





Abstract
Adenosine kinase (ADK) is supposed to be a schizophrenia susceptibility gene based on the findings that ADK is an enzyme that catalyzes transfer of the gamma-phosphate from ATP to adenosine, which interacts with dopamine and glutamate neurotransmitters. However, no reports of schizophrenia cases with loss of function variants in the ADK region have been published. In our previous study investigating copy number variants in schizophrenia, we detected a copy number variant in the ADK region in 1 of 1699 schizophrenia patients.
We validated the ADK deletion by determining the breakpoint. Then, we compared the relative expression of ADK in 32 schizophrenia patients, including a schizophrenia patient with deletion of ADK, with 29 healthy controls using lymphoblastoid cell lines. Furthermore, we evaluated the clinical phenotypes of the schizophrenia with ADK deletion.
We validated the copy number variants with Sanger sequencing and predicted that this copy number variant results in loss of function of ADK. Furthermore, expression analysis of mRNA from peripheral blood in this schizophrenia patient with the ADK deletion showed an extremely low level of ADK. Here we describe a case report of a patient with ADK deletion with phenotypes (schizophrenia, parkinsonism, epilepsy) that are predicted when ADK function is disrupted.
Considering that the patient had a low ADK mRNA level and showed a phenotype that may be related to ADK deficiency, the copy number variants in the region of ADK may be strongly related to the phenotypes described here, such as schizophrenia, Parkinsonism, and epilepsy.
Adenosine kinase (ADK) is an enzyme that catalyzes transfer of the gamma-phosphate from ATP to adenosine and is known to be related to neurological phenotypes such as epilepsy, Alzheimer’s disease, Parkinson’s disease, and sleep disorder. Furthermore, ADK is supposed to be one of the schizophrenia (SCZ) susceptibility genes, because adenosine modulates neurotransmission through activation of adenosine receptors at synaptic sites. However, no reports of SCZ cases with loss of function variants in the ADK region have been published. This study firstly describes the relationship between the loss of function variant of ADK and the phenotypes of SCZ. Considering that the patient had a low ADK mRNA level and showed a phenotype that may be related to ADK deficiency, the ADK deletion may be strongly related to the phenotypes described here, such as SCZ, Parkinsonism, and epilepsy.
Introduction
Schizophrenia (SCZ), a devastating mental disease characterized by hallucinations and delusions, is a highly complex neurodevelopmental disorder with heritability of up to 80% (Sullivan et al., 2003; Lichtenstein et al., 2009). Recently, rare variants such as copy number variants (CNVs) (Malhotra and Sebat, 2012) and single nucleotide variants (Kim et al., 2017) have been a focus of research because of their large effect size on increased susceptibility to SCZ and because of the possibility of elucidating the pathophysiology of this mental disorder and identifying new drug targets through functional analysis of the discovered rare variants (Legge et al., 2016).
The genotype first approach (Stessman et al., 2014) is a gene-centric methodology for complex disorders such as SCZ and has been applied for subtyping this mental disorder and understanding the etiopathophysiology of SCZ. For example, loss of function variants in SETD1A, which encodes a subunit of histone methyltransferase and is related to the histone H3K4 methylation pathway, are associated with SCZ and are mutated in 0.13% of SCZ cases; carriers of these variants have common phenotypic features (Takata et al., 2014; Singh et al., 2016). Moreover, carriers of the disruptive variant of chromodomain-helicase-DNA binding protein 8 (CHD8) show phenotypes such as autistic features, intellectual disability, insomnia, and gastrointestinal problems (Bernier et al., 2014; Kimura et al., 2016). Thus, discovering variants associated with characteristic phenotypes and investigating the functional mechanism is a promising method for subtyping and elucidating the etiopathophysiology of neurodevelopmental disorders.
In this study, we investigated the role of adenosine kinase (ADK) in the etiopathophysiology of SCZ. ADK converts adenosine to adenosine monophosphate and is closely related to several physical phenotypes, such as liver dysfunction and dysmorphic features (Staufner et al., 2016), and neurological phenotypes such as epilepsy, Alzheimer’s disease, Parkinson’s disease, and sleep disorder (Boison and Aronica, 2015). Furthermore, ADK is a highly ranked haploinsufficient gene in the DECIFER database (https://decipher.sanger.ac.uk/browser) (Firth et al., 2009; Huang et al., 2010). In this database, 5 of 10 registered CNVs in the ADK region are de novo variants, and patients with a CNV in the ADK region have phenotypes related to neurodevelopment such as intellectual disability, developmental delay, and seizures.
Adenosine modulates neurotransmission through activation of adenosine receptors at synaptic sites and is related to the pathophysiology of SCZ by regulating the release of both dopamine and glutamate (Boison et al., 2012). Extracellular levels of endogenous adenosine in the brain mainly depend on the astrocyte-based adenosine cycle, which involves ADK (Boison et al., 2010, 2013). ADK deficiency in the brain may result in dysregulation of synaptic plasticity (Sandau et al., 2016). ADK transgenic mice show impaired attention, which is closely linked to SCZ. Augmentation of adenosine by pharmacologic inhibition of ADK ameliorates cognitive function and exerts antipsychotic-like activity in mice (Shen et al., 2012). Furthermore, adenosine modulator adjuvant therapy is beneficial for treating the symptoms of SCZ (Hirota and Kishi, 2013).
Although ADK may contribute to SCZ susceptibility, no reports of SCZ cases with loss of function variants in the ADK region have been published. In our previous CNV analysis with a large number of Japanese individuals (Kushima et al., 2017), we discovered a deletion in the ADK region that was present in only one SCZ case and not in control samples.
Here we present a case study describing the details of this SCZ patient with ADK deletion. We identified a frame shift mutation in ADK, resulting in low ADK mRNA. Moreover, this carrier of the ADK deletion had not only the SCZ phenotype but also other neurological phenotypes such as epilepsy and Parkinsonism that were previously suggested to be caused by ADK deficiency. Therefore, we conclude that the CNV in the ADK region could result in susceptibility to the phenotypes described in this case.
Methods
Sample and Clinical Assessments
In our previous genome-wide CNV analysis using high-resolution oligonucleotide array comparative genomic hybridization (Kushima et al., 2017), we identified a SCZ patient with deletion in the ADK region (Figure 1A) among Japanese cohort samples comprising 1699 SCZ and 824 controls (CON). The details of the patients’ profiles and the method of the CNV analysis are available elsewhere (Kushima et al., 2017). This study protocol was approved by the Ethics Committees of the Nagoya University Graduate School of Medicine and other participating institutes and hospitals.
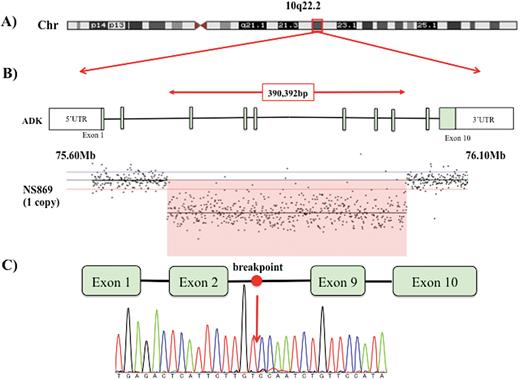
The region of the ADK deletion and the result of the breakpoint search. (A) High-resolution array comparative genomic hybridization data of the schizophrenia (SCZ) case with the adenosine kinase (ADK) deletion. The orange box (NCBI36/hg18) represents the copy number variants (CNV) detected in the present study. Results revealed a loss on the long arm of chromosome 10q22.2. (B) Information about the transcript isoform and genomic coordinates corresponding to transcript ID ENST00000541550.5; human genome GRCh38.p7. Red arrows represent the deleted exons of ADK (Chr10: 74292670-chr10: 74623062). The exonic region of ADK deletion in this study encompasses exons 3 to 8. (C) Breakpoint search by Sanger sequencing. The position of the breakpoint is marked with a red arrow.
The Search for the Breakpoint for the Deletion at the Site of ADK
We performed PCR to search for the breakpoint of the deletion. We designed forward and reverse primers that aligned with the region of the breakpoint (supplementary Figure 1). Genomic DNA was extracted from peripheral blood using standard methods, and amplicons were generated using standard PCR conditions. After PCR amplification, aliquots of PCR products were purified using Illustra Exonuclease I and alkaline phosphatase (GE Healthcare & Life Science) and sequenced using the Sanger method and a 3130XL Genetic Analyzer (Applied Biosystems).
mRNA Expression Analysis
To investigate the effect of deletion of ADK mRNA transcripts, we compared the relative expression of ADK in 32 SCZ patients (44.5 ± 11.5 years of age), including a SCZ patient with deletion of ADK, with 29 healthy controls (43.9 ± 11.4 years of age) using lymphoblastoid cell lines (LCLs). LCLs were established by Epstein-Barr Virus transformation of lymphocytes and cultured in RPMI-1460 medium containing 20% fetal bovine serum, penicillin, and streptomycin. Total RNA was extracted from LCLs using the RNAqueous kit (Invitrogen), treated with DNase using the TURBO DNA-free kit (Invitrogen), and then reverse transcribed to cDNA with the high-capacity RNA-to-cDNA kit (Invitrogen). Two housekeeping genes, beta-2-microglobulin (B2M) and glucuronidase-beta (GUSB), were selected as internal control genes to normalize the PCR. Real-time quantitative PCR was performed with predesigned probes from the TaqMan Gene Expression Assay (Hs01097293_ml for ADK, Hs99999907_ml for B2M, and Hs99999908_ml for GUSB; Applied Biosystems) using Applied Biosystems 7900HT. Measurement of the cycle threshold was performed in duplicate. The data, including the amplifying efficiency and relative quantitative expression, were analyzed using the 2−ΔΔCT method (Livak and Schmittgen, 2001). The Mann-Whitney U test was used for the comparison of expression levels of ADK between SCZ patients and controls, because this test is robust when deviation from normal distribution is present. P < .05 (the P value of directional test) was considered significant.
Results
Search for the Breakpoint of the CNV Deletion
We validated the CNV in the region of ADK to evaluate the pathophysiology of the SCZ patient in detail. The results of the breakpoint search are presented in Figure 1 and supplementary Figure 1. The CNV regions were chr10: 74292670-chr10: 74623062 according to the ensemble database (transcript ID ENST00000541550.5; human genome GRCh38.p7), and the deleted length was 390392 bp. The deleted mRNA was predicted to result in a frame shift because exon 3 to exon 8 of ADK were deleted (supplementary Figure 2).
mRNA Expression Analysis
We analyzed ADK mRNA expression in peripheral blood (Figure 2). We found no significant association between ADK relative expression in the 31 SCZ and 29 CON. However, the mRNA transcript level of the patient with ADK deletion was lower than the first quartile of the 31 SCZ patients (Figure 2).
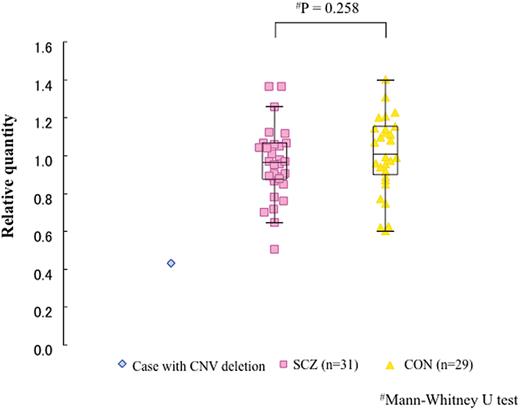
The results of expression analysis with a patient of ADK deletion. Boxplot; box represents the middle 50% of observations. The middle bold line represents the median gene expression. Whiskers represent the minimum and maximum observations. Each dot represents the relative expression of an individual sample calculated by the 2−ΔΔCT method.
Case Presentation of the Patient with ADK Deletion
This patient was a 64-year-old Japanese female at the time of study recruitment. She was diagnosed with SCZ at 38 years of age based on auditory hallucinations, persecutory delusions, and psychomotor excitation. Her mother was also diagnosed with SCZ. Her developmental milestones were normal. After she graduated from junior high school, she worked in manufacturing. She was married and had 3 children. When she was 38 years old, she suffered from persecutory delusions and auditory hallucinations and was then diagnosed with SCZ, hospitalized, and started medication treatment with antipsychotics. An electroencephalogram abnormality was discovered during her first hospitalization, but she was not diagnosed with epilepsy at that time. She was hospitalized repeatedly after failing to report to the mental hospital and for discontinuing her medications. She was hospitalized for a long time after she was 52 years old, because she was abandoned by her family. Her psychotic symptoms were improved by small amounts of antipsychotics such as 2 mg risperidone. When she was 53 years old, she suffered from short-stepping gait, finger tremors, bradykinesia, and a dexterity movement disorder despite a reduction in the amount of risperidone. She was diagnosed with Parkinson’s disease and prescribed l-dopa. When she was 63 years old, she suffered from an episode of unconsciousness and was diagnosed with epilepsy after electroencephalography, magnetic resonance angiography, and brain magnetic resonance imaging. She was then prescribed the antiepileptic drugs valproic acid and clonazepam. However, the seizure discharges were not detected by electroencephalography. When she was 73 years old, she suffered from sepsis with unknown etiology despite several diagnostic tests.
Discussion
To the best of our knowledge, this is the first case report of an SCZ patient with a loss of function variant in the ADK region identified in a whole-genome CNV study in a Japanese cohort of samples comprising 1699 SCZ and 824 controls (Kushima et al., 2017). The search for the breakpoint revealed that 6 exons (from exon 3 to exon 8) were deleted and that deletion of ADK is a result of the loss of function variant (frame shift). The protein resulting from this deletion may be a nonsense protein that is degraded. Considering that ADK expression in this patient with ADK deletion was low (Figure 2) and that the pattern of expression in the Human Transcriptome database (http://hbatlas.org/) (Kang et al., 2011) indicates that ADK is highly expressed from the posterior brain regions in the human fetus to the adult human brain (supplemental Figure 3), ADK in this SCZ patient may not have been expressed from the early fetal period. Furthermore, considering that ADK had a high haploinsufficiency score (3.41) in the DECIFER database (Firth et al., 2009; Huang et al., 2010), the ADK protein may not be fully functional in this schizophrenic patient.
Interestingly, this patient with ADK deficiency presented with several phenotypes such as SCZ, epilepsy, and Parkinsonism. Therefore, considering the loss of function mutation and the reduced expression of ADK mRNA, the phenotypes described here may have arisen in part because of this deletion in the region of ADK. The patient did not have a clear phenotype related to liver dysfunction as a former study about ADK deficiency showed (Staufner et al., 2016).
Our study has several limitations. First, because this variant is rare, detecting a statistical association in the loss of function variant of ADK will be difficult. Second, we did not perform biochemical assays with samples from this patient with ADK deletion to evaluate the ability of the remaining ADK enzyme to convert adenosine to adenosine monophosphate (Staufner et al., 2016). Third, we did not have sufficient DNA from family members of the patient, which prevented us from analyzing segregation of the variant. Fourth, in the case summary of this carrier with the ADK deletion, we were unable to obtain detailed clinical information for the developmental period and electroencephalography data; therefore, we could not fully evaluate the effect of the discovered rare mutation.
In conclusion, we identified and reported a case with a loss of function variant of ADK with the same phenotypes predicted by a study of adenosine (Boison et al., 2012). For future studies, we should collect many more cases with the loss of function variant of ADK to subtype and elucidate the etiopathophysiology of this neuropsychiatric disorder.
Statement of Interest
Dr. N. Ozaki has received research support or speakers’ honoraria from, or has served as a consultant to, Astellas, Dainippon Sumitomo, Eisai, Eli Lilly, Glaxo Smith Kline, Janssen, Meiji Seika Pharma, Mochida, MSD, Nihon Medi-Physics, Novartis, Ono, Otsuka, Pfizer, Shionogi, Takeda, Taisho, Tanabe Mitsubishi, Teijin, Tsumura, and, Yoshitomi.
Acknowledgments
We are grateful to all the patients, their families, and control individuals who contributed to this study. We thank Yukari Mitsui, Mami Yoshida, and Hiromi Noma for their technical assistance, discussion, and contributions to creating and managing the database.
This work was supported by research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; the Ministry of Health, Labour, and Welfare of Japan; the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development (AMED); the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) project of AMED; Grant-in-Aid for Scientific Research on Innovative Areas “Glial assembly: a new regulatory machinery of brain function and disorders”; and Innovative Areas “Comprehensive Brain Science Network.”

{kind=link}
{kind=link}