation that DENV replication is enhanced by FGF2 treatment suggests that blocking FGF receptors or MEK signaling may have broad antiviral activity against flaviviruses.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Notes
Acknowledgments. We thank Dr Matthew J. Evans (Icahn School of Medicine at Mount Sinai, New York) and Dr David Safronetz (Public Health Agency of Canada), for providing the ZIKV strains; the Applied Genomics Centre, the Cell Imaging Centre and the HistoCore facilities in the Faculty of Medicine and Dentistry at the University of Alberta; and Brittany Fraser, Eileen Reklow, Valeria Mancinelli, Jordan Patterson, and Sandra O’Keefe, for excellent technical support.
Disclaimer. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Financial support. This work was funded by the Canadian Institutes of Health Research (grants PJT-148699 and ZV1-149782 to T. C. H.), the Women and Children’s Health Research Institute, and the Li Ka Shing Institute of Virology.
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}