




Abstract
Widespread provision of human embryonic stem cells (hESCs) for therapeutic use, drug screening and disease modelling will require cell lines sustainable over long periods in culture. Since the short-term, in vitro culture of mammalian embryos can result in DNA methylation changes, the epigenetic stability of hESCs warrants investigation. Existing hESC lines have been derived and cultured under diverse conditions, providing the potential for programming differential changes into the epigenome that may result in inter-line variability over and above that inherited from the embryo. By examining the DNA methylation profiles of > 2000 genomic loci by Restriction Landmark Genome Scanning, we identified substantial inter-line epigenetic distance between six independently derived hESC lines. Lines were found to inherit further epigenetic changes over time in culture, with most changes arising in the earliest stages post-derivation. The loci affected varied between lines. The majority of culture-induced changes (82.3–87.5%) were stably inherited both within the undifferentiated cells and post-differentiation. Adapting a line to a serum-free culture system resulted in additional epigenetic instability. Overall 80.5% of the unstable loci uncovered in hESCs have been associated previously with an adult tumour phenotype. Our study shows that current methods of hESC propagation can rapidly programme stable and unpredictable epigenetic changes in the stem cell genome. This highlights the need for (i) novel screening strategies to determine the experimental utility and biosafety of hESCs and (ii) optimization and standardization of procedures for the derivation and culture of hESC lines that minimize culture-induced instability.
INTRODUCTION
Human embryonic stem cells (hESCs) have generated considerable excitement as a potential new therapeutic tool for regenerative medicine. Under appropriate differentiation conditions, hESCs have the potential to make all cell types of the human body and so could be used to treat a wide range of degenerative diseases (1). Since hESCs and their derivatives represent human primary cell types, they also offer considerable advantages over the transformed cell lines currently used for disease modelling and drug screening.
Differences between hESC lines all positive for classical pluripotency markers are increasingly evident, even between lines isolated in the same laboratory using similar procedures (2). In addition to variation in doubling time (2), karyotypic stability (3) and transcriptome profile (4), lineage-specific differentiation protocols do not appear to be universally applicable (5,6). Since, within most lines, the effect of culture variables and stability over time has not been investigated, more extensive characterization is required before the potential for hESC-based applications can be realized in a safe and efficient manner.
The transcriptome profile characterizing a particular cell type is regulated in part by epigenetic modifications to DNA and associated histone proteins, in conjunction with re-modelling of chromatin structure. In particular, DNA cytosine methylation is thought to stabilize epigenetic gene silencing during lineage specification and to maintain gene silencing patterns between cell cycles (7). Regulation of gene expression can be mediated by methylation of promoter-associated CpG islands. Although the majority of CpG islands are unmethylated in all cell types, to allow the constitutive expression of housekeeping genes, a subset (designated tissue-specific differentially methylated regions or T-DMRs) have been identified as developmentally regulated in a lineage specific manner (8,9). Disrupted DNA methylation is now well established in tumourigenesis and in some assisted reproduction technologies (10). Since epigenetic reprogramming of the embryonic genome after fertilization results in dynamic changes in DNA methylation that are still ongoing in the blastocyst stage embryo from which ESCs are derived (11,12), we reasoned that culture-induced instability in DNA methylation may well confer a variable ability of hESC cells artificially maintained in the undifferentiated state to gain methylation changes in gene-related loci over time that can be programmed into the epigenome post-differentiation. Such instability could contribute to variations in phenotype, differentiation potential and genetic stability of hESCs (6,10). Thus epigenotype characterization could provide an important means of safety evaluation as a prerequisite to therapeutic applications. By examining genome-wide DNA methylation profiles of NotI sites in gene-rich CpG islands (13) using restriction landmark genome scanning (RLGS), our studies revealed substantial epigenetic variation between hESC lines that cannot be accounted for by genetic variation. Epigenetic changes within undifferentiated hESC cells were induced by culture conditions and inherited both by undifferentiated cells over time and post-differentiation. Furthermore, many of the affected genomic loci were associated with differentiation and/or have been demonstrated to change their methylation status in adult tumours.
RESULTS
Epigenetic variations between hESC lines
The degree of epigenetic variation between six independently-derived hESC lines (Table 1) at the lowest available number of population doublings (PD) was determined by RLGS. Each hESC RLGS fragment display was compared with a reference human peripheral blood lymphocyte RLGS ‘master’ profile (14). The majority (97.5%) of the RLGS fragments in the lymphocyte profile were in common with at least one hESC line, indicating an abundant set of constitutively unmethylated CpG islands in hESCs. This is broadly consistent with the prevalence of unmethylated housekeeping gene loci previously observed by RLGS for a range of tissues (8) and the prevalence of housekeeping genes in hESC microarray profiles (15). Overall 152 loci were found hypermethylated in the six hESC lines compared with the lymphocyte profile (Fig. 1A), whereas 109 novel loci were identified that were unmethylated (i.e. were absent from the lymphocyte profile; Fig. 1B). The 39 sequences with consistent differences in methylation for all six hESC lines relative to adult human lymphocyte DNA (Fig. 1A and B) likely represent T-DMRs (8,9).
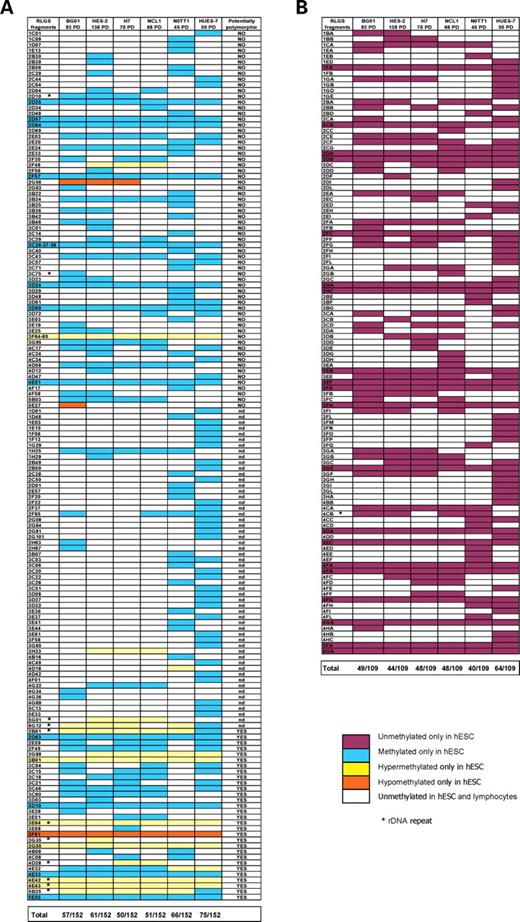
RLGS comparison of six independently-derived hESC lines. (A) One hundred and fifty-two RLGS fragments identified from our lymphocyte RLGS ‘master profile’ that varied between the hESC lines and lymphocytes. Fragments in common between the lymphocyte profile and all six hESC lines are not shown. Whether (YES) or not (NO) fragments were previously designated as potentially polymorphic (14) from RLGS studies is indicated; nd represents loci where the potentially polymorphic status was not determined. Fragment numbers designated 3C36–37–38 and 3F64–65 represent ambiguous RLGS gel spots where the precise identity could not be distinguished. (B) One hundred and nine fragments absent from the lymphocyte ‘master profile’ that were present in hESCs; these have been assigned a new identifying code. PD, population doublings.
Culture conditions and characteristics of cells used for RLGS
| hESC line | Mouse strain | MEF density (×104/cm2) | Passage interval and split ratio | PD harvested for RLGSa (p) | Karyotypeb (% clonally aneuploid cells) | Basal medium | Supplements | Observed phenotypic variations (unpublished) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FBS (%) | KSR (%) | TIS (%) | NEAA (%) | Gln (mm) | βME (mm) | bFGF(ng/ml) | LIF (U/ml) | Pen–Strep (%) | ||||||||
| NCL1 | ||||||||||||||||
| Feeders/manual + collagenase | Swiss MF1 | 7.5 | 5d, 1:3 | 68 (p43) | 46, XX | KO | — | 10 | — | 1 | 1 | 0.1 | 4 | — | 1 | 1 mm diameter colonies |
| H7 | ||||||||||||||||
| Feeders/collagenase | MF1 | 0.6 (CM + 8 ng/ml bFGF) | 5.5d, 1:3 | 70 (p44) | 46, XX | KO | — | 20 | — | 1 | 1 | 0.1 | 4 | — | — | Increasing growth rate and plating efficiency at p141 |
| 3.5d, 1:7.5 | 361 (p141) | 47, XX,+1, der (6)t(6;17q) (100%) | ||||||||||||||
| HES-2 | ||||||||||||||||
| Feeders/manual+dispase | 129/Sv | 7.3 | 7d, 1:8 | 138 (p46) | 46, XX | DMEM | 20 | — | 1 | 1 | 2 | 0.1 | — | — | 0.5 | 2 mm diameter colonies |
| 219 (p73) | 46, XX | Reducing propensity to form cardiomyocytes at p > 70 | ||||||||||||||
| BG01 | ||||||||||||||||
| MEFH | CD1 | 6 | 4d, 1:8 | 93 (p31) | 46, XY | |||||||||||
| 165 (p55) | ND | DF12 | 15 | 5 | — | 1 | 2 | 0.1 | 4 | — | — | 0.5–1 mm diameter colonies | ||||
| 207 (p69) | 46, XY | |||||||||||||||
| MEFKSR | 6 | 4d, 1:8 | 111 (p37) | ND | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Less efficiently maintained undifferentiated than MEFH | |
| MC | 6 (CM + 4 ng/ml bFGF) | 6d, 1:2 | 130c (p48) | 46, XY | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Elevated mortality during passage | |
| MT | 3d, 1:3 | 121d (p44) | 46, XY | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer | ||
| 152e (p64) | 46, XY | Increased growth rate at p84 | ||||||||||||||
| 192f (p84, BG01v) | 48, XY,+12,+17 (26%) | |||||||||||||||
| 49, XXY,+12,+17 (74%) | ||||||||||||||||
| NOTT1 | ||||||||||||||||
| MEFD | CD1 | 7.5 | 5d, 1:8 | 45 (p15) | 46, XX | KO | 20 | — | — | 1 | 2 | 0.1 | 4 | 1000 | — | 1 mm diameter colonies |
| MEFH | 7.5 | 5d, 1:8 | 45 (p15) | 46, XX | KO | 15 | 5 | — | 1 | 2 | 0.1 | 4 | 1000 | — | 1 mm diameter colonies | |
| MT | 6 (CM + 4 ng/ml bFGF | 3d, 1:2 | 52g (p22) 62h (p32) | 46, XX 46, XX | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer | |
| HUES-7 | ||||||||||||||||
| MT | CD1 | 6 (CM + 4 ng/ml bFGF | 3d, 1:3 | 50i (p23) 65j (p32) 82k (p43) | 46, XY ND 46, XY (97%) | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer |
| 47, XY, +12 (3%) | ||||||||||||||||
| hESC line | Mouse strain | MEF density (×104/cm2) | Passage interval and split ratio | PD harvested for RLGSa (p) | Karyotypeb (% clonally aneuploid cells) | Basal medium | Supplements | Observed phenotypic variations (unpublished) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FBS (%) | KSR (%) | TIS (%) | NEAA (%) | Gln (mm) | βME (mm) | bFGF(ng/ml) | LIF (U/ml) | Pen–Strep (%) | ||||||||
| NCL1 | ||||||||||||||||
| Feeders/manual + collagenase | Swiss MF1 | 7.5 | 5d, 1:3 | 68 (p43) | 46, XX | KO | — | 10 | — | 1 | 1 | 0.1 | 4 | — | 1 | 1 mm diameter colonies |
| H7 | ||||||||||||||||
| Feeders/collagenase | MF1 | 0.6 (CM + 8 ng/ml bFGF) | 5.5d, 1:3 | 70 (p44) | 46, XX | KO | — | 20 | — | 1 | 1 | 0.1 | 4 | — | — | Increasing growth rate and plating efficiency at p141 |
| 3.5d, 1:7.5 | 361 (p141) | 47, XX,+1, der (6)t(6;17q) (100%) | ||||||||||||||
| HES-2 | ||||||||||||||||
| Feeders/manual+dispase | 129/Sv | 7.3 | 7d, 1:8 | 138 (p46) | 46, XX | DMEM | 20 | — | 1 | 1 | 2 | 0.1 | — | — | 0.5 | 2 mm diameter colonies |
| 219 (p73) | 46, XX | Reducing propensity to form cardiomyocytes at p > 70 | ||||||||||||||
| BG01 | ||||||||||||||||
| MEFH | CD1 | 6 | 4d, 1:8 | 93 (p31) | 46, XY | |||||||||||
| 165 (p55) | ND | DF12 | 15 | 5 | — | 1 | 2 | 0.1 | 4 | — | — | 0.5–1 mm diameter colonies | ||||
| 207 (p69) | 46, XY | |||||||||||||||
| MEFKSR | 6 | 4d, 1:8 | 111 (p37) | ND | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Less efficiently maintained undifferentiated than MEFH | |
| MC | 6 (CM + 4 ng/ml bFGF) | 6d, 1:2 | 130c (p48) | 46, XY | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Elevated mortality during passage | |
| MT | 3d, 1:3 | 121d (p44) | 46, XY | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer | ||
| 152e (p64) | 46, XY | Increased growth rate at p84 | ||||||||||||||
| 192f (p84, BG01v) | 48, XY,+12,+17 (26%) | |||||||||||||||
| 49, XXY,+12,+17 (74%) | ||||||||||||||||
| NOTT1 | ||||||||||||||||
| MEFD | CD1 | 7.5 | 5d, 1:8 | 45 (p15) | 46, XX | KO | 20 | — | — | 1 | 2 | 0.1 | 4 | 1000 | — | 1 mm diameter colonies |
| MEFH | 7.5 | 5d, 1:8 | 45 (p15) | 46, XX | KO | 15 | 5 | — | 1 | 2 | 0.1 | 4 | 1000 | — | 1 mm diameter colonies | |
| MT | 6 (CM + 4 ng/ml bFGF | 3d, 1:2 | 52g (p22) 62h (p32) | 46, XX 46, XX | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer | |
| HUES-7 | ||||||||||||||||
| MT | CD1 | 6 (CM + 4 ng/ml bFGF | 3d, 1:3 | 50i (p23) 65j (p32) 82k (p43) | 46, XY ND 46, XY (97%) | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer |
| 47, XY, +12 (3%) | ||||||||||||||||
d, days; ND, not determined; DMEM, Dulbecco's Modified Eagle Medium; KO, knockout DMEM; DF12, DMEM/F12; FBS, fetal bovine serum; KSR, knockout serum replacement; ITS, insulin–transferrin–selenium; LIF, leukaemia inhibitory factor; NEAA, non-essential amino acids; Gln, glutamine; bFGF, human basic fibroblast growth factor; Pen, penicillin; Strep, streptomycin.
aDenotes first harvest passage (see Supplementary Materials and Methods Online for full details); PD: since passage numbers (p) are arbitrary periods (partially dependent on the proportion of cells reseeded at passage which varies considerably between lines), estimated population doubling (PD) times were calculated using the formula: log (number of subcultures established at split)/log2 (assuming 100% cell survival at each passage).
bKaryotype summary does not include non-clonal changes observed only in single metaphase spreads.
cBased on 41 manual passages (123 PD)+7 collagenase passages (7 PD)=130 PD.
dBased on 36 manual passages (108 PD)+8 trypsin passages (13 PD)=121 PD.
eBased on 36 manual passages (108 PD)+28 matrigel passages (44 PD)=152 PD.
fBased on 42 manual passages (126 PD)+31 cell dissociation buffer passages (49 PD)+11 trypsin passages (17 PD)=223 PD.
gBased on 15 manual passages (45 PD)+7 matrigel passages (7 PD)=52 PD.
hBased on 15 manual passages (45 PD)+17 matrigel passages (7 PD)=62 PD.
iBased on 17 trypsin on MEFs passages (41 PD)+6 matrigel passages (9 PD).
jBased on 17 trypsin on MEFs passages (41 PD)+15 matrigel passages (24 PD).
kBased on 17 trypsin on MEFs passages (41 PD)+26 matrigel passages (41 PD).
Culture conditions and characteristics of cells used for RLGS
| hESC line | Mouse strain | MEF density (×104/cm2) | Passage interval and split ratio | PD harvested for RLGSa (p) | Karyotypeb (% clonally aneuploid cells) | Basal medium | Supplements | Observed phenotypic variations (unpublished) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FBS (%) | KSR (%) | TIS (%) | NEAA (%) | Gln (mm) | βME (mm) | bFGF(ng/ml) | LIF (U/ml) | Pen–Strep (%) | ||||||||
| NCL1 | ||||||||||||||||
| Feeders/manual + collagenase | Swiss MF1 | 7.5 | 5d, 1:3 | 68 (p43) | 46, XX | KO | — | 10 | — | 1 | 1 | 0.1 | 4 | — | 1 | 1 mm diameter colonies |
| H7 | ||||||||||||||||
| Feeders/collagenase | MF1 | 0.6 (CM + 8 ng/ml bFGF) | 5.5d, 1:3 | 70 (p44) | 46, XX | KO | — | 20 | — | 1 | 1 | 0.1 | 4 | — | — | Increasing growth rate and plating efficiency at p141 |
| 3.5d, 1:7.5 | 361 (p141) | 47, XX,+1, der (6)t(6;17q) (100%) | ||||||||||||||
| HES-2 | ||||||||||||||||
| Feeders/manual+dispase | 129/Sv | 7.3 | 7d, 1:8 | 138 (p46) | 46, XX | DMEM | 20 | — | 1 | 1 | 2 | 0.1 | — | — | 0.5 | 2 mm diameter colonies |
| 219 (p73) | 46, XX | Reducing propensity to form cardiomyocytes at p > 70 | ||||||||||||||
| BG01 | ||||||||||||||||
| MEFH | CD1 | 6 | 4d, 1:8 | 93 (p31) | 46, XY | |||||||||||
| 165 (p55) | ND | DF12 | 15 | 5 | — | 1 | 2 | 0.1 | 4 | — | — | 0.5–1 mm diameter colonies | ||||
| 207 (p69) | 46, XY | |||||||||||||||
| MEFKSR | 6 | 4d, 1:8 | 111 (p37) | ND | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Less efficiently maintained undifferentiated than MEFH | |
| MC | 6 (CM + 4 ng/ml bFGF) | 6d, 1:2 | 130c (p48) | 46, XY | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Elevated mortality during passage | |
| MT | 3d, 1:3 | 121d (p44) | 46, XY | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer | ||
| 152e (p64) | 46, XY | Increased growth rate at p84 | ||||||||||||||
| 192f (p84, BG01v) | 48, XY,+12,+17 (26%) | |||||||||||||||
| 49, XXY,+12,+17 (74%) | ||||||||||||||||
| NOTT1 | ||||||||||||||||
| MEFD | CD1 | 7.5 | 5d, 1:8 | 45 (p15) | 46, XX | KO | 20 | — | — | 1 | 2 | 0.1 | 4 | 1000 | — | 1 mm diameter colonies |
| MEFH | 7.5 | 5d, 1:8 | 45 (p15) | 46, XX | KO | 15 | 5 | — | 1 | 2 | 0.1 | 4 | 1000 | — | 1 mm diameter colonies | |
| MT | 6 (CM + 4 ng/ml bFGF | 3d, 1:2 | 52g (p22) 62h (p32) | 46, XX 46, XX | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer | |
| HUES-7 | ||||||||||||||||
| MT | CD1 | 6 (CM + 4 ng/ml bFGF | 3d, 1:3 | 50i (p23) 65j (p32) 82k (p43) | 46, XY ND 46, XY (97%) | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer |
| 47, XY, +12 (3%) | ||||||||||||||||
| hESC line | Mouse strain | MEF density (×104/cm2) | Passage interval and split ratio | PD harvested for RLGSa (p) | Karyotypeb (% clonally aneuploid cells) | Basal medium | Supplements | Observed phenotypic variations (unpublished) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FBS (%) | KSR (%) | TIS (%) | NEAA (%) | Gln (mm) | βME (mm) | bFGF(ng/ml) | LIF (U/ml) | Pen–Strep (%) | ||||||||
| NCL1 | ||||||||||||||||
| Feeders/manual + collagenase | Swiss MF1 | 7.5 | 5d, 1:3 | 68 (p43) | 46, XX | KO | — | 10 | — | 1 | 1 | 0.1 | 4 | — | 1 | 1 mm diameter colonies |
| H7 | ||||||||||||||||
| Feeders/collagenase | MF1 | 0.6 (CM + 8 ng/ml bFGF) | 5.5d, 1:3 | 70 (p44) | 46, XX | KO | — | 20 | — | 1 | 1 | 0.1 | 4 | — | — | Increasing growth rate and plating efficiency at p141 |
| 3.5d, 1:7.5 | 361 (p141) | 47, XX,+1, der (6)t(6;17q) (100%) | ||||||||||||||
| HES-2 | ||||||||||||||||
| Feeders/manual+dispase | 129/Sv | 7.3 | 7d, 1:8 | 138 (p46) | 46, XX | DMEM | 20 | — | 1 | 1 | 2 | 0.1 | — | — | 0.5 | 2 mm diameter colonies |
| 219 (p73) | 46, XX | Reducing propensity to form cardiomyocytes at p > 70 | ||||||||||||||
| BG01 | ||||||||||||||||
| MEFH | CD1 | 6 | 4d, 1:8 | 93 (p31) | 46, XY | |||||||||||
| 165 (p55) | ND | DF12 | 15 | 5 | — | 1 | 2 | 0.1 | 4 | — | — | 0.5–1 mm diameter colonies | ||||
| 207 (p69) | 46, XY | |||||||||||||||
| MEFKSR | 6 | 4d, 1:8 | 111 (p37) | ND | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Less efficiently maintained undifferentiated than MEFH | |
| MC | 6 (CM + 4 ng/ml bFGF) | 6d, 1:2 | 130c (p48) | 46, XY | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Elevated mortality during passage | |
| MT | 3d, 1:3 | 121d (p44) | 46, XY | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer | ||
| 152e (p64) | 46, XY | Increased growth rate at p84 | ||||||||||||||
| 192f (p84, BG01v) | 48, XY,+12,+17 (26%) | |||||||||||||||
| 49, XXY,+12,+17 (74%) | ||||||||||||||||
| NOTT1 | ||||||||||||||||
| MEFD | CD1 | 7.5 | 5d, 1:8 | 45 (p15) | 46, XX | KO | 20 | — | — | 1 | 2 | 0.1 | 4 | 1000 | — | 1 mm diameter colonies |
| MEFH | 7.5 | 5d, 1:8 | 45 (p15) | 46, XX | KO | 15 | 5 | — | 1 | 2 | 0.1 | 4 | 1000 | — | 1 mm diameter colonies | |
| MT | 6 (CM + 4 ng/ml bFGF | 3d, 1:2 | 52g (p22) 62h (p32) | 46, XX 46, XX | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer | |
| HUES-7 | ||||||||||||||||
| MT | CD1 | 6 (CM + 4 ng/ml bFGF | 3d, 1:3 | 50i (p23) 65j (p32) 82k (p43) | 46, XY ND 46, XY (97%) | DF12 | — | 15 | — | 1 | 2 | 0.1 | 4 | — | — | Grows as a uniform monolayer |
| 47, XY, +12 (3%) | ||||||||||||||||
d, days; ND, not determined; DMEM, Dulbecco's Modified Eagle Medium; KO, knockout DMEM; DF12, DMEM/F12; FBS, fetal bovine serum; KSR, knockout serum replacement; ITS, insulin–transferrin–selenium; LIF, leukaemia inhibitory factor; NEAA, non-essential amino acids; Gln, glutamine; bFGF, human basic fibroblast growth factor; Pen, penicillin; Strep, streptomycin.
aDenotes first harvest passage (see Supplementary Materials and Methods Online for full details); PD: since passage numbers (p) are arbitrary periods (partially dependent on the proportion of cells reseeded at passage which varies considerably between lines), estimated population doubling (PD) times were calculated using the formula: log (number of subcultures established at split)/log2 (assuming 100% cell survival at each passage).
bKaryotype summary does not include non-clonal changes observed only in single metaphase spreads.
cBased on 41 manual passages (123 PD)+7 collagenase passages (7 PD)=130 PD.
dBased on 36 manual passages (108 PD)+8 trypsin passages (13 PD)=121 PD.
eBased on 36 manual passages (108 PD)+28 matrigel passages (44 PD)=152 PD.
fBased on 42 manual passages (126 PD)+31 cell dissociation buffer passages (49 PD)+11 trypsin passages (17 PD)=223 PD.
gBased on 15 manual passages (45 PD)+7 matrigel passages (7 PD)=52 PD.
hBased on 15 manual passages (45 PD)+17 matrigel passages (7 PD)=62 PD.
iBased on 17 trypsin on MEFs passages (41 PD)+6 matrigel passages (9 PD).
jBased on 17 trypsin on MEFs passages (41 PD)+15 matrigel passages (24 PD).
kBased on 17 trypsin on MEFs passages (41 PD)+26 matrigel passages (41 PD).
Most loci that were hypermethylated relative to the ‘master’ profile were found in HUES-7 (HUES-7: 74; NOTT1: 65; HES-2: 59; BG01: 54; NCL1: 50; H7: 48). Interestingly, treatment with non-toxic doses of 5-aza-2′-deoxycytydine failed to demethylate any of the 74 hypermethylated loci within this line (data not shown). However, after 12 PD of 5-aza-2′-deoxycytydine treatment, eight novel loci were demethylated (Fig. 2).
DNA methylation changes in HUES-7 treated with 5-aza-2′-deoxycytidine. Cells were treated with 10 nm 5-aza-2′-deoxycytidine for 12 PD and then RLGS performed. Data corresponding to untreated and 5-aza-2′-deoxycytidine treated cells are expressed relative to HUES-7 50 PD RLGS profile. PD, population doublings.
Overall, we observed interline variation in 10.39% of the loci examined, comprising 133 fragments known from the lymphocyte master profile and 89 novel, unmethylated fragments (Fig. 1A and B). Our RLGS profiles represent ∼10% of the CpG islands in the human genome (13), this suggests a potential for interline variation in ∼2200 CpG islands. Notably, pairwise comparisons of the four lines revealed a surprisingly high epigenetic distance of 1.64–6.55% of the RLGS profile, given that the consistent difference observed between all six lines and differentiated, adult lymphocytes was 1.82% (Fig. 1A and B). Bootstrap and randomization test statistics were used to determine whether the loci known from the ‘master profile’ that varied between lines were randomly altered among all of the DNA fragments in the RLGS profiles or represented ‘hotspot’ loci that were more likely to be epigenetically unstable. Using a χ2 goodness-of-fit test statistic, P-values (P < 0.0001, bootstrap; P < 0.0001, randomization) obtained from 25 000 simulations clearly indicated that the 152/2028 variable loci showed a non-random profile of altering their methylation status.
hESCs have been described previously as a ‘unique version of the human genome traced to the embryo’, attributing inter-line variation to allelic differences (4). Since RLGS differences between lines can reflect genetic variability where there is a homozygous or heterozygous restriction enzyme site polymorphism in one of the three enzymes (NotI, EcoRV or HinfI) used, or a length polymorphism within the NotI-EcoRV fragment, we included previously identified potentially polymorphic fragments present in the lymphocyte RLGS-Master profile (14) in our analysis in order to estimate the degree of non-epigenetic inter-line variability. By analysing pairwise comparisons of hESC lines, there was no significant difference in the proportion of RLGS changes in fragments previously identified as potentially polymorphic within humans and those considered non-polymorphic (14) (Supplementary Material, Fig. S1, χ2=2.42, P = 0.12). Of the 133 identified loci that vary between the six lines at lower PD, 15% are potentially polymorphic (Fig. 1A). However, 33.3% of the potentially polymorphic loci that varied between hESCs and lymphocytes showed the same RLGS fragment intensities in all six lines, suggesting that significantly fewer than 15% of the potentially polymorphic interline variations were in fact likely to be due to genetic loss of the RLGS fragment. Thus, our data indicated that at least 85% of the variation we observed between lines is truly epigenetic and cannot be accounted for by genetic differences between originating embryos. Differences between the lines occurred regardless of sex (Table 1, Fig. 1).
Acquisition of epigenetic instability over time and in different culture conditions
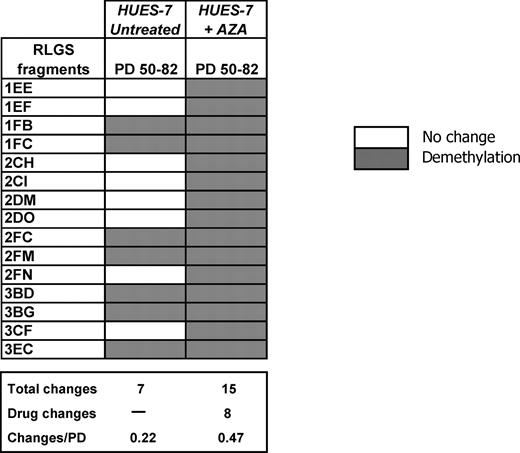
We then investigated whether the inter-line epigenetic variation was likely to be inherent to the originating embryos, or whether at least some of the variation was culture-induced. All four of the lines analysed by RLGS over time demonstrated epigenetic instability in between 0.33 and 0.79% of loci for 15–291 PD (Fig. 3), a substantial proportion when compared with the 1% observed in CpG islands during the human lifespan (16) and 0.22–3.39% mean changes observed between tumours and their primary tissue equivalents (13).
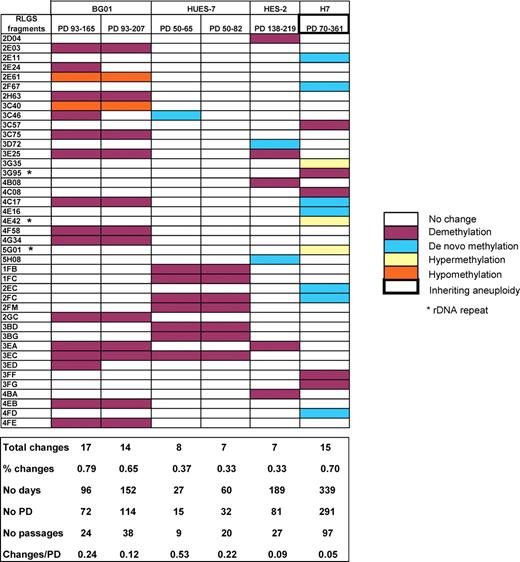
DNA methylation changes over time in culture. RLGS fragments that vary over time in HES-2 (manual passage culture on feeders), H7 (collagenase passage on feeders), BG01 (manual passage on feeders; MEFH) and HUES-7 (trypsin passage on Matrigel) are represented. BG01 and HUES-7 culture data are expressed relative to the MEFH 93 PD and MT 50 PD RLGS profile, respectively. The percentage of changes represents the proportion of changing loci relative to the 2137 examined RLGS fragments. PD, population doublings.
Next the effects of switching mouse embryonic fibroblast (MEF) feeder-based cultures of the BG01 line to serum-free or feeder-free conditions were tested, since these modifications are both common approaches to improving the clinical utility of hESC lines (17). The same basal culture medium was used in all cases, removing the confounding of variable composition that complicates interpretation of the interline data presented earlier (Table 1). Our baseline was the BresaGen protocol provided to us upon purchase of BG01 cells at 93 PD, i.e. manual passage on MEF feeder layers with 15% fetal bovine serum (designated MEFH). Serum-free cultures of cells manually passaged on MEFs with 15% KnockOut Serum Replacement (KSR; MEFKSR; 111 PD), trypsin-passaged on Matrigel (MT; 121, 152 and 192 PD) and collagenase-passaged on Matrigel (MC; 130 PD) were analysed for comparison (Fig. 4A). Morphologically, MC cells retained a similar colony appearance to manually passaged cells, whereas MT cultures grew as a continuous monolayer (Table 1 and Fig. 4B). However, all cell cultures showed the expected immunocytochemistry pattern of a range of hESC markers (i.e. SSEA-3+, SSEA-4+, TRA-1–60+ and TRA-1–81+ and SSEA-1−; data not shown) in the majority of cells in the cultures.
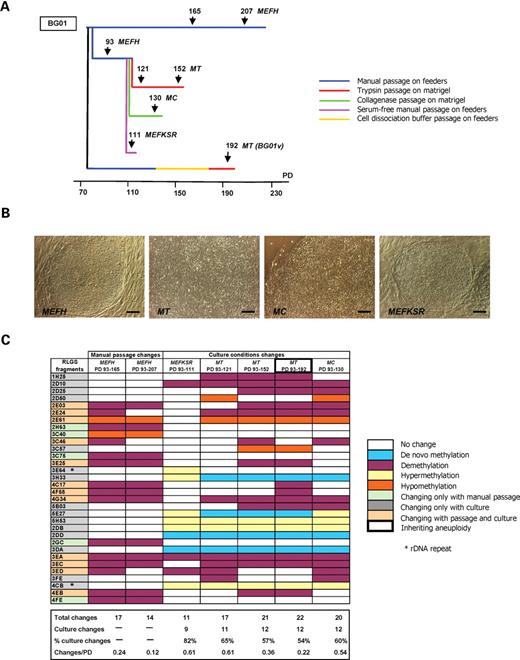
DNA methylation changes in BG01 adapted to serum-free and feeder-free culture conditions. (A) Schematic of adapting BG01 to different culture conditions and sampling periods used for RLGS. (B) Morphology of BG01 cells in MEFH, MT, MC and MEFKSR conditions (160 × magnification; bar 100 µm). (C) RLGS fragments that vary over time in manual passage culture on feeders (MEFH) relative to changes in serum-free culture on feeders (MEFKSR), trypsin passage on Matrigel (MT) or collagenase passage on Matrigel (MC). All BG01 culture data are expressed relative to the MEFH 93 PD RLGS profile. PD, population doublings.
Twelve of the RLGS fragments that differed between MT or MC cells relative to 93 PD MEFH also changed with increasing time in MEFH cultures. However, 15 loci unaffected over time in MEFH culture were observed to change when switching MEFH cells to serum-free and/or feeder-free cultures (Fig. 4C). Thus even changing culture method within a hESC line could account for an extra 90–120 CpG island differences throughout the genome over only a few PD. Significant commonality in RLGS profile changes with the three serum-free culture treatments was observed. Notably, of the 11 changes observed in cells collected after only 1–2 serum-free manual passage PD (MEFKSR; 111 PD), 10 RLGS sequences were also in common with both trypsin and collagenase passaged cells on Matrigel, suggesting a rapid effect of either increasing concentration of KSR, lack of serum or general effect of environmental change. Furthermore, since no fragments were identified which changed only in the two Matrigel cultures (trypsin and collagenase), a greater effect of serum-free rather than feeder-free conditions is indicated.
We then investigated the relationship between epigenetic stability and genomic alterations in hESC cultures. Our observed number of RLGS changes was not increased between 100% aneuploid cultures of H7 or BG01 and their earlier passage euploid parent cultures relative to euploid Matrigel (MT) or feeder (MEFH) BG01 cultures over time (Table 1, Fig. 3 and 4C). In addition, the known loci identified as susceptible to epigenetic variation within this study (Supplementary Material, Table S1) map to 18 different chromosomes (Fig. 5), with no statistical evidence of over-representation of individual chromosomes based on randomization and bootstrap tests (P = 0.551). These data confirm the association of epigenetic instability with culture conditions, rather than simply as a result of aneuploidy.
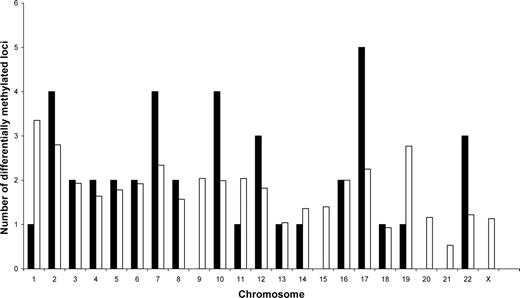
Observed versus expected number of differentially methylated loci per chromosome for 41 identified RLGS-DNA fragments. For the fixed observed number of methylation events on each chromosome, we calculated the null expected number of events as proportional to the respective NotI sites. The graph shows no over-representation of methylation events on specific chromosomes based on randomization and bootstrap tests (P = 0.551). Black columns represent observed events, white columns represent expected events.
More epigenetic alterations occur at the earliest stages post-derivation
Of note from the analysis of MT BG01 and MT HUES-7 over 3 and 2 time intervals, respectively, was an apparent decrease in rate of number of changing loci with time in culture (BG01 changes/PD: 0.61 for 28 PD, 0.36 for 59 PD, 0.22 for 99 PD; HUES-7 changes/PD: 0.53 for 15 PD, 0.22 for 32 PD; Fig. 3 and 4C). Since the earliest PD numbers we were able to examine with the BG01, HES-2 and H7 cells from independent providers were 93–138 (passages 31–46), this raised the possibility that the cells we analysed would have already undergone significant and rapid epigenetic changes. We therefore derived a new line, NOTT1, to enable us to collect cells at lower passage number than typically distributed by other providers. After 45 PD (15 passages), the newly-derived NOTT1 cells had expanded sufficiently to allow collection of the > 1 million cells required for RLGS. To provide comparable culture conditions to the BG01 and HUES-7 MT cells, NOTT1 cells also derived by manual passaging on MEFs were adapted to MT conditions. As in BG01 and HUES-7, the number of changes/PD relative to 45 PD NOTT1 MEFH cells decreased at 62 PD relative to 52 PD (1.53 versus 2.29; Fig. 6). This indicates that the greatest epigenetic instability occurs in hESC lines, at least in the conditions tested, at the earliest stages post-derivation.
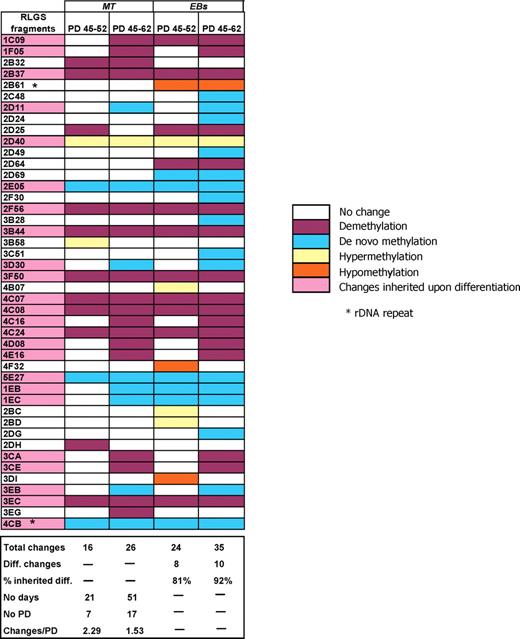
DNA methylation changes in NOTT1 cultures and their inheritance in day 12 EBs. RLGS fragments that vary over time in culture and after 12 days post-differentiation via EB formation are indicated. All NOTT1 culture data are expressed relative to the MT 45 PD RLGS profile. Culture-induced methylation changes inherited upon differentiation are highlighted. PD, population doublings.
Stability of acquired epigenetic alterations
DNA methylation changes in gene promoters induced in undifferentiated cells are only likely to be phenotypically relevant if they are inherited long-term by cultures (i.e. they are not selected against as detrimental changes) and if they are also inherited upon differentiation to transplantable cell types. Figs 3, 4 and 6 demonstrate that in BG01, HUES-7 and NOTT1 cultures, the majority (82.3–87.5%) of RLGS changes detected at the lowest passage number analysed are stably inherited within the cell populations.
It is likely that many of the observed stable changes may offer a selective advantage of some kind, since at least the majority of the cells within a culture must exhibit the same changes in order for them to be detected by RLGS. In cultured hESCs, variable RLGS fragment intensity is most likely to represent the proportion of methylated cells in the population (18) and would be expected to change DNA methylation over time in culture due to clonal penetrance rather than gene deletion/ amplification (14,18,19). This is consistent with the scenario observed by bisulphite sequencing analysis of fragments 4C17, 2E24 and 5B03, all of which showed an increase in the proportion of cells changing methylation over time in culture and directly correlated with RLGS spot intensity (Fig. 7A).
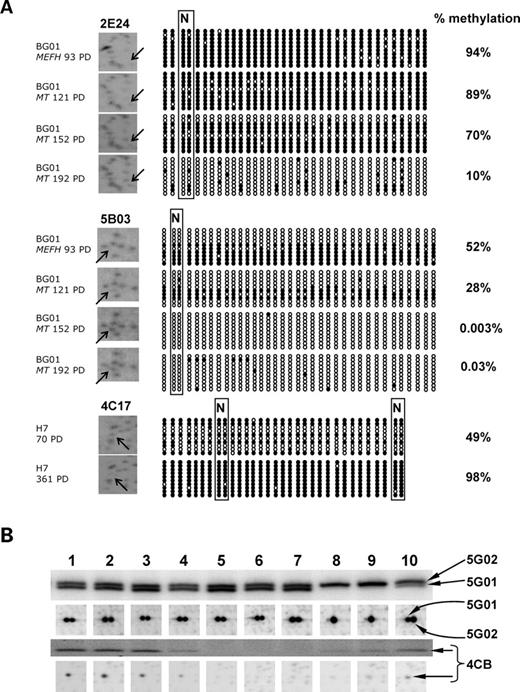
Verification of RLGS fragment differences as changes in DNA methylation. (A) RLGS profiles and corresponding bisulphite sequencing for CpG island fragments 2E24, 5B03 and 4C17. Each bisulphite sequence is represented by a row of circles representing CpG dinucleotides and each row represents a single cloned DNA molecule. Filled circles represent methylated CpG dinucleotides; open circles represent unmethylated CpG dinucleotides. N and boxed circles represent the methylation-sensitive, NotI restriction enzyme site analysed in the RLGS fragment. Bisulphite sequences indicate a change in clonal penetrance of demethylation (2E24, 5B03) or methylation (4C17) events over time. (B) RLGS profiles (lower boxes) and corresponding Southern blots (upper boxes) of NotI/EcoRV digested DNA using 28S and INTERGENIC SPACER 2 rDNA probes representing fragments 5G01, 5G02 and 4CB (F2, G1 and N1 as identified by Kuick et al. (22). Lanes 1, BG01 MEFH 93 PD; 2, BG01 MEFH 165 PD; 3, BG01 MEFH 207 PD; 4, BG01 MEFKSR 111 PD; 5, BG01 MT 121 PD; 6, BG01 MT 152 PD; 7, BG01 MT 192 PD; 8, HES-2 138 PD; 9, HES-2 219 PD; 10, NCL1 68 PD. PD, population doublings.
Strikingly, 81 and 92% of the NOTT1 52 PD and 62 PD changes, respectively, were still apparent in embryoid bodies (EBs) 12 days post-differentiation (Fig. 6).
We next studied the relationship between differential methylation and gene expression for three affected loci inherited by NOTT1 EBs that were associated with 5′ CpG islands (1C09, 2D25, 4C16). Bisulfite sequencing was first performed in the CpG island region including the NotI site to confirm the RLGS-determined differential methylation and then gene expression was studied by quantitative polymerase chain reaction (Q-PCR). The 1C09 RLGS fragment associated with the ZC3HAV1L gene showed a real-time PCR profile that correlated directly with the variation observed by RLGS in undifferentiated cells and EBs, whereas gene expression for the fragment 2D25 associated with the gene SPRN was correlated only in EBs (Fig. 8). No correlation was found for fragments 4C16.

Gene expression and DNA methylation changes in NOTT1 undifferentiated cells and EBs. Relative changes in mRNA expression for affected loci were measured using the ΔΔCt method with the NOTT1 MT PD 45 as calibrator sample (error bar, SD of ΔΔCt). RLGS-determined differential methylation was confirmed by direct bisulfite sequencing. Circles represents methylation of individual CpGs and the methylation sensitive NotI site (N) is indicated by boxed circles; black, methylated; white, unmethylated; back/white, hemi-methylated; back/grey, predominantly methylated; grey/white, predominantly unmethylated. PD, population doublings; ND, not determined.
Epigenetically modified gene sequences in hESC lines
Dissecting the functional relevance of these methylation changes will require identification of affected loci. Some unstable hESC RLGS fragments that were present in the human lymphocyte ‘master’ profile could be identified from the corresponding arrayed library of cloned NotI/EcoRV fragments (19) (Supplementary Material, Table S1). The majority (80%) were gene-associated CpG islands, indicating loci where the DNA methylation status could potentially determine transcriptional activity (9,14). In fact, 24/41 (58%) loci had previously been identified as variably expressed in hESC gene expression databases, indicating the relevance of analysed loci to hESC biology. Twenty-one out of 41 (51.2%) identified sequences contained LINE and/or SINE elements and six contained LTR, cis-acting regions previously implicated in the spreading of aberrant methylation (20).
Comparisons of all available hESC databases indicating differentiation-associated gene expression revealed that all six undifferentiated hESC lines we examined showed loss of methylation in some genes that are normally upregulated upon cellular differentiation (Supplementary Material, Table S1). This was the case even in H7 cells that had been FACS sorted for SSEA3+ cells prior to RLGS to remove differentiated cells (21). Since all other lines examined also stained positively for a range of stem cell markers in the majority of cells, the presence of differentiated cells in the cultures cannot account for the observed RLGS DNA methylation changes.
Perhaps the most important observation of this study is that 23/41 of sequenced loci (and a further 10 unidentified RLGS fragments) had previously been reported as changing their DNA methylation status in at least one human tumour type by comparable RLGS (Supplementary Material, Tables S1 and S2). Virtually all of the RLGS-detected changes reported in human tumours involved de novo methylation of normally unmethylated CpG islands (13) and 21 of these ‘tumour-associated’ loci were also methylated in at least one hESC line at the lowest PD number examined (Fig. 1A, Supplementary Material, Tables S1 and S2), but not in normal human lymphocytes. Seven out of 24 of the culture-induced changes inherited by NOTT1 EBs were also identified as tumour-associated CpG island loci. In addition, hypermethylation of at least 12 loci previously identified as ribosomal DNA (rDNA) repeats (22) was typically observed in the hESC lines relative to lymphocytes (Fig. 1A), a scenario only previously reported for embryonal carcinoma (EC) cells relative to somatic tissues (23). For instance, the 28S and INTERGENIC SPACER 2 rDNA regions (represented by fragments 4CB and 5G01) were hypermethylated in our lowest passage HES-2 cells relative to NCL1, BG01 or NOTT1 (Figs 1A, 5 and 7B). Although the consequences for ribosomal function are largely unknown, hypermethylation of rDNA has been associated with cancer (24) and ageing (25).
Since several of the loci we identified with temporally-varying methylation status were previously reported as genes variably expressed in a range of undifferentiated hESC lines (Supplementary Material, Table S1), we scrutinized the six lines in the present study for consistently unstable DNA methylation loci that could be used as a panel of markers for wider epigenetic screening of all hESC lines. No common locus changes were observed between HES-2 and H7 over time in culture and only 13.8–37.5% of time-related spot changes in H7 (4/15), BG01 (7/24), HES-2 (2/7), NOTT1 (4/29) and HUES-7 (3/8), respectively, were in common with another line (Figs 2 and 6). Within BG01, 39% of temporally-changing loci were common to both MEFH and MT cultures. These results indicate that the majority of time-dependent changes identified thus far are unlikely to be predictable for any new line, emphasizing the challenge of developing a high-throughput reliable epigenetic screening tool that can predict the biosafety of hESC lines.
DISCUSSION
We have used the RLGS technique to assess patterns of DNA methylation in different hESC lines and to determine the effects of time in culture, specific culture conditions and differentiation into embryoid bodies on the stability of the hESC epigenome. The RLGS technique was chosen since it does not involve operator selection of candidate gene regions to analyse and is able to investigate a representative sample of around 10% of the 29 000 CpG islands in the human genome (13). Overall, 142 unstable loci were identified, a number likely to increase as more cell lines and culture conditions are examined. Thus long-term maintenance of pluripotency characteristics of cells in vitro, that would differentiate rapidly in vivo, results in programming of epigenetic changes in at least some loci that are expected to be functionally significant for downstream applications. To ensure the biosafety of stem cell therapies, this study highlights the urgent need for further definition of the significance of the observed changes in advance of considering clinical trials. Although no hESC RLGS library currently exists to identify the novel RLGS fragments that we found to be unmethylated in hESCs but not in lymphocytes, these merit further study as epigenetically regulated T-DMRs potentially involved in the regulation of pluripotency. This is consistent with recent data showing that hESCs have a unique epigenetic signature of a subset of T-DMRs that is distinct from more differentiated cell types (26,27).
We found that the majority of epigenetic changes induced in vitro were not transient but were stably inherited by the cultures, many even after differentiation. For the ZC3HAV1L gene, we found a direct correlation between loss of methylation and increased gene expression in the undifferentiated cells, which was maintained post-differentiation. This demonstrates that at least some of the observed epigenetic instability may be functionally significant. However, in the SPRN locus, gene expression only correlated with loss of methylation in embryoid bodies. Although microarray transcriptional profiling of hESCs provides a snapshot of the gene expression patterns at the time of analysis, the possibility that epigenetic changes may not be manifested as altered gene expression until post-differentiation (e.g. when appropriate transcription factors are expressed) further highlights the importance of this level of analysis. Loss of methylation at the AFG3L1 locus did not correlate with gene expression at the time of analysis. It may be that this gene is not regulated by DNA methylation or that the core CpGs regulating gene expression were not affected (28). Moreover, since initial methylation of isolated CpGs can lead to spreading in the surrounding regions over time (28), the possibility of later methylation changes affecting gene expression cannot be excluded.
Since we observed epigenetic changes in karyotypically normal cell lines across many chromosomes, alteration in DNA methylation provides a clear potential mechanism for underlying the interline differences in hESC line properties and differentiation capability, which are increasingly reported (6,29). De novo methylation in hESCs could silence genes that are non-essential in culture (30), whereas demethylation provides an alternative mechanism to karyotype change to upregulate genes that confer a growth advantage (31). Previously, a generalized methylation increase in around half of CpG islands has been reported in transformed cell lines in culture relative to their primary tissue counterparts (30), although this effect was not observed in pluripotent EC cells. Also in human adult tumours, the majority of CpG island DNA methylation changes detected relative to the corresponding primary tissue are gains in methylation (32). Consistent with this, we observed some culture-related hypermethylation changes in gene loci previously associated with adult neoplasia. These could be simply features of highly proliferative cells and have no detrimental consequences. However, the possibility of making transplantable cells more tumourigenic in the host merits urgent attention, especially in the light of recent reports of human mesenchymal stem cells inheriting functional tumourigenic potential in culture (33). Unlike in transformed cell lines (30), there was not a significant probability of all genes with CpG islands becoming methylated, but only hotspot loci were affected by hESC culture. Moreover, in hESCs, these loci could either be methylated or demethylated, in marked contrast to the tendency for hypermethylation in transformed cells. That ES and EC cells appear to differ in their epigenetic instability from transformed cell lines is consistent with the unique epigenomic regulatory mechanisms being uncovered in pluripotent cells (34–36). DNA methylation inhibitors, like 5-aza-2′deoxycytidine, are commonly used for demethylating hypermethylated gene promoters in transformed cells. However, currently available drugs are unlikely to improve the epigenetic instability of undifferentiated hESCs since most of the observed culture-induced changes were losses of methylation. Moreover, both 5-aza-2′-deoxycytidine and Zebularine (data not shown) proved highly toxic to hESCs at the effective doses previously reported for cancer cell lines (37,38).
Despite searching for commonalities in sequence features or chromosomal location, no association with the susceptibility for hotspot loci has yet been identified (C. Allegrucci, L. Young and C. Bock, unpublished data). Interestingly, many are associated with cellular differentiation. Around 16% of total murine CpG islands known from RLGS studies have been shown to alter their methylation status during production of various differentiated tissues (8) and differentiation of mouse ESCs into embryoid bodies was associated with methylation and demethylation of 2.1 and 1.9% of loci, respectively (39). This is consistent with the scenario we observed upon differentiation of the hESC line, NOTT1, into embryoid bodies and a recent DNA methylation microarray comparison of hESCs and more differentiated cell types (26). However, in the six hESC lines we examined (including SSEA3+ sorted H7 cells), there was no morphological or pluripotency marker evidence of changes in the proportion of spontaneously differentiated cells with culture conditions or time in culture. Furthermore, since RLGS changes need to occur in at least 30% of the cells in cultures to be scored as a change, we do not believe that increased cellular differentiation underlies the observed epigenetic instability in hotspot loci. Instead, our demonstration of changes in differentiation-associated loci is more consistent with the notion of altering the full developmental potential of undifferentiated hESCs that exist in an ‘epigenetically-primed’ state to rapidly form a wide range of derivatives dependent on developmental signals (34). Consistent with the aforementioned hypothesis, a recent microarray study identified 1417 genes exhibiting differential hESC cultures switched to serum-free conditions including a range of differentiation-associated transcription factors and, interestingly, the embryonic DNA methyltransferase, DNMT3B (40). In addition, differential methylation at specific loci between hESC-derived neural progenitors and human fetal brain neural progenitors has been observed, suggesting epigenetic instability during or after the differentiation process (27).
We have demonstrated that at least part of the epigenetic variability between independently-derived hESC lines is culture induced and that the rate of change is higher in the earlier phases of culture. This suggests that although hESCs can be cultured long-term for therapeutic use, pharmaceutical screening and disease modelling, more future emphasis may have to be placed on expanding maximally newly derived cell lines at the earliest possible passages. This is of relevance to the debate over how many lines are needed worldwide to represent the immune haplotypes of potential patients (41), thus optimization of methods to minimize the degree of instability is required. Indeed, our studies of serum-free cultures indicate that developing methods compatible with producing clinical grade cells needs to occur in association with genetic and epigenetic stability measures. As yet, it is not clear whether instability is due to the supraphysiological levels of methyl group substrates present in human embryo and hESC culture medium (42), to other variable media components, to specific passage methods, to an interaction of culture with specific genotypes or to other unknown factors. The heterogeneous population of typically poor quality embryos donated from patients with many causes of infertility raises the possibility of an effect of factors inherited from the oocyte or sperm genomes. Interestingly, DNMT3B is expressed at very high levels in some, but not all hESC lines (6), providing another potential source of variable instability.
The implications of the identified epigenetic instability for safe development of stem cell technologies urge investigation of which unstable loci are most functionally significant for each intended application.
MATERIALS AND METHODS
Culture of hESC lines
Composition of the culture media used for BG01, NCL1, H7, HES-2, NOTT1 and HUES-7 is described in Table 1. Culture components were from Invitrogen and chemicals from Sigma unless otherwise specified. hESC culture was carried out at 37°C in a humidified atmosphere containing 5% CO2 and medium was changed daily.
Owing to inter-line variation in both the proportion of cells reseeded at passage and the passage interval, PD times were calculated from the information in Table 1 to provide more comparable measures than passage numbers. This also takes into account the various passage/culture conditions each line has been exposed to since derivation.
BG01 cells and the subline BG01v were purchased from BresaGen (43). Cultures were maintained at the University of Nottingham by manual passaging on mitomycin C-inactivated MEF feeder layers and by collagenase (1 mg/ml) or trypsin (0.05%) passaging on Matrigel (BD Biosciences) in feeder-free conditions employing MEF-conditioned medium (CM), as previously described (44).
The NOTT1 line was derived at the University of Nottingham in accordance with Human Fertilization and Embryology Authority licence R0141-2-a. A fresh grade two-three IVF embryo supernumerary to fertility treatment was cultured for 5 days in Vitrolife GIII sequential media as indicated by the manufacturer. The whole embryo was plated onto MEFs and the zona pellucida was removed after 2 days with acid Tyrode's. An hESC outgrowth was isolated after 5 days in culture and manually passaged every 7 days. Cultures were maintained by manual dissection on MEFs and by trypsin (0.05%) passaging on Matrigel in CM. Differentiation was induced in NOTT1 via embryoid body (EB) formation using the method described by Denning et al. (44), harvesting at day 12 for RLGS analysis.
The HUES-7 line (2) was kindly gifted by Harvard University and maintained at the University of Nottingham by trypsin (0.05%) passaging on Matrigel in CM.
Cultures of HES-2 (5) were maintained on MEFs at the Hubrecht Laboratory by using manual dissection coupled with dispase treatment (10 mg/ml).
H7 cells (45) were maintained on MEFs at the University of Sheffield and passaged by collagenase. The sublines H7-S14 and H7-S6 were used (21,31). Since S14 and S6 H7 cultures showed relatively high proportion of SSEA3− cells (21,31) that were not identified in the other hESC lines examined in this study, cells were FACS sorted prior to collection for RLGS analysis, gating the 20% most fluorescent cells using a Beckman–Coulter Altra flow cytometer at 1000 cell/s.
The NCL1 line was derived and maintained on MEFs at the University of Newcastle by manual dissection coupled with collagenase treatment (46).
5-aza-2′-deoxycytydine treatment
HUES-7 cultures in exponential growth phase were incubated with 0.01 µm, 0.025 µm, 0.05 µm, 0.1 µm, 0.5 µm, 1 µm of 5-aza-2′-deoxycytidine (Sigma). Drug-containing media were replaced every day for up 22 days and DNA was isolated at different time points (0.8–12, PD) for RLGS analysis. 5-aza-2′deoxycytidine treatments at 1 µm, 0.5 µm, 0.1 µm, 0.5 µm and 0.25 µm were terminated at 0.8 PD and 7.5 PD, respectively, due to drug toxicity.
Karyotype assessment
Exponentially growing cells from at least 50 colonies or a monolayer of 17 × 105 cells were treated with 100 ng/ml colcemid (Karyomax) for 30–45 min (BGO1, H7, NOTT1, HUES-7) or 60 min (HES-2, NOTT1). BGO1/H7/NCL1/NOTT1, HUES-7 cells were harvested (including feeders where used) with 0.05% Trypsin–EDTA. HES-2 colonies were harvested with 10 mg/ml dispase (37°C for 7 min) and washed twice in PBS before reducing to a single cell suspension using CDB (37°C for 15 min). Pelleted cells from all six lines (200g for 4 min) were resuspended as a single cell suspension in 0.6% sodium citrate and incubated at 37°C for 20 min. Cells were then recentrifuged (300g for 4 min) and fixed by resuspension in 16.7% glacial acetic acid in methanol and were washed with two further changes of fixative. Chromosome spreads were prepared by dropping cells onto glass slides which were air dried and heated to 70°C overnight. Chromosomes were G-banded with trypsin (BD Difco) and stained with Leishmans. For each analysis, chromosomes in 30 (BG01; HES-2, HUES-7), 20 (H7) and 10 (NCL1, NOTT1) metaphase cells were counted and visually examined for gross abnormalities. We used the designation of clonal aberrations in karyotype reported in Table 1 (chromosomal gain in two cells/loss in three cells), in accordance with ISCN Human Cytogenetic Nomenclature International Guidelines.
Restriction landmark genome scanning
For analysis by RLGS, 200–1200 hESC colonies, 1.5–5 × 106 cells in feeder-free conditions or 800 EBs were harvested and snap frozen. DNA was extracted and RLGS performed based on a previous method (47). Briefly, randomly sheared genomic DNA ends (1.5 µg) were blocked by incubation with DNA polymerase I (2U, Roche) and a mixture of nucleotide analogues (αS-dGTP 1.6 µm, αS-dCTP 0.8 µm, ddATP 1.6 µm, ddTTP 1.6 µm, Amersham Biosciences) for 20 min at 37°C. The reaction was stopped by incubation at 65°C for 30 min and DNA was digested with the methylation sensitive enzyme, NotI (20U, Promega) for 2 h at 37°C. DNA was then end-labelled with 1.3U Sequenase (USB), 1 µl [α32-P]-dGTP (6000 Ci/mmol, New England Nuclear) and 1 µl [α32-P]-dCTP (3000 Ci/mmol; Amersham Biosciences) for 30 min at 37°C. Labelled DNA was subjected to a second digestion with EcoRV (20U, Promega) for 2 h at 37°C and then electrophoresed through a 0.8% agarose tube gel at 230 V for 19 h. Agarose tube gels were then digested with HinfI (700U, NEB) for 2 h at 37°C and electrophoresed in a 5% non-denaturing polyacrylamide gel second dimension at 180 V overnight. Gels were then dried and exposed to X-ray films (Kodak X-OMAT-AR) in the presence of intensifying screens (Quanta III, Dupont) for up to 14 days.
The high background evident in some RLGS profiles was reduced by a second round of DNA dialysis using a Slide A-Lyzer Mini Dialysis unit (10 000 MWCO, Pierce).
RLGS profile analysis
In hESCs, we analysed a set of 2028 NotI/EcoRV/HinfI DNA fragments that corresponded to 84% of the reference human fragments in our normal human peripheral blood lymphocyte RLGS ‘master’ profile (14) (http://pandora.med.ohio-state.edu/masterRLGS). hESC fragments corresponding to the lymphocyte ‘master profile’ were identified by a previously published code (14), allocating a second dimension (NotI/HinfI) coordinate (from 1 to 6), a first dimension (NotI/EcoRV) coordinate (from A to H) and a fragment number to each spot (e.g. 5G01). RLGS fragments unique to the hESC profiles were allocated a new three character code, representing the same first number and second letter as the ‘master profile’ and a new letter identifying the fragment (e.g. 4CB). Changes in fragment intensity were designated as gain or loss of methylation when at least 30% intensity difference was observed. The arrayed library of NotI/EcoRV fragments, which represents 75% of the ‘master profile’ (19) was used to identify hESC genomic loci, whenever possible, and homology to a gene, or EST defined from the Human Genome database (http://genome.ucsc.edu/; Build 36.1, Supplementary Material, Table S1).
Bisulphite sequencing and Southern blot
A subset of the differences observed in RLGS profiles were verified by bisulphite sequencing of PCR-amplified cDNA, either by sequencing of 10 independent clones or by PCR product direct sequencing. Primers spanning CpG island sequences including NotI sites were designed using Methprimer software (http://www.urogene.org/methprimer): 2E24 forward 5′-TTAGTAGGGTTAGGTTGAATAGATA-3′ and reverse 5′-CCTACTAAACTTAACAATATACACC-3′; 5B03 forward 5′-AGTATTAAATATTTTTTTAGAAGGG-3′ and reverse 5-AACTAACCAAAATAAAAATCCAAAC-3′; 4C17 forward 5′-TAAGTTGTATAAATTTGGGTGTTTT-3′ and reverse 5′-CCCTTTCTAAACAATTTAATAACAAAC-3′; 1C09 forward GTTTTTATTTTTTGTAGTAGGAAA and reverse AAACCCACAATATACTCCTTCCTC; 2D25 forward AAAAACTAAAAACCCTTTCCCTCTCTTC and reverse GGGGGTTAGATGTGGTTTT; 4C16 forward GGGTTTTTATGGGTAGGTTTAGTTT and reverse TATTCTCTAAAACCCCAAAACCC. Genomic DNA (1 µg) from PBL (0% methylated control), BG01 93–121–152–192 PD, H7 70–361 PD, NOTT1 45–52–62 PD was treated with sodium bisulfite as previously described (48). PCR reactions were performed using 1 µl of bisulfite-treated DNA and 2.5 U of Platinum Taq polymerase (Invitrogen) for 35 cycles with annealing temperature ranging 57–60°C. PCR products were visualized on 8% polyacrylamide gels. Products of interest were then either directly purified from the PCR reaction (Qiagen Gel Extraction Kit) or electrophoresed in a 1.5% agarose gel and then purified in the same way. Purified PCR products were cloned using TOPO TA-cloning kit (Invitrogen). Either purified PCR products or 10 clones for each sample were subjected to sequencing. Complete bisulfite conversion was confirmed by the lack of cytosines in non-CpG dinucleotides after sequencing.
In addition, Southern blot analysis (19) for spots 5G01, 5G02 and 4CB was performed using probes generated from restriction fragments of the relevant NotI–EcoRV clones and labelled with the Prime-It II Random primer labeling kit (Stratagene).
Quantitative polymerase chain reaction
Real-time Taqman PCR was utilized to determine relative expression of ZC3HAV1L (1C09), SPRN (2D25) and AFG3L1 (4C16). Reverse transcription was carried out using the First Strand cDNA synthesis kit (Amersham Biosciences) with 0.2 µg RNA, according to manufacturers' instructions. The resulting cDNA was diluted to a final volume of 50 µl. Taqman PCR of samples was carried out using Applied Biosystems Assay on Demand primer/probe sets to ZC3HAV1L (Hs00293283_m1), SPRN (Hs00928414_s1), AFG3L1 (Hs00327853_m1) and 18S (internal control Hs99999901_ s1) in conjunction with Taqman Universal PCR Master Mix, (No AmpErase UNG, Applied Biosystems) and 2 µl cDNA (diluted to 1:10 for 18S amplification) in a total reaction volume of 20 µl. Cycle conditions were 95°C for 10 min for 1 cycle, followed by cycles of 95°C for 15 s/60°C for 1 min. Each PCR reaction was performed in triplicate, with relative quantification performed using the 7500 Fast Real-Time PCR System and SDS sequence detection software v. 1.3.1 (Applied Biosystems).
Statistical analysis
In order to test whether there was a consistent bias in the 152/2028 fragments that varied between the six hESC lines in Figure 1A and the lymphocyte ‘master’ profile, we examined the Pearson χ2 statistic comparing the observed frequency with expected frequencies (assuming each locus within a line has a ‘k’/2028 chance of varying from the master profile, where there are ‘k’ observed variations within that line). Using 25 000 simulations, we examined whether each locus is affected at random across the four hESC lines assuming either (i) the observed counts of variable loci are Poisson random variables with a mean total of ‘k’ (bootstrap test) or (ii) the total observed count is fixed at ‘k’ (randomization test) (49).
Pearson's χ2 test with a P-value simulated 100 000 times and the Yates' continuity correction were used to compare the counts of total variable loci versus only those designated potentially polymorphic in pairwise comparisons of hESC lines.
In order to test whether identified loci (Supplementary Material, Table S1) are randomly distributed across the 22 autosomal chromosomes and the X chromosome, we compared the observed frequencies with expected frequencies calculated from normalizing each chromosome for number of NotI sites (downloaded from the UCSC genome Build 35, May 2004 assembly by PERL script; http://genome.ucsc.edu/). The Y chromosome was not included in this analysis since the lymphocyte library from which clones were identified was prepared from female DNA. Pearson χ2 statistic comparison was performed using 100 000 simulations.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
Generous support from the University of Nottingham and BBSRC grant no BBS/B/06164 made this project possible (L.E.Y.). C.D. is an MRC Fellow, C.P. is a Leukemia Lymphoma Society Scholar and P.W.A. is funded by BBSRC and MRC. We thank Chad Cowan and Doug Melton for the HUES-7 cell line, Jayson Bispham for assistance with Q-PCR, Sonia Sebastian for assistance with bisulphite sequencing, Stephen Lee for assistance with RLGS and Ramana Davulurui for statistical advice. We acknowledge all the staff in NURTURE for facilitating the provision of donated embryos for the derivation of NOTT1, particularly Bruce Campbell (Scientific Director and HFEA licence holder), Cecilia Sjoblom (Director of Embryology), James Hopkisson (Clinical Director), Sarah Chamberlain and Lyndsey Devlin (Embryologists).
Conflict of Interest statement. None declared.
REFERENCES
Author notes
Present address: Cellular Reprogramming Laboratory, Principe Felipe Centro de Investigacion, Valencia, Spain.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}