




Abstract
Mutations in ZIC3 cause X-linked heterotaxy, a disorder characterized by abnormal lateralization of normally asymmetric thoracic and abdominal organs. Animal models demonstrate an early role for ZIC3 in embryonic left–right (LR) patterning. ZIC3 mutations have also been described in patients with isolated cardiovascular malformations. We wished to address the hypothesis that ZIC3 has plieotropic effects in development and may regulate cardiac development independent of its role in LR patterning. We observed significantly reduced expression of several markers of cardiac lineage commitment in Zic3 null/y embryonic stem cells including atrial natriuretic factor (ANF), Nkx2.5 and Tbx5. Likewise, ANF expression—a molecular marker of trabecular myocardium and a direct target of multiple cardiac-specific transcription factors—was severely reduced in E9.5 Zic3 null hearts. Trabecular myocardium was reduced in these embryos. This finding was similar to that observed in embryos with cardiac-specific ablation of serum response factor (SRF), a direct transcriptional regulator of ANF expression. While ZIC3 by itself had no effect on the ANF promoter, it could bind to and inhibit a cardiac α-actin promoter through its zinc finger domains. We observed that ZIC3 could function as a coactivator of SRF on both cardiac α-actin and ANF promoters. The zinc fingers of ZIC3 and the mcm1, agamous deficiens SRF (MADS) box motif of SRF were found to mediate their physical and functional interactions. These findings reveal a novel role of ZIC3 in regulating cardiac gene expression and may explain, in part, the association of ZIC3 mutation with cardiovascular malformations.
INTRODUCTION
Cardiovascular malformations are among the most common abnormalities of human development and are the most common cause of infant mortality attributable to birth defects ( 1 , 2 ). It has become increasingly clear that mutations in developmental control genes are responsible for some cardiac defects once thought to have multifactorial etiologies ( 1 ). Mutations in ZIC3, an X chromosome encoded zinc finger transcription factor, cause X-linked heterotaxy (HTX1, MIM 306955). ZIC3 is one of a small number of genes associated with isolated or non-syndromic cardiovascular malformations in humans. To date, five nonsense, five missense and one frameshift ZIC3 mutations have been identified in subjects with heterotaxy or isolated complex cardiovascular malformations. Although the majority of the ZIC3 mutations have been associated with X-linked heterotaxy, a syndrome best explained by abnormal left–right (LR) patterning during early embryogenesis, three ZIC3 mutations have been identified in patients with isolated congenital heart defects including transposition of the great arteries, atrial septal defect, ventricular septal defect and pulmonary valve stenosis ( 3–5 ). The mechanisms by which normal cardiac development is altered by ZIC3 mutations are unknown. It is possible that ZIC3 is required in early steps of embryogenesis, including LR patterning, and that cardiac defects are a consequence of imbalances in a secondary cascade of cell growth and differentiation defects. Alternatively, ZIC3 may have multiple roles in development and might directly affect cardiogenesis. The finding of ZIC3 mutations in patients with isolated cardiovascular malformations led us to examine the hypothesis that it may play a role in cardiac development independent of the LR patterning.
Murine Zic3 is expressed in the newly formed mesoderm at early gastrulation. As gastrulation proceeds, the pattern of Zic3 expression remains relatively constant, with transcripts located in the embryonic mesoderm, primitive streak and adjacent ectoderm ( 6 ). Zic3 deficiency in mice typically gives a phenotype that is strikingly similar to human heterotaxy ( 7 ), but a wide range of phenotypic variation from abnormal early gastrulation to mildly kinked tail in otherwise healthy adults is also observed. Ware et al . ( 8 ) recently demonstrated that Zic3 can play a role in mesoderm differentiation and migration. No specific role for ZIC3 in precardiac mesoderm or in committed cardiac lineages has been defined, however.
Serum response factor (SRF) belongs to an ancient DNA binding protein family that shares a highly conserved DNA-binding/dimerization domain containing 90 amino acids termed the MADS box ( 9 ). SRF is required for embryonic mesoderm formation in mouse and Xenopus models ( 10 , 11 ). SRF null mutants fail to fully develop mesoderm ( 12 ), giving an embryonic morphology superficially similar to that found in a much smaller percentage of Zic3 null embryos ( 8 ). Conditional inactivation of SRF in the developing heart of mice led to significantly downregulated atrial natriuretic factor (ANF) expression. The mutant embryos also displayed poor trabeculation and thinning of the ventricular compact layer ( 13 ). Over 70 novel SRF gene targets were recently reported and many of these genes have been shown to play important roles in mesoderm formation and cardiovascular development ( 14 ).
Numerous studies show that SRF acts as a versatile protein that binds to the serum response element (SREs) with a consensus sequence CC(A/T) 6 GG. SREs occur in a multitude of promoters apparently acting as a platform to recruit accessory transcriptional factors and ultimately altering specific gene expression programs ( 15–20 ). Cardiac α-actin promoter and ANF promoter are two heart-specific promoters that contain combinations of SREs with strong and weak affinities that bind SRF in a highly cooperative manner. Other cardiac regulatory proteins, including Nkx2.5 and GATA4, are recruited to multiple SREs in the cardiac α-actin promoter by SRF, allowing the formation of higher ordered DNA binding complexes and leading to greatly enhanced SRF transactivation ( 15 , 16 ).
In the present study, we observed dramatically decreased expression levels of Nkx2.5, Tbx5, ANF and Nkx2.3 in Zic3 −/y ES cells. The expression level of SRF remained unchanged in the absence of Zic3. Absent or scant expression of ANF in developing heart and reduced trabecular myocardium were observed in E9.5 Zic3 null embryos without heart looping defects. ZIC3 could bind to multiple sites in the cardiac α-actin promoter and physically interact with SRF through the ZIC3 zinc finger domains. ZIC3 by itself had an inhibitory effect on the cardiac α-actin promoter and no effect on ANF promoter, but could synergistically activate both the cardiac α-actin and ANF promoters with SRF. Using SRF deletion constructs, the SRF–ZIC3 interaction domain in SRF was mapped to its MADS box. Investigation of the ZIC3 mutants identified in patients with cardiovascular malformations suggests that both N′-domain and zinc finger domains in ZIC3 are required for ZIC3–SRF functional interaction, while the C′-domain functions as an inhibitory domain. These studies indicate a potential novel role of ZIC3 in regulating cardiac development.
RESULTS
Zic3 is weakly expressed in embryonic heart tissue and consequences of Zic3 deficiency for cardiac lineage-specific gene expression
Zic3 is known to be expressed in the mesoderm during gastrulation; and in the dorsal central nervous system and somites at later stages ( 6 , 21 , 22 ). Cardiac-specific expression of Zic3 appeared weak in our previous in situ hybridization studies. Whether it is expressed in specific domains of the developing heart is unknown. We used quantitative RT–PCR to examine the expression of Zic3 in the embryonic heart. Cardiac tissue was carefully dissected from E10.5 wild-type embryos and RNA was extracted from this material and from the remaining parts of the embryos. The extracted RNA from Zic3 null/y embryos served as a negative control. As shown in Figure 1 A, Zic3 is expressed in the cardiac tissue of wild-type embryos, however, the expression level is low when compared with that found in the non-cardiac portion of the embryo.
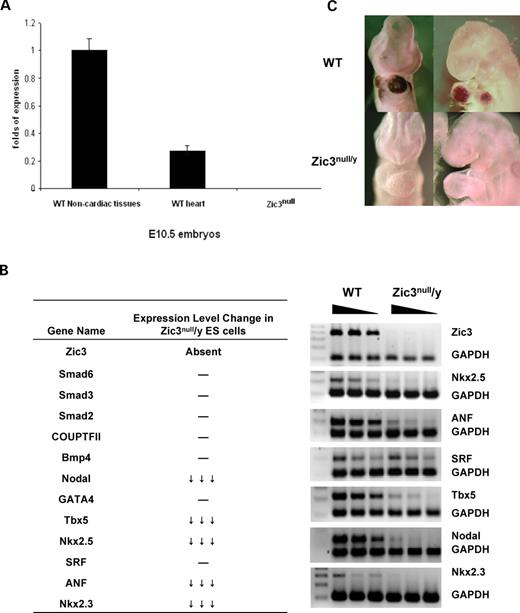
Expression analyses. ( A ) Expression of Zic3 in E10.5 mouse embryos. RNA was extracted from dissected and pooled cardiac tissue and the remaining embryonic tissue. The results were normalized by reference to GAPDH analyzed in parallel for each sample. ( B ) Expression of candidate growth factors and transcription factors in Zic3 null ES cells. Wild-type and Zic3 null/y ES cell cDNA (25, 50, 75 ng) was used for semi-quantitative RT–PCR. Relative gene expression levels were obtained by normalization to GAPDH. The left table summarizes expression levels of all genes tested and the right panel shows the semi-quantitative RT–PCR results of selected genes. ( C ) Embryonic expression of ANF. The top two panels are the in situ hybridization results of wild-type embryos and the lower two panels are the in situ hybridization results of Zic3 null/y embryos.
Murine embryonic stem (ES) cell cultures are a model system widely used for investigation of the function of developmental genes in embryogenesis. Figure 1 B shows that Zic3 was expressed in undifferentiated ES cells. In Zic3 null/y ES cells, Zic3 expression was absent. We wished to use these cells as a limited in vitro model for gene expression alterations that might occur as a consequence of Zic3 deficiency. Many loci that play critical roles in early cardiac development are expressed in ES cell cultures. Aberrations in ES cell gene expression would help to formulate hypotheses about which pathways are actually impacted in the early embryo. Expression levels of markers of early cardiac lineage differentiation, including transcription factors SRF, GATA4, Coup-TFII, SMAD2, SMAD3, SMAD6 and BMP4, were unaffected by the absence of Zic3 (Fig. 1 and Supplementary Material, Fig. S1). However, significant reduction in ANF (>90%), a direct SRF target, was observed in Zic3 null/y ES cells, as well as three SRF direct interaction partners important for cardiogenesis, Nkx2.5 (>90%), Tbx5 (>85%) and Nkx2.3 (>90%). We also observed reduced transcription of Nodal, a downstream target of Zic3 ( 23 ).
In order to evaluate whether embryonic expression of these genes is altered by Zic3 deficiency, we investigated several of them in the developing hearts of Zic3 mutants. Using whole-mount in situ hybridization, we observed that ANF expression was absent or scant in E9.5 Zic3 null embryos (Fig. 1 C). Previous studies have shown that there are an Nkx2.5 binding site (NKE), two GATA sites, a T-box binding site (TBE) and two SRF binding sites (SREs) in the ANF promoter region, and the ANF gene is regulated by combinatorial interactions between Nkx2.5, Gata4, Tbx5 and SRF (see review in ( 24 )). To examine whether the absent expression of ANF in Zic3 null embryos could be correlated with decreased cardiac expression of Tbx5, Nkx2.5 or GATA4, in situ hybridization was performed. The expression of Tbx5, Nkx2.5 and GATA4 in cardiac crescent or heart looping stage Zic3 null embryos appeared normal (data not shown).
Reduced trabecular myocardium and thinner myocardium in Zic3 null embryos
ANF is a molecular marker for trabecular myocardium, as it is strongly expressed in the trabecular layer of the left ventricle and weakly expressed in the atrium and the trabecular layer of the right ventricle ( 25 ). We wished to determine whether lack of ANF expression in the developing heart of E9.5 Zic3 null embryos was due to the absence of trabecular myocardium. Examination of the transverse sections of E9.5 wild-type and Zic3 null/y embryos with no heart looping defects revealed less trabecular myocardium in the ventricular walls of the mutant (Fig. 2 ). The breadth of the ventricular wall in the mutant was slightly reduced and the ventricular chamber was dilated, compared with the wild-type.
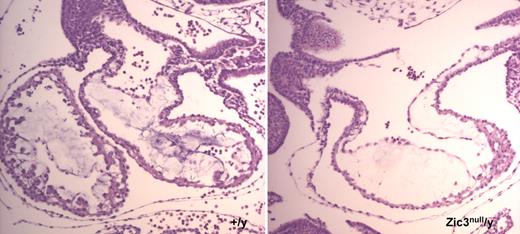
Reduced trabecular myocardium and thinner myocardium observed in a Zic3 null embryo. Transverse sections of E9.5 embryos were hemotoxylin and eosin-stained.
ZIC3 binds to and inhibits the cardiac α-actin promoter through the zinc finger domains
Since thinner trabecular myocardium was observed in Zic3 null/y embryos compared with the wild-type, we reasoned that it was likely that ZIC3 directly or indirectly regulated the cardiac α-actin promoter. We scanned the cardiac α-actin promoter region and detected multiple potential GLI consensus binding sites. Zic3 has previously been reported to bind to the GLI consensus binding site ( 26 ). Here, we used electrophoresis mobility shift assay (EMSA) and luciferase reporter assays to investigate the interaction between ZIC3 and the cardiac α-actin promoter. The result of the EMSA assay, shown in Figure 3 A, demonstrated that wild-type ZIC3 bound to multiple regions in the cardiac α-actin promoter, including a 60 bp element that also contains the SRF binding site (SRE4). The Q249X, a nonsense mutant lacking all five zinc finger domains, was detected in an individual with congenital heart defects ( 5 ). The mutant protein did not bind to the probe, demonstrating that zinc finger domains are required for efficient binding to the cardiac α-actin promoter in this assay.
![ZIC3 binds to and inhibits cardiac α-actin promoter through its zinc finger domains. ( A ) The left panel shows the sequence of the cardiac α-actin promoter region. The potential Zic3 binding elements with high similarity to GLI consensus binding sites are indicated in gray. The solid rectangular shows the sequence of probe 1, and the dotted rectangle shows the sequence of probe 2. The right upper panel shows the EMSA assay performed with γ- 32 P ATP 5′ end-labeled probes and in vitro translated unlabeled wild-type and mutant ZIC3 proteins. The right lower panel shows the input proteins. ( B ) Luciferase reporter assays. The luciferase activity was assessed by cotransfecting 100 ng of cardiac α-actin promoter-luciferase construct with 100 ng wild-type or mutant ZIC3 expression plasmids. Firefly luciferase activities were normalized to Renilla luciferase activity. Data shown are expressed as mean ± SD. ( C ) EMSA assay. Input proteins were simultaneously generated by in vitro transcription and translation assay using [ 35 S]methionine labeling. The expected band is marked. A schematic diagram at the bottom of the figure shows wild-type ZIC3 and a series of nonsense and missense ZIC3 mutants.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/16/14/10.1093_hmg_ddm106/2/m_ddm10603.jpeg?Expires=1716610684&Signature=toplriGsiC4UHqb-TRPCNkiR6rXUGrHCyZoBrBjf5bynBaHTGEZCUnzTuZhFoVEG6hrH8ti6CNig9~AieYNE9WfVVAufN6fwbQeaEXNTa92S5EYwtLxQ0UjHI2DfA1gVvCSsEv2zXmpZCX7Knp1ryMSR6jWHFvMTG9YXpbDgFUaf-oVpC5m58JJK5f0ILO7CWeBJwVOWLSildXYiKcRRBai640OOF1VESLWG0yRvKHtUJP-NxW6QEAvkK58jTTrf3KWmlHPUs8VeIkcuaHPTC9lNk4p78shds~Cw5WyYa3K8VwqyNtCSN~vq9isu0knZMKULOQ4TMDxiwquStnXwdQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
ZIC3 binds to and inhibits cardiac α-actin promoter through its zinc finger domains. ( A ) The left panel shows the sequence of the cardiac α-actin promoter region. The potential Zic3 binding elements with high similarity to GLI consensus binding sites are indicated in gray. The solid rectangular shows the sequence of probe 1, and the dotted rectangle shows the sequence of probe 2. The right upper panel shows the EMSA assay performed with γ- 32 P ATP 5′ end-labeled probes and in vitro translated unlabeled wild-type and mutant ZIC3 proteins. The right lower panel shows the input proteins. ( B ) Luciferase reporter assays. The luciferase activity was assessed by cotransfecting 100 ng of cardiac α-actin promoter-luciferase construct with 100 ng wild-type or mutant ZIC3 expression plasmids. Firefly luciferase activities were normalized to Renilla luciferase activity. Data shown are expressed as mean ± SD. ( C ) EMSA assay. Input proteins were simultaneously generated by in vitro transcription and translation assay using [ 35 S]methionine labeling. The expected band is marked. A schematic diagram at the bottom of the figure shows wild-type ZIC3 and a series of nonsense and missense ZIC3 mutants.
Luciferase reporter assays were used to investigate the functional consequences of the physical interaction between ZIC3 and the cardiac α-actin promoter. In this experiment, we took advantage of the mutations found in patients with cardiovascular malformations. Figure 3 B shows that wild-type ZIC3 inhibits basal transcription of the cardiac α-actin promoter, compared with the S43X mutant [served as protein negative control ( 5 )]. The Q249X, C268X, Q292X and 1477insTT mutants lack three, four or five zinc finger domains, respectively. Their transactivational activities on the cardiac α-actin promoter were similar to the S43X mutant, i.e. no inhibition was observed. In contrast, the P217A mutant, which is a missense mutation located in the N′-terminal region, retained the ability to inhibit the cardiac α-actin promoter. These observations implied that zinc finger domains also played an important role in the inhibitory effect of ZIC3 on the cardiac α-actin promoter. We performed EMSA assays to examine whether this inhibitory effect of ZIC3 correlated with its binding ability to the cardiac α-actin promoter in vitro . We found that Q249X, C268X, Q292X and 1477insTT mutants all failed to bind the cardiac α-actin promoter probe, while P217A mutant retained the binding ability (Fig. 3 C).
ZIC3 physically interacted with SRF and synergistically activated cardiac α-actin promoter and ANF promoter with SRF
Previous studies have shown that SRF can activate the cardiac α-actin promoter ( 15 ) through its SREs. Specific cardiac myocyte ablation of SRF resulted in reduced trabecular myocardium ( 13 ), similar to that observed in Zic3 null/y embryos. We asked whether ZIC3 and SRF can physically associate with each other. In vitro -synthesized [ 35 S]methionine-labeled ZIC3 protein was used to examine its binding ability to (glutathione-S transferase) GST–SRF. The results, shown in Figure 4 A, suggest that ZIC3 has a strong, specific interaction with SRF. We also performed GST pull-down assay using GST–ZIC3 and in vitro -synthesized [ 35 S]methionine-labeled SRF protein. The results again confirmed that ZIC3 could physically interact with SRF (Fig. 5 B). We also tested the binding ability of ZIC3 to other cardiac-specific transcription factors interacting with ANF or cardiac α-actin promoters, including GATA4, Nkx2.5 and dHAND, by GST pull-down assay. ZIC3 did not physically interact with them in this assay (Fig. 4 B).
![ZIC3 physically interacts with SRF and synergistically activates cardiac α-actin and ANF promoters with SRF. ( A ) GST–SRF pull-down assay. In vitro -translated [ 35 S]methionine-labeled wild-type ZIC3 was incubated with GST or GST–SRF fusion protein immobilized on glutathione-agarose beads. ( B ) Additional GST pull-down assays. In the top panel, in vitro -translated 35 S-ZIC3 was incubated with GST, GST–Nkx2.5, GST–dHAND and GST–GLI3 (547–639) fusion proteins immobilized on glutathione-agarose beads, respectively; while in the lower panel, in vitro -translated 35 S-GATA4 was incubated with GST or GST–ZIC3. For all GST pull-down assays, the beads were washed extensively, and the bound proteins were resolved on 4–12% Bis–Tris gel and visualized by autoradiography. In these studies, GST–GLI3 (547–639) is served as a positive control. ( C ) Co-activation assay. HeLa cells were cotransfected with or without 100 ng SRF expression vector (pCGN-SRF), 100 ng cardiac α-actin promoter-luciferase reporter construct and with and without ZIC3 expression vector (+, 100 ng and ++, 200 ng). ( D ) HeLa cells were cotransfected with 100 ng PCGN-SRF, 100 ng ZIC3 expression vector and 100 ng ANF-luciferase reporter construct. Firefly luciferase activities were normalized to Renilla luciferase activity. Data shown are expressed as mean ± SD.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/16/14/10.1093_hmg_ddm106/2/m_ddm10604.jpeg?Expires=1716610684&Signature=Ik8Y0sftHIjLHZKzbuitjoF1J3rwrQgVFsLpUpIPkwmZBa3OkwQiSHHhkZ-OokuqPFX9KAKP7IKIrFrkq9rZI4TGgpvQz6UZw5YnRqFXHJhW8djEybV7ZkwXA1V1S6g~0v7V4aTPqC0-IDxm417MArUgZhosmgRPC34oloI8DIhPM9STFnJQhPZKg0Kajin2Zgez-JbqFmFkNLeQs833uEPUjrn7MLpT8dZGhBg2kCi87soF3kr~y6pXv64Db5hm8g-l3WKdZ0o8BNX10pGQjD34hPkSh5fTOUH3aT7PVHxn9F27Qtww9LcCQ2ltCRBfHbajjFIkBBGTj59Z1fWd2w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
ZIC3 physically interacts with SRF and synergistically activates cardiac α-actin and ANF promoters with SRF. ( A ) GST–SRF pull-down assay. In vitro -translated [ 35 S]methionine-labeled wild-type ZIC3 was incubated with GST or GST–SRF fusion protein immobilized on glutathione-agarose beads. ( B ) Additional GST pull-down assays. In the top panel, in vitro -translated 35 S-ZIC3 was incubated with GST, GST–Nkx2.5, GST–dHAND and GST–GLI3 (547–639) fusion proteins immobilized on glutathione-agarose beads, respectively; while in the lower panel, in vitro -translated 35 S-GATA4 was incubated with GST or GST–ZIC3. For all GST pull-down assays, the beads were washed extensively, and the bound proteins were resolved on 4–12% Bis–Tris gel and visualized by autoradiography. In these studies, GST–GLI3 (547–639) is served as a positive control. ( C ) Co-activation assay. HeLa cells were cotransfected with or without 100 ng SRF expression vector (pCGN-SRF), 100 ng cardiac α-actin promoter-luciferase reporter construct and with and without ZIC3 expression vector (+, 100 ng and ++, 200 ng). ( D ) HeLa cells were cotransfected with 100 ng PCGN-SRF, 100 ng ZIC3 expression vector and 100 ng ANF-luciferase reporter construct. Firefly luciferase activities were normalized to Renilla luciferase activity. Data shown are expressed as mean ± SD.
![Mapping domains in SRF and ZIC3 required for their physical interaction. ( A ) GST pull-down assay was performed using in vitro translated 35 S-labeled ZIC3 mutants and GST or GST–SRF fusion protein immobilized on glutathione-agarose beads. The input proteins generated by in vitro transcription and translation assay internally labeled by incorporation of [ 35 S]methionine. The expected band is marked. ( B ) GST pull-down assay was performed using in vitro translated 35 S-labeled wild-type SRF protein and GST or GST–ZIC3 or GST–ZIC3ΔN fusion protein. ( C ) In vitro translated 35 S-labeled wild-type ZIC3 protein was incubated with various SRF deletion fusion GST proteins and subsequent GST pull-down assay was performed. A schematic diagram at the bottom of the figure shows the structure of SRF protein. MADS box is indicated by light gray color.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/16/14/10.1093_hmg_ddm106/2/m_ddm10605.jpeg?Expires=1716610684&Signature=aeFRvrSrSTVjsmOiTOS4iwYCEl43~gilFgPL-C~oSdlnUoSm~6Nn6pNiNqEaFby5pnvpd1k1JQHVzmYJMX1WeZl5HiWPK3z4u0l9GuSbw-zAVoCTV8--JG4oGhq1bia2nQTv~nTuX37Efk3wacC8zalr9Fw8DrYHdEF8YRhO8Z4aCl2A2jYTE2KN~qMjcOWfZ3KQ22e0DQ-iMjLinM3eDzgWKLseoUK8Sw24TPhNbkMjaFWtlgvKFrmnlN0PQcP-P2-1XYDVagvxdG9nqTzqQW1P36yUlFCGPEAjb-4eWMihiFGzmr5a~GVgYuc~uKFefRAs46m31HedQqf7rBv1Zw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Mapping domains in SRF and ZIC3 required for their physical interaction. ( A ) GST pull-down assay was performed using in vitro translated 35 S-labeled ZIC3 mutants and GST or GST–SRF fusion protein immobilized on glutathione-agarose beads. The input proteins generated by in vitro transcription and translation assay internally labeled by incorporation of [ 35 S]methionine. The expected band is marked. ( B ) GST pull-down assay was performed using in vitro translated 35 S-labeled wild-type SRF protein and GST or GST–ZIC3 or GST–ZIC3ΔN fusion protein. ( C ) In vitro translated 35 S-labeled wild-type ZIC3 protein was incubated with various SRF deletion fusion GST proteins and subsequent GST pull-down assay was performed. A schematic diagram at the bottom of the figure shows the structure of SRF protein. MADS box is indicated by light gray color.
We next tested the functional interaction between ZIC3 and SRF using luciferase reporter assays using a cardiac α-actin promoter construct as a model system. As noted above, ZIC3 alone inhibited and, as previously reported, SRF activated the cardiac α-actin promoter ( 15 ). ZIC3 combined with SRF synergistically activated the cardiac α-actin promoter reporter construct, although a higher amount of ZIC3 resulted in the reduction of transactivation (Fig. 4 C).
Although the trabecular myocardium is thinner in Zic3 null/y embryos, ANF expression is virtually absent. Given the observation that the ANF expression was also dramatically decreased in Zic3 null/y ES cells, we speculated that the absent or scant expression of ANF in the developing heart of mutant embryos was not a sole secondary effect of the reduced trabecular myocardium but might be explained by a more direct effect. Previous studies have indicated that SRF activates the ANF promoter through two SRF binding sites ( 27 ). We asked whether ZIC3 could activate ANF promoter with and without SRF. As shown in Figure 4 D, ZIC3 alone did not affect ANF promoter-controlled reporter expression, but could synergistically activate the ANF promoter with SRF. This result suggests that ZIC3 can increase the expression level of ANF through co-operation with SRF.
Mapping the domains for SRF and ZIC3 physical interaction
To begin the process of mapping the domains in ZIC3 involved in the physical interaction with SRF, various in vitro -synthesized [ 35 S]methionine-labeled ZIC3 deletion mutants found in human patients with cardiovascular malformations were used to examine their binding ability to bind GST–SRF. Figure 5 A shows that Q249X which lacks all the zinc finger domains and only contains the N′-terminal domain retains binding to SRF in vitro . Other ZIC3 mutants lacking some of the zinc finger domains also interacted with SRF. In order to investigate whether the zinc finger domains in ZIC3 are also required for binding to SRF, a GST fusion protein without N′-terminal (GST–ZIC3ΔN) was generated and incubated with in vitro -synthesized [ 35 S]methionine-labeled SRF protein. After extensive washings, the bound material was eluted and analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) and autoradiography. As shown in Figure 5 B, the GST–ZIC3ΔN can also bind to SRF, suggesting the existence of multiple SRF-binding domains in ZIC3.
The minimal interactive motifs in SRF required for physical interaction with ZIC3 were mapped by evaluating the avidity of various GST–SRF deletion mutant proteins for pull-out of the in vitro -synthesized [ 35 S]methionine-labeled ZIC3 protein. As shown in Figure 5 C, deletions in the N′- and C′-terminals of SRF (amino acids 1–142; amino acids 1–171; amino acids 142–245; amino acids 142–171) did not compromise the ability of SRF to interact with ZIC3. In contrast, deletion of the MADS box of SRF (Δ46-244) resulted in loss of the physical interaction with ZIC3. A sub-domain of the MADS box, containing part of the α-I helix and its N-terminal extension (amino acids 142–171), was necessary and sufficient for binding ZIC3. GST alone, as a negative control, did not interact with ZIC3. These experiments show that the MADS box is required for ZIC3–SRF interaction in vitro .
Both zinc finger and N′-terminal domains in ZIC3 are required for the synergistic activation with SRF on cardiac α-actin promoter, while the C′-terminal domain in ZIC3 serves as an inhibitory domain
Given the observation that ZIC3 physically interacted with SRF through its zinc finger domains and N′-terminal region, we asked which domains in ZIC3 were indispensable for the synergistic activation with SRF on the cardiac α-actin promoter. As shown in Figure 6 , ZIC3 lacking one or more zinc finger domains has reduced synergistic activation with SRF. No synergistic activation was observed with the Q249X mutant (almost the same activity as the S43X mutant, which serves as a control), although it could still physically associate with SRF. The number of remaining zinc finger domains are increased in the C268X, Q292X and 1478InsTT mutants, and interestingly, their synergistic activity with SRF on the cardiac α-actin promoter is also increased. The missense mutation P217A, located in the N′-terminal domain, had mildly decreased synergistic activity, compared with the wild-type ZIC3. These results suggest that both the N′-terminal region and the zinc finger domains of ZIC3 can affect its synergistic activation with SRF.
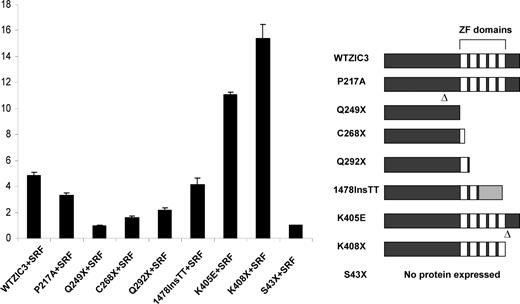
Functional interactions of various ZIC3 mutants with SRF on cardiac α-actin promoter. HeLa cells were cotransfected with 100 ng PCGN-SRF and 100 ng various mutant ZIC3 expression vectors along with 100 ng cardiac α-actin promoter-luciferase reporter construct. Firefly luciferase activities were normalized to Renilla luciferase activity. Data shown are expressed as mean ± SD. The right panel shows wild-type ZIC3 and a series of nonsense and missense ZIC3 mutant alleles.
Surprisingly, K408X and K405E dramatically activated the cardiac α-actin promoter in the presence of SRF. K408X is a nonsense mutation located in the last third amino acid of the fifth zinc finger domain, while K405E is a missense mutation located in the last sixth amino acid of the fifth zinc finger domain. It is possible that these same structures are altered due to the substitution of a basic amino acid lysine by an acidic amino acid glutamic acid in the K405E mutant. The results from these two mutants raise the possibility that the C′-terminal and the last zinc finger domains of ZIC3 may function as inhibitory domains for the co-activation of ZIC3 and SRF on the cardiac α-actin promoter. This study provides the first evidence for a potential function of the C′-terminal domain of ZIC3.
MADS box in SRF is necessary for the synergistic activation of the cardiac α-actin promoter
As shown above, the SRF MADS box was required for SRF–ZIC3 physical interaction in vitro . We asked whether this co-activation of SRF by ZIC3 was mediated by MADS box. In order to address this question, we studied the cardiac α-actin reporter activity by cotransfection of wild-type and individual SRF deletion mutant expression vectors with or without the ZIC3 expression construct. Because of the different transcriptional activation capacity of the various SRF proteins on the cardiac α-actin promoter, we used the ratio of the average luciferase activity in the presence and absence of ZIC3 as a measure of the ZIC3 effect. We observed that the ratio of reporter activity of wild-type SRF plus ZIC3 to SRF alone was above 1 (Fig. 7 ), consistent with the previous result that SRF and ZIC3 exhibit synergistic activation on the cardiac α-actin promoter. The SRF-ΔC lacks 266 amino acids–508 amino acids, but still contains the N′-terminal region and MADS box. The ratio of reporter activity of SRF-ΔC plus ZIC3 to SRF-ΔC itself is similar to the situation of wild-type SRF, i.e. above 1. However, the ratio of reporter activity of SRF-Δ46-244 plus ZIC3 to SRF-Δ46-244 itself is only 0.65, close to the control, in which an empty vector pCGN was used. This observation shows that ZIC3 and SRF can no longer co-activate the cardiac α-actin promoter when the MADS box is deleted and the protein–protein interaction is disrupted. In that circumstance, ZIC3 regains the inhibitory function on the cardiac α-actin promoter.
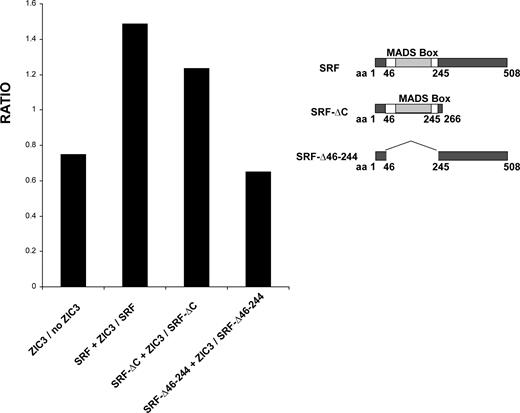
Functional interactions of various SRF deletion mutants with ZIC3 on cardiac α-actin promoter. HeLa cells were cotransfected with or without 100 ng wild-type ZIC3 expression vector and 100 ng various SRF deletion expression vectors along with 200 ng cardiac α-actin promoter-luciferase reporter construct. Firefly luciferase activities were normalized to Renilla luciferase activity. The average luciferase activity in the presence of ZIC3 was divided by the average luciferase activity in the absence of ZIC3 and the ratio plotted. The average activity was calculated from triplicates. The right panel shows wild-type SRF and a series of SRF deletion mutant alleles.
DISCUSSION
Cardiac development is controlled by spatially and temporally coordinated combinatorial interactions among various classes of transcription factors. The heart is the first organ to exhibit LR asymmetry during embryonic development and human cardiovascular malformations are closely associated with LR patterning defects ( 28 ). Both human clinical studies and Zic3 deficiency mouse models establish Zic3 as an important regulator of LR patterning during embryonic development. Its deficiency is also associated with cardiovascular malformations both in human and mouse ( 3 , 5 , 7 ). However, it remains unknown whether heart defects are the sole consequence of abnormal LR patterning or whether there is an additional function of Zic3 in heart development. Here, we showed that Zic3 is expressed in the embryonic heart and that its deficiency is associated with decreased expression of cardiac-specific genes, including Nkx2.5, Tbx5 and ANF, which are not known to be directly involved in LR patterning. Defective myocardial development as evidenced by absence of cardiac ANF expression, thin myocardium and decreased trabeculation was observed in Zic3 null embryos without heart looping defects. The combined evidence is consistent with a model in which Zic3 regulates cardiac gene expression independent of its role in LR patterning. Based on the previous reports that ANF was a direct target of SRF, as well as a molecular marker for trabecular myocardium, our data suggest that the decreased expression of ANF observed in the developing heart of Zic3 null embryos could result from a direct effect of ZIC3–SRF interaction and/or an indirect effect of thinner trabecular myocardium.
Zic3 contains five highly conserved C2H2-type zinc finger motifs and belongs to the GLI transcription factor superfamily. It can bind to the GLI-consensus DNA binding site and function as a weak transcriptional coactivator ( 26 ). Previous studies have shown that SRF activates the cardiac α-actin promoter by interaction with the SREs ( 17 ). Here, we found that multiple elements with high similarity to GLI binding sites were located in the cardiac α-actin promoter, including one close to an SRF binding site (SRE4). Further investigation revealed that ZIC3 itself can bind to the cardiac α-actin promoter through its zinc finger domains and can act as an inhibitor of the cardiac α-actin promoter in luciferase reporter assays. However, ZIC3 behaves as a transcriptional coactivator in these same assays when coexpressed with SRF. ZIC3 can also synergistically activate the ANF promoter with SRF, although it has no effect by itself. Since ZIC3 requires its zinc finger domains for binding to both the cardiac α-actin promoter and SRF protein, we hypothesize that SRF may compete with double stranded DNA for binding and we propose the following model: when the ratio of ZIC3 to SRF dosage is lower, ZIC3 favorably binds to SRF and functions as a co-activator of SRF on the cardiac α-actin promoter. In the presence of higher levels of ZIC3, excess ZIC3 molecules directly bind to the cardiac α-actin promoter and convey an inhibitory transcriptional activity. A similar phenomenon was recently observed by Acar et al . in which the Drosophila zinc finger protein Senseless inhibits gene targets at low levels of expression but at higher levels may bind to basic helix-loop-helix (bHLH) proneural proteins resulting in target gene activation ( 29 ). This was interpreted as evidence for differential affinity of the Zn-finger for binding to DNA and bHLH interacting proteins. The Drosophila neural development process appears to exploit this differential affinity to attain regulation of proneural gene expression.
SRF plays an important role in the commitment of cardiac progenitors by virtue of its indispensable role in mesoderm formation and by its ability to activate downstream target genes via specific protein–protein associations with other early cardiac-enriched transcription factors. In this study, we found that ZIC3 could physically associate with the SRF MADS box motif in vitro . The MADS box is an evolutionarily conserved domain thought to involve in various protein–protein interaction ( 30 ). For example, the physical interaction between SRF and GATA4, a C4 zinc finger transcription factor requires the SRF MADS box domain ( 15 ). We observed that the synergistic interaction between ZIC3 and SRF on the cardiac α-actin promoter was lost when the SRF MADS box was deleted, a finding consistent with previous observations that this motif is necessary for the GATA4 coactivation on this same promoter ( 15 ).
Koyabu et al . ( 31 ) showed that the zinc finger domains of ZIC3 are required for GLI-consensus site DNA binding and GLI protein interaction. The ZIC3 Q249X mutant that was identified in a family with X-linked heterotaxy, retains specific binding to SRF in vitro . Functions of the ZIC3 N-terminal domain may be mediated in part by this interaction. P217A is a missense mutation located in the N-terminal domain. We previously reported that P217A significantly increased transactivation in an SV40 promoter luciferase transcription assay ( 5 ). Here, we have shown that P217A maintains an inhibitory activity and DNA binding on the cardiac α-actin promoter. These observations further support the idea that the inhibitory effect of ZIC3 on cardiac α-actin promoter is mediated through the zinc finger domains. P217A can physically interact and cooperate with SRF to synergistically activate cardiac α-actin promoter. However, compared with wild-type ZIC3, the level of transcriptional activity is lower. These results suggest that the N-terminal domain participates in both the physical and functional interaction between ZIC3 and SRF.
The ZIC3 K408X mutant contains a premature stop codon in the last third amino acid of the last zinc finger domain and results in a mutant protein lacking the C′-terminal domain. The ZIC3 missense mutation, K405E, is characterized by the substitution of a neutral amino acid (lysine) by an acidic moiety (glutamic acid) within the fifth zinc finger domain. The consequences for the zinc finger and the C-terminal domain conformation are unknown, but the fact that they both exhibit superactivation with SRF suggests that there are important common features. Both of these mutations were found in subjects with isolated cardiovascular malformations rather than heterotaxy ( 4 , 5 ). We speculate that these two mutations result in a different pathogenesis when compared with other ZIC3 mutations. Here, we studied their functional interactions with SRF on cardiac α-actin promoter and surprisingly found that K405E and K408X have markedly enhanced SRF cooperative activity. Given the fact that all the other identified ZIC3 mutations result in decreased synergistic activity with SRF, we reasoned that the partial fifth zinc finger domain and C-terminal domain of ZIC3 functions differently from its N-terminal domain and other zinc fingers and acts with an inhibitory activity in the interaction with SRF. SRF has also been shown to activate cardiac α-actin promoter in association with other cofactors, such as Nkx2-5, GATA4 ( 15 , 16 ), which do not physically interact with ZIC3 (Fig. 4 B). It is very likely that ZIC3 is recruited by SRF and plays an indispensable role in this complex transcriptional regulatory network. Both over activation and lack of cooperative activity of ZIC3 and SRF may result in disruption of required transcriptional regulatory networks and cause cardiovascular malformations.
Ventricular trabeculation and compaction are important morphogenetic processes during cardiac development. Cardiac trabeculation initiates at E9.0, the final stage of cardiac looping. The trabecular myocardium is essential for cardiac function as demonstrated in mice that lack of trabecular myocardium die at E10.5 because of heart failure ( 32 ). We observed reduced trabeculation and thinner myocardium in Zic3 null embryos without heart looping defects and gross morphological abnormalities. Interestingly, depletion of SRF in the developing heart also resulted in a hypoplastic cardiac wall with reduced trabecular myocardium ( 13 , 33 ).
ANF is one of the direct targets of SRF. Multiple transcriptional factors, including SRF, Tbx5, GATA4 and Nkx2.5, associate with each other and regulate the ANF promoter ( 24 ). ZIC3 by itself had no effect on the ANF promoter in the transcriptional activation assay, but did function as a transcriptional coactivator with SRF. In E9.5 Zic3 null/y embryos, ANF expression is absent or barely detected, which could be a result of loss of the interaction between ZIC3 and SRF. In addition, since less trabecular myocardium is observed in the E9.5 Zic3 null/y embryos and ANF is a molecular marker for trabecular myocardium ( 25 ), the absence of ANF expression could also be a secondary effect of thinner trabecular myocardium.
In conclusion, these results provide evidence for a novel role of ZIC3 in cardiac development. This function may act directly in myocardium at a level that is independent of the LR patterning. Future studies using in vivo models of Zic3 domain-specific mutations and conditional elimination of Zic3 in cardiac mesoderm may directly address this hypothesis. The fact that ZIC3 can both physically and functionally associate with SRF in vitro suggests that it may play a role as a mediator of its activities in mesoderm and committed cardiac lineages. Future study will focus on characterization of the function of Zic3 and its interaction with SRF in precardiac mesoderm and the developing heart.
MATERIALS AND METHODS
Plasmid construct
The HA-tagged wild-type ZIC3 and various ZIC3 mutation expression vectors were constructed as previously described ( 5 ). Western blotting analysis using mouse anti-HA monoclonal antibody (Roche) confirmed the expression of HA-tagged wild-type ZIC3 and various ZIC3 mutants, except for the S43X mutant that did not make any detectable protein. A 1.4-kb Kpn 1– Dra I fragment of the human ZIC3 cDNA, encompassing the entire ORF, was subcloned in frame into the Bam HI– Eco RI sites of the pGEX-2T vector (Amersham Biosciences), to create an N-terminal GST fusion protein. A GST-tagged ZIC3 deletion construct (GST–ZIC3Δ1-252) containing all the five zinc finger domains and the C-terminal domain, as well as GST–Nkx2.5 and GST–dHAND, were constructed in the same way. cDNA corresponding to the third and fifth zinc finger domains (547–639) of GLI3 was prepared by PCR using the human GLI3 cDNAs (kindly provided by Dr. Bert Vogelstein at John Hopkins University) as template, and subcloned in frame into the Bam HI– Eco RI sites of the pGEX-2T vector. HA-tagged full-length SRF (pCGN-SRF), SRF-N (residues 1–254, including MADS box; pCGN-SRF-N), SRF-C (residues 255–508; pCGN-SRF-C), SRF-Δ46-244 (residues 45–244 were deleted, pCGN-SRF-Δ46-244), GST-fused full-length SRF and various deletion mutants of SRF, GATA4 expression vector and luciferase reporter plasmids ANF promoter-Luc, cardiac α-actin promoter-luc, have been described earlier ( 15–17 , 34 , 35 ).
ES cell culture, isolation of RNA, quantitative and semi-quantitative RT–PCR
ES cells were grown in 10 µg/ml LIF off feeder cells to ensure that the isolated RNA was originated only from the wild-type or Zic3 null /y ES cells. Mice were maintained on a 0600–2000 h light–dark cycle, with noon of the day of observation of a vaginal plug defined as E0.5. RNeasy Mini Kit (QIAGEN) was used to extract RNA from ES cells and desired embryonic tissues. cDNA was synthesized from total RNA using the SuperScript First-Strand synthesis system for RT–PCR (Invitrogen) following the manufacturers' instructions. A series dosage of wild-type and Zic3 null/y ES cell cDNA (25, 50, 75 ng) were used for semi-quantitative RT–PCR analysis for GAPDH, Zic3, SRF, Coup-TFII, Nodal, SMAD2, SMAD3, SMAD6, BMP4, Nkx2.5, Nkx2.3, ANF, Tbx5 and GATA4. GAPDH was served as a loading control. ImageJ Software was used to quantify the expression level of genes. Relative gene expression levels were obtained by normalization to the expression level of GAPDH. For the quantitative RT–PCR, all primers were optimized on the ABI 7000 SDS using SYBR Green dye incorporation. The following cycling parameters were used for quantitative PCR using ABI 7000 SDS: 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min. Expression of Zic3 was compared with an endogenous RNA control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The following PCR primers were used: Zic3 F:cgaaggctgtgacaga cggt R:catgtgcttcttgcggtcg; GAPDH F:tgtgtccgtcgtggatctga R:cctgcttcaccaccttcttga.
Whole mount in situ hybridization, histology and imaging
Embryos were collected at desired embryonic developmental stages and embryonic yolk sacs were used to extract DNA for genotyping. Probes were labeled using a DIG RNA Labeling kit (Roche). Whole-mount in situ hybridization was performed using the same protocol described ( 7 ). For histology studies, embryos were fixed in 4% paraformaldehyde in 1× phosphate-buffered saline (PBS) at 4°C for 4 h overnight, dehydrated through a series of EtOH, embedded in paraffin and sectioned at a thickness of 7 µm. Sections were stained with hemotoxylin and eosin. Images were captured using the Zeiss Axioplan II microscope.
Transfection and luciferase reporter assay
HeLa cells were maintained in DMEM with 10% fetal bovine serum in 24-well plates. All transfections were performed using lipofectamine (Invitrogen) according to the manufacturer's recommendations. Reporter constructs (100 ng) and different expression vectors if needed were co-transfected into HeLa cells, as well as a pRL-TK luciferase vector (40 ng) serving as a control for transfection efficiency. Appropriate empty expression vectors were used to keep the total amount of DNA constant per well. Cells were harvested 24 h after transfection, and luciferase activities were determined using the Dual Luciferase Reporter Assay System (Promega). Firefly luciferase activities were normalized to Renilla luciferase activity. Data shown are expressed as mean ± SD from the assay carried out in replicates.
In vitro transcription and translation assay and EMSA
In vitro translation of various cold protein products were performed with rabbit reticulocyte lysates from TNT T7 Coupled Reticulocyte Lysate System (Promega). The DNA probes were synthesized by Invitrogen and annealed to double-stranded oligonucleotides, or amplified by PCR. The GLIBS and non-GLIBS were labeled by α- 32 P dCTP by Klenow fragment DNA polymerase I (USB). The DNA probes located in cardiac α-actin promoter were 5′ end-labeled by γ- 32 P ATP using T4 polynucleotide kinase (Invitrogen). EMSA was performed in a binding buffer consisting of 25 m m HEPES, pH 7.5, 50 m m KCl, 5 m m MgCl 2 , 10 µ m ZnSO 4 , 1 m m dithiothreitol, 0.1% Nonidet P-40, 12% glycerol 32 P-labeled, double-stranded oligonucleotides, 150 ng of poly(d I –d C ) and in vitro transcription and translated ZIC3 protein. The binding reaction was resolved by 4% native polyacrylamide gel and visualized by autoradiography.
GST pull-down assay
The GST-tagged expression constructs were transformed into Escherichia coli BL-21. GST recombinant proteins were produced and were affinity-purified by glutathione-Sepharose 4B (Amersham Pharmacia Biotech) or purified using the Pierce GST purification kit according to the protocol provided by the company. 10 µg GST recombinant proteins were immobilized on glutathione sepharose 4B (Amersham Biosciences) and then incubated with 5 µl of 35 S-labeled in vitro translated proteins at room temperature in the immunoprecipitation buffer for 1 h. The beads were washed, resuspended in 1X NuPage LDS Sample Buffer (Invitrogen) and boiled for 5 min. The bound proteins was resolved by NuPAGE 4–12% Bis–Tris Gel (Invitrogen) and visualized by autoradiography.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health grants HD39056 and HL067155 (J.W.B.), American Heart Association Postdoctoral Fellowship Award 0625227Y (L.Z.) and the NIH grants and a grant from the Fondation Leducq (R.J.S.).
Conflict of Interest statement . None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}