




Abstract
Wolfram syndrome, an autosomal recessive disorder associated with diabetes mellitus and optic atrophy, is caused by mutations in the WFS1 gene encoding an endoplasmic reticulum (ER) membrane protein. Herein, we report that pancreatic islets of wfs1 -deficient mice exhibit increases in phosphorylation of RNA-dependent protein kinase-like ER kinase, chaperone gene expressions and active XBP1 protein levels, indicating an enhanced ER stress response. We established wfs1 -deficient MIN6 clonal β-cells by crossing wfs1 -deficient mice with mice expressing simian virus 40 large T antigen in β-cells. These cells show essentially the same alterations in ER stress responses as wfs1 -deficient islets, which were reversed by re-expression of WFS1 protein or overexpression of GRP78, a master regulator of the ER stress response. In contrast, these changes are not observed in heart, skeletal muscle or brown adipose tissues with WFS1-deficiency. The increased ER stress response was accompanied by reduced BrdU incorporation and increased caspase-3 cleavage, indicating impaired cell cycle progression and accelerated apoptotic processes in the mutant islets. These changes are associated with increased expression of the cell cycle regulator p21 CIP1 in wfs1 -deficient islets and clonal β-cells. Treatment of islets with thapsigargin, an ER stress inducer, caused upregulation of p21 CIP1 . In addition, forced expression of p21 CIP1 resulted in reduced MIN6 β-cell numbers, suggesting the ER stress-induced increase in p21 CIP1 expression to be involved in β-cell loss in the mutant islets. These data indicate that WFS1-deficiency activates the ER stress response specifically in β-cells, causing β-cell loss through impaired cell cycle progression and increased apoptosis.
INTRODUCTION
Type 2 diabetes is caused by complex interactions between insulin resistance in peripheral tissues and impaired insulin secretion from pancreatic β-cells. There is a general consensus that the latter results from both impaired β-cell function and decreased β-cell mass ( 1 – 3 ). Adult β-cell mass is maintained by a balance between generation and death of β-cells. In patients with type 2 diabetes, new islet formation and β-cell replication are reportedly normal, and an increased rate of apoptosis has been suggested to underlie the loss of β-cell mass ( 4 ).
Recent studies using novel mutant mice have led to new insights into endoplasmic reticulum (ER) stress and maintenance of β-cell mass ( 5 , 6 ). The ER stress response, also known as the unfolded protein response (UPR), involves translational attenuation, transcriptional induction of chaperones and folding enzymes, as well as degradation of misfolded proteins, a process called ER-associated degradation (ERAD). When ER stress is strong and cellular survival mechanisms fail to correct the protein-folding defects, an ER stress-mediated apoptotic process is initiated ( 5 – 7 ). Mice with a homozygous null mutation of RNA-dependent protein kinase-like ER kinase (PERK) lose their ability to phosphorylate eukaryotic initiation factor 2α (eIF2α) and fail to attenuate translation in response to ER stress. These mice develop diabetes owing to reduced β-cell mass ( 8 ). Importantly, mutations of the EIF2AK3 gene encoding PERK in humans have been recognized as causing Wolcott-Rallison syndrome with diabetes mellitus in early infancy ( 9 ). A mouse model in which a Ser51Ala mutation of eIF2α prevents the protein from being phosphorylated by PERK and other eIF2α kinases, also displays a β-cell defect and impaired gluconeogenesis leading to lethal hypoglycemia ( 10 ). Mice with a deletion mutation of P58 IPK , a cytosolic chaperone, were recently reported to exhibit β-cell failure and diabetes ( 11 ). These examples suggest that β-cells, producing large quantities of insulin and thus a greater load on the ER, are especially sensitive to ER stress.
Wolfram syndrome is a rare autosomal recessive disorder characterized by juvenile-onset diabetes mellitus, optic atrophy, diabetes insipidus and sensorineural deafness ( 12 ). This syndrome is caused by mutations in the WFS1 gene ( 13 , 14 ), which encodes an ER resident membrane protein ( 15 ). Post-mortem studies of the pancreas from subjects with Wolfram syndrome have shown β-cell loss ( 16 ). We recently established a line of mutant mice with a disrupted wfs1 gene and found that these mice also exhibited impaired glucose homeostasis accompanied by a progressive reduction of β-cell mass ( 17 ). Thus, the wfs1 -deficient mouse is a model for studying mechanisms of β-cell loss during the development of diabetes in Wolfram syndrome. We and others have also shown expression of WFS1 protein to be up-regulated by ER stress-inducing agents ( 18 – 20 ). A recent study employing IRE1α knockout and PERK knockout cells suggested that WFS1 is a component of the IRE1 and PERK signaling pathways ( 20 ). In addition, wfs1 -deficient islets have been shown to exhibit increased DNA fragmentation in response to ER stress inducers ( 17 ), suggesting β-cell loss in Wolfram syndrome to be attributable to an inability to handle ER stress. A very recent study of islets conditionally lacking the wfs1 gene in β-cells, demonstrated an increased GRP78 mRNA to GLUT2 mRNA ratio. This observation was interpreted as evidence of an enhanced ER stress response, on the assumption that GLUT2 mRNA levels represented the β-cell number in islets ( 21 ).
To further investigate the mechanisms underlying β-cell loss in Wolfram syndrome, we conducted a systematic study of the UPR in wfs1 -deficient islets as well as other tissues. We also created β-cell lines with WFS1-deficiency and studied UPR. We found all three UPR subpathways to be activated in wfs1 -deficient islets and β-cell lines. Furthermore, we demonstrated increased cleavage of caspase-3, a hallmark of apoptosis, and impaired proliferation associated with enhanced expression of the cell cycle regulator p21 CIP1 .
RESULTS
UPR activation in wfs1 -deficient islets
A systematic study of the UPR was conducted using islets isolated from 6-week-old male wfs1 -deficient mice with the C57Bl/6 background. At 6 weeks of age, the β-cell mass of these mice begins to decrease ( 17 ). Accumulation of unfolded proteins in the ER is well known to induce dissociation of GRP78 from PERK, resulting in oligomerization and subsequent auto-phosphorylation of PERK. Activated PERK then phosphorylates eIF2α and suppresses general protein translation to reduce the ER load ( 5 – 7 ). In freshly isolated wfs1 -deficient islets, PERK phosphorylation was increased (Fig. 1 A). In addition, eIF2α phosphorylation was slightly but significantly enhanced with no alteration in total eIF2α levels in mutant islets (Fig. 1 A). Thus, the ratio of phosphorylated eIF2α over total eIF2α levels analyzed by densitometry was increased by 27±7% ( n =4 experiments, P <0.05). These data indicate that one of three subpathways of the UPR arising from PERK phosphorylation is initiated in response to WFS1-deficiency in islets.
ER stress is also sensed by other ER resident proteins, IRE1 and ATF6, in addition to PERK ( 5 – 7 ). Activation of ATF6 via GRP78 dissociation and subsequent cleavage is known to induce the expressions of various chaperone genes, constituting another subpathway of the UPR ( 5 – 7 ). In wfs1 -deficient islets, GRP94 mRNA levels were increased and those of GRP78 and P58 IPK also tended to rise (Fig. 1 B). Correspondingly, although the differences failed to reach statistical significance, levels of these chaperone proteins tended to be increased (Fig. 1 C), suggesting that the ATF6 subpathway of the UPR is activated in response to WFS1-deficiency.
As shown in Figure 1 D, a shorter form of XBP1 mRNA was increased. This form is produced by 26-nucleotide splicing from primary XBP1 mRNA by the ribonuclease activity of IRE1, increasing active XBP1 protein levels in mutant islets (Fig. 1 E). HRD1, a ubiquitin ligase involved in ERAD, is one of the XBP1 target genes ( 22 ). In wfs1 -deficient islets, levels of HRD1 protein were markedly increased (Fig. 1 E). In addition, mRNA levels of ER-associated degradation-enhancing α-mannosidase-like protein (EDEM) ( 23 ), another target of XBP1, were significantly increased in mutant islets [100±5 arbitrary units (wild-type) versus 136±18 (mutant), n =6, P <0.05]. These data indicate that the IRE1-initiated subpathway of the UPR is also activated in wfs1 -deficient islets.
Establishment of MIN6 β-cell lines deficient in WFS1
To examine the influence of WFS1-deficiency specifically in a homogenous β-cell population, β-cell lines were established by crossing wfs1+/− and wfs1−/− mice ( 17 ) with IT6 mice expressing simian virus 40 (SV40) large T antigen under the insulin promoter ( 24 ) and were designated MIN6wfs1 +/− and MIN6wfs1 −/− , respectively (see Materials and Methods). IT6 mice were previously reported to develop insulinoma, from which the MIN6 cell line ( 24 ), one of the most highly differentiated β-cell lines, was generated. We established two cell lines each for the wfs1+/− and wfs1 −/− genotypes. As shown in Figure 2 A, the two cell lines with the wfs1 −/− genotype (MIN6wfs1 −/− −1 and 2) show similar UPR characteristics. Similarly, characteristics of two cell lines with the wfs1+/− genotype (MIN6wfs1 +/− ) were indistinguishable (data not shown). Therefore, only one line of each genotype was used for subsequent analyses. We compared MIN6wfs1 −/− with MIN6wfs1 +/− at the same passage numbers (passages 5–8), but not with the original MIN6 cells. This is because we were concerned that a difference in passage number between the original MIN6 and MIN6wfs1 −/− cells, irrespective of WFS1-deficiency, might affect the protein expression profile, rendering the former an inappropriate control for the latter. After completion of a series of experiments, MIN6wfs1 +/− cells reached passages 15–20, the same passage of original MIN6 cells we have. The function and survival of MIN6wfs1 +/− cells are similar to those of wild-type MIN6 cells at similar passage numbers (data not shown).
Effects of WFS1-deficiency on UPR in β-cell lines
As shown in Figure 2 A, altered expressions of UPR-related proteins observed in wfs1 -deficient islets were reproduced in MIN6wfs1 −/− cells; PERK phosphorylation, as well as expressions of active XBP1 and HRD1, were increased in wfs1 -deficient MIN6 cells. ATF4 levels were also shown to be increased in these cells. Furthermore, although GRP78 and GRP94 protein levels were similar (Fig. 2 B, upper panel), the activity of the GRP78 promoter containing three ER stress response elements was greater in MIN6wfs1 −/− cells than in MIN6wfs1 +/− cells (Fig. 2 B), strongly suggesting activation of the ATF6 subpathway of the UPR in MIN6wfs1 −/− cells. To confirm that alterations in UPR-related proteins are due to WFS1-deficiency, wild-type human WFS1 protein was expressed in MIN6wfs1 −/− cells. We took advantage of the tetracycline-inducible expression system. MIN6wfs1 −/− cells were infected with the Tet-repressor expressing virus (AdCAG-TR) together with a recombinant adenovirus bearing wild-type human WFS1 cDNA under the CMV promoter containing the Tet-operator (AdCTO-WFS1). The cells were then treated with doxycycline (2 µg/ml). As shown in Figure 2 C, when WFS1 expression was restored to levels comparable to those of the original MIN6 cells, the increase in PERK phosphorylation was prevented. In addition, overexpression of GRP78, a master regulator of the ER stress response, also resulted in normalization of PERK phosphorylation levels (Fig. 2 D), clearly indicating the observed alteration in UPR-related proteins to be due to exacerbation of ER stress caused by WFS1-deficiency.
No UPR induction in heart, skeletal muscle or brown adipose tissues from wfs1 -deficient mice
WFS1 protein is expressed in a variety of non-pancreatic tissues, though less abundantly than in islets (Fig. 3 A). Therefore, we also examined expressions of UPR genes in tissues other than pancreatic islets. Cardiac function is reportedly not impaired in subjects with Wolfram syndrome ( 12 ) or in wfs1 -deficient mice ( 17 ). Skeletal muscle and brown adipose tissue also appear essentially normal in mutant mice (data not shown). In contrast to islets, no UPR alterations were observed in these tissues from wfs1 -deficient mice (Fig. 3 B–D). Thus, UPR activation is tissue-specific in WFS1-deficiency.
Increased β-cell apoptotic response in wfs1 -deficient islets
ER stress induces apoptosis through activation of various signaling molecules including JNK and pro-apoptotic proteins, such as CHOP ( 5 – 7 ). CHOP expression was increased at both the mRNA (Fig. 4 A) and the protein level (Fig. 4 B), in mutant when compared with wild-type islets. In contrast, JNK expression levels and phosphorylation states were not altered in wfs1 -deficient islets (Fig. 4 B). We also found increased levels of cleaved caspase-3, a hallmark of apoptosis, in mutant islets (Fig. 4 B). CHOP expression and cleaved caspase-3 levels were also increased in wfs1 -deficient MIN6 cells (Fig. 4 C), whereas no such changes were observed in heart, skeletal muscle or adipose tissue (data not shown).
We also measured apoptosis in MIN6wfs1 −/− and MIN6wfs1 +/− cells by counting adherent cells positive for annexin V staining under fluorescent microscope. We found 1–2% cells to be annexin V positive for both the wfs1−/− and the wfs1+/− genotype cultured under standard conditions, i.e. no differences between MIN6wfs1 −/− and MIN6wfs1 +/− cells. An increase in the number of apoptotic cells was observed when MIN6wfs1 −/− cells were challenged with 0.5 µ m thapsigargin (TG) for 24 h, as compared with MIN6wfs1 +/− cells under the same conditions [2.7±1.0% (MIN6wfs1 +/− ) versus 6.2±1.1% (MIN6wfs1 −/− ) n =3, P <0.05]. Therefore, MIN6wfs1 −/− cells exhibited increased apoptosis susceptibility. These data, together, indicate that an ER stress mediated-apoptotic process is activated in wfs1 -deficient β-cells.
Impaired β-cell proliferation in wfs1 -deficient islets
In addition to increased apoptosis, decreased proliferation may contribute to loss of β-cell mass in wfs1 -deficient mice. When β-cell proliferation activity was assayed by 5-bromodeoxyuridine (BrdU) incorporation in pancreases from wild-type and mutant mice, BrdU incorporation was found to be significantly reduced in wfs1 -deficient β-cells (Fig. 5 A and B). This observation suggested impaired proliferation, along with increased apoptosis, to contribute to β-cell loss in wfs1 -deficient islets.
We next explored possible causes of the decreased β-cell proliferation in wfs1 -deficient islets. The link between the UPR and cell cycle arrest was previously reported to be mediated by down-regulation of cyclin D1 because of general translational suppression via eIF2α phosphorylation ( 25 ). However, neither expression of cyclin D1 nor that of cyclin D2, major isoforms of the D type cyclins in β-cells ( 26 , 27 ), was changed in mutant islets (data not shown). CHOP has also been recognized as causing cell cycle arrest and apoptosis ( 28 , 29 ). As GADD34 is reportedly a target of CHOP ( 30 ) and is involved in cell growth and survival ( 31 ), GADD34 expression was examined. GADD34 transcript levels were found to be increased in wfs1 -deficient islets (100±11 versus 151±14, P <0.05). Recent studies have demonstrated that cell cycle regulation is critical for maintenance of β-cell mass ( 25 , 26 ). As GADD34 reportedly induces p53 phosphorylation and enhances expression of the cell cycle inhibitor p21 CIP1 ( 32 ), p53 and p21 CIP1 expressions were assessed. We found phosphorylation of p53 to be increased, though total p53 was not elevated (Fig. 5 C). In addition, increased expressions of p21 CIP1 mRNA (100±11 versus 413±32, P <0.01) and p21 CIP1 protein (Fig. 5 C) were observed in wfs1 -deficient islets. We also examined the expression of another cell cycle inhibitor, p27 KIP1 , and found no difference between wild-type and mutant islets (Fig. 5 C). Increased expression of p21 CIP1 protein was also observed in wfs1 -deficient MIN6 cells, SV40 large T antigen-transformed cells in which p53 activity was considered to be suppressed (Fig. 5 D). Expression of p21 CIP1 protein was not increased in heart, skeletal muscle or brown adipose tissues from wfs1 -deficient mice (data not shown).
In order to determine whether increased expression of p21 CIP1 is attributable to ER stress, wild-type islets were treated with TG (0.5 µ m ) for 12 h. As shown in Figure 5 E, expression of p21 CIP1 was significantly increased. In addition, expression of p21 CIP1 was markedly increased in MIN6 cells treated with TG (Fig. 5 F) or tunicamycin (data not shown). These data suggest 21 CIP1 expression to be induced by ER stress in β-cells.
Finally to assess the effects of p21 CIP1 expression on β-cell proliferation, p21 CIP1 was expressed in wild-type MIN6 cells in a tetracycline-inducible manner (Fig. 6 A). Overexpression of p21 CIP1 suppressed a MIN6 cell number increase (Fig. 6 B), suggesting that increased p21 CIP1 expression contributes to the reduced β-cell mass in wfs1 -deficient islets.
DISCUSSION
We systematically investigated UPR in wfs1 -deficient islets and MIN6 β-cells as well as heart, skeletal muscle and brown adipose tissues from the mutant mice in this study. Enhanced UPR was specifically observed in β-cells but not in other tissues examined. These findings indicate that diabetes in Wolfram syndrome is caused by increased ER stress in β-cells and establish Wolfram syndrome as an ER stress-based disease, as is the case in Wolcott-Rallison syndrome with PERK-deficiency ( 9 ). Furthermore, we found enhanced UPR to be associated with not only activation of the apoptotic pathway but also impaired cell cycle progression in β-cells. These observations provide evidence of novel mechanisms underlying ER stress-mediated β-cell loss.
We demonstrated activation of the PERK and IRE1 subpathways of the UPR. Increased activation of the GRP78 promoter indicates the ATF6 subpathway to be induced as well. GRP78 expression was also reportedly increased by knockdown of WFS1 expression in INS1 insulinoma β-cells ( 20 ). Collectively, these data indicate that all three UPR subpathways are activated by WFS1-deficiency in β-cells. The UPR is activated when ER homeostasis is perturbed by defective ER calcium homeostasis, mutations in ER resident proteins and/or abnormalities of the ERAD system. Disturbed ER homeostasis is also induced by defect(s) in components of the UPR system, as is the case in Wolcott-Rallison syndrome with PERK-deficiency. The present data suggest that impaired ER homeostasis does not result from defect(s) in a specific pathway(s) of the UPR. Our previous study demonstrated an abnormal cytosolic Ca 2+ response in wfs1 -deficient β-cells ( 17 ), suggesting that impaired ER Ca 2+ homeostasis is a possible cause of ER stress associated with WFS1-deficiency.
We found that WFS1 protein is highly expressed in heart, skeletal muscle and brown adipose tissues. However, there is no UPR activation in these tissues from mutant mice. Thus, the UPR is tissue-specific in wfs1 -deficient mice. One possible explanation of this tissue specificity is that a protein(s), compensating for loss of WFS1 protein function is present in these tissues but not in β-cells. This interesting possibility merits further investigation and elucidation of WFS1 function is necessary to resolve the tissue-specific effects of WFS1-deficiency.
Our results demonstrate, in addition to the augmented apoptotic process evidenced by increased caspase-3 cleavage, that β-cell proliferation is decreased in wfs1 -deficient mice. Impaired proliferation was also reported in BRIN-BD11 cells expressing the human WFS1 antisense transcript ( 33 ). Our observation is in contrast to that by Riggs et al . ( 21 ) who detected no changes in the numbers of BrdU-positive cells in islets from β-cell specific wfs1 knockout mice. The reason for this discrepancy is currently unclear, but may reflect differences in the ages of the mice studied: 6-week-old mice were used in the present versus 12- or 24-week-old animals in their study ( 21 ). Cell cycle dysregulation in wfs1 -deficient islets was associated with increased expression of p21 CIP1 , a cell cycle regulator. p21 CIP1 can serve, depending on which tissues or cells it is activated in, as both an inhibitor and an agonist of cell cycle progression ( 34 ). Our observation that forced expression of p21 CIP1 suppressed MIN6 β-cell proliferation suggests that p21 CIP1 operates as a cell cycle inhibitor in β-cells, although our results must be interpreted cautiously, as forced overexpression of p21 CIP1 may produce effects different from those occurring in mutant β-cells with increased p21 CIP1 levels. A very recent study, demonstrating that p21 CIP1 acts as a molecular brake on mitogenic stimuli in β-cells ( 35 ), supports the notion of p21 CIP1 functioning as a cell cycle inhibitor in β-cells. ER stress inducers were recently reported to cause p21 CIP1 expression and cell cycle arrest in chondrocytes ( 36 ) and prostatic cancer cells ( 37 ), suggesting that cell cycle arrest associated with increased p21 CIP1 expression is a common feature in cells under ER stress. Furthermore, reduced proliferation associated with increased expression of p21 CIP1 , in wfs1 -deficient β-cells (the present study) and β-cells transgenic for hepatocyte growth factor and/or placental lactogen ( 35 ), highlights an important role for p21 CIP1 in regulation of β-cell mass in addition to the roles of p27 KIP1 recently reported ( 38 ).
CHOP induces GADD34 expression ( 30 ), which then activates p53 phosphorylation and p21 CIP1 transcription ( 32 ). Therefore, the CHOP→GADD34→p53 pathway is a candidate for ER stress-mediated p21 CIP1 expression. Indeed, an increase in p21 CIP1 expression was associated with increased GADD34 expression and p53 phosphorylation in wfs1 -deficient β-cells. However, induction of p21 CIP1 expression by TG was observed in MIN6 cells transformed with SV40 large T antigen, a well-known suppressor of p53. In addition, an ER stress-induced increase in p21 CIP1 expression was observed in p53-deficient prostatic cancer cells ( 37 ). Thus, ER stress appears to induce p21 CIP1 expression through both p53-dependent and -independent mechanisms.
As β-cells are apparently much more sensitive to ER stress than other types of cells and tissues ( 39 ), ER stress might be a more common cause of β-cell failure than previously thought, especially in terms of the increased insulin demands of modern lifestyles. Our data indicate that both increased apoptosis and impaired proliferation, in β-cells, are mechanisms leading to β-cell loss in wfs1 -deficient islets, a model of ER-stress mediated β-cell failure. Further studies designed to elucidate the molecular mechanisms of β-cell loss under chronic ER stress are anticipated to contribute to future treatments for type 2 diabetes.
MATERIALS AND METHODS
Antibodies
The monoclonal antibody against P58 IPK was a generous gift from Prof. M.G. Katze (University of Washington). Other antibodies were purchased from the indicated sources: anti-GRP94, anti-KDEL (Stressgen Biotechnologies), anti-GRP78, anti-XBP1, anti-p21 CIP1 , anti-CHOP, anti-p53, anti-phosphorylated p53 and anti-ATF4 (Santa Cruz Biotechnology), anti-HRD1 (Abgent), anti-phosphorylated PERK, anti-JNK, anti-phosphorylated JNK, anti-eIF2α, anti-phosphorylated eIF2α, and anti-cleaved caspase-3 (Cell Signaling), and anti-p27 KIP1 (BD Transduction Laboratories).
Mouse islet isolation, real-time RT–PCR and western blot
The wfs1 -deficient mice used had a C57Bl/6 background and were described previously ( 17 ). All animal experiments were approved by the Tohoku University Institutional Animal Care and Use Committee (#15–45). Islets were isolated by collagenase infusion through the common bile duct and harvested by hand. Total RNA was prepared immediately after islet isolation using an RNAeasy kit (Qiagen). For real-time RT–PCR analysis, cDNA was synthesized by reverse transcription using the oligo d(T) 16 primer and subjected to PCR amplification with gene-specific primers (Table 1 ) using a SYBR Green 1 kit (Roche). Data are presented as relative values to actin mRNA. For detection of the spliced form of XBP1 mRNA, the primers were: 5′-TGAGAACCAGGAGTTAAGAAACGC-3′ and 5′-TTCTGGGTAGACCTCTGGGAGTTCC-3′. For immunoblotting, islets from three to four mice were pooled, dissolved immediately after isolation in a lysis buffer (∼100 islets/15 µl) and subjected to SDS–polyacrylamide gel electrophoresis. In several experiments, isolated islets were cultured overnight and treated with 0.5 µ m TG for 12 h. All western blot experiments were repeated at least three times, with different sets of samples, throughout this study. Immunoblot band intensities were analyzed using Scion image software (Scion Corporation) and normalized with those of actin.
Establishment of MIN6wfs1 −/− and MIN6wfs1 +/− cell lines
The wfs1−/− mice ( 17 ) were bred with IT6 mice expressing SV40 large T antigen under the human insulin promoter ( 24 ) and the resulting wfs1+/− :SV40Tag/+ mice were further bred with wfs1−/− mice. Tumors from pancreases of 10- to 12-week-old wfs1+/− :SV40Tag/+ and wfs1−/− : SV40Tag/+ mice were carefully excised and placed in Dulbecco's Modified Eagle's Medium containing penicillin and streptomycin. Cells were expanded and frozen at passages 3 and 4. We used these cells at 5–8 passages in this study. For study of apoptosis, MIN6 cells were infected with AdRIPeGFP expressing enhanced green fluorescent protein under the insulin promoter to facilitate detection of cells under fluorescent microscope. Apoptosis was examined by staining with annexin V using the Annexin V-Cy3 apoptosis detection kit (Medical and Biological Laboratories). At least 1000 cells per sample were counted for annexin V positive cells.
GRP78 promoter assay
The pGL3-promoter, pTK-RLuc and pGL3-basic plasmids were purchased from Promega. The mouse GRP78 promoter fragment spanning −172 to −21 (positions relative to the transcription start site) was amplified by PCR using oligonucleotides 5′-GACTCGAGGCCGCTTCGAATCGGCAG-3′ and 5′-TCAAGCTTGGCCAGTATCGAGCGCGC-3′. This fragment contains three ER stress response elements ( 40 ) and the corresponding regions of human ( 40 ) and rat ( 41 ) GRP78 genes were shown to respond to ATF6 activation. A GRP78 promoter-driving luciferase reporter plasmid (designated pmGRP78pro(−172)-Luc) was constructed by subcloning this fragment into the Xho I and Hind III sites of the pGL3-basic vector. MIN6wfs1 +/− or MIN6wfs1 −/− cells were co-transfected with pGL3-promoter or pGRP78pro(−172)-Luc together with pTK-RLuc using the LipofectAMINE reagent (Invitrogen). Luciferase activities were assayed with Dual-Luciferase reporter system (Promega) using a Lumat LB9507 luminometer (Berthold).
BrdU incorporation assay
BrdU (100 mg/kg) was injected into the mice intraperitoneally. Six hours later, the mice were sacrificed and their pancreases were fixed with 4% paraformaldehyde. Immunohistochemical analyses were performed with a Cell Proliferation Assay kit (BD Pharmingen). Sections were also stained with anti-insulin. BrdU-positive β-cells were counted in at least 50 sections per mouse.
Recombinant adenovirus experiments
Human GRP78 cDNA was purchased from Open Biosystems. Human WFS1 cDNA was a generous gift from Prof. Y. Tanizawa (Yamaguchi University). The CMV promoter containing two Tet-operator sequences (designated CTO) was excised from pcDNA5/TO (Invitrogen) and ligated to these cDNAs. The Tet-repressor cDNA was excised from pcDNA6/TR (Invitrogen) and ligated to the CAG promoter unit ( 42 ). These expression units were used to generate recombinant adenoviruses by a previously described method ( 43 ). The resulting viruses were designated AdCAG-TR for the Tet-repressor expressing virus and AdCTO-GRP78 for the GRP78 expressing virus under the CTO promoter, and so on. MIN6 and its derivative cells were infected with AdCAG-TR at a multiplicity of infection (m.o.i.) of 30 together with viruses with the CTO promoter at an m.o.i. of 100. One day after infection, cells were reseeded and divided into two groups. Two days thereafter, the cells were fed media with or without doxycycline (2 µg/ml). We have observed no adverse effects of infection of a control recombinant adenovirus expressing green fluorescence protein at an m.o.i. of less than 250 on MIN6 cell function in terms of cell proliferation and glucose-stimulated insulin secretion (data not shown). For the cell number assessment, MIN6 cells infected with AdCAG-TR and AdCTO-p21 CIP1 were seeded in six-well plates at 2×10 5 per well, cultured in media with or without doxycycline (2 µg/ml) and harvested after the indicated intervals. Cells were then stained with trypan blue and counted.
Statistical analysis
Data are presented as means±S.E. Differences between groups were assessed by Student's t -test.
ACKNOWLEDGEMENTS
We thank Profs. M.G. Katze and Y. Tanizawa for their generous gifts of the monoclonal antibody against P58 IPK and human WFS1 cDNA, respectively. We are also grateful to Y. Nagura and K. Tanaka for their expert assistance. This research was supported by Grants-in-Aid for Scientific Research (17590264 to H.I. and 17390258 to Y.O.) from the Ministry of Education, Science, Sports and Culture of Japan.
Conflict of Interest statement . None declared.
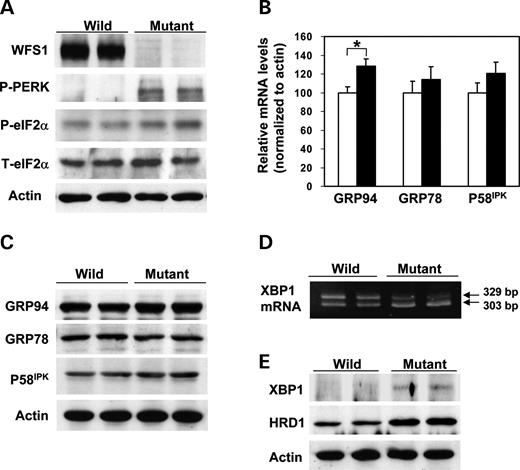
Figure 1. Activation of three subpathways of the UPR in wfs1 -deficient islets. ( A ) Activation of the PERK/eIF2α pathway. Islets isolated from wild-type and wfs1 -deficient mice were subjected to SDS–PAGE and probed with the indicated antibodies: P-PERK, phosphorylated-PERK; P-eIF2α, phosphorylated-eIF2α; T-eIF2α, total eIF2α. ( B ) Real-time RT–PCR analysis of GRP94, GRP78 and P58 IPK gene expressions in wild-type (open columns) and wfs1 -deficient (closed columns) mice. Relative mRNA levels were obtained after normalization to actin mRNA. * P <0.05, n =6. ( C ) Expressions of chaperone proteins in wfs1 -deficient islets. Lysates of isolated islets were probed with the indicated antibodies. ( D ) Increased XBP1 mRNA splicing in wfs1 -deficient islets. Amplification of XBP1 mRNA from islet total RNA with specific primers yields spliced (303 bp) and non-spliced (329 bp) XBP1 transcripts. ( E ) Activation of the IRE1/XBP1 pathway. Lysates of isolated islets were probed with the indicated antibodies. Western blot data shown are representative of at least three experiments with different sets of samples.
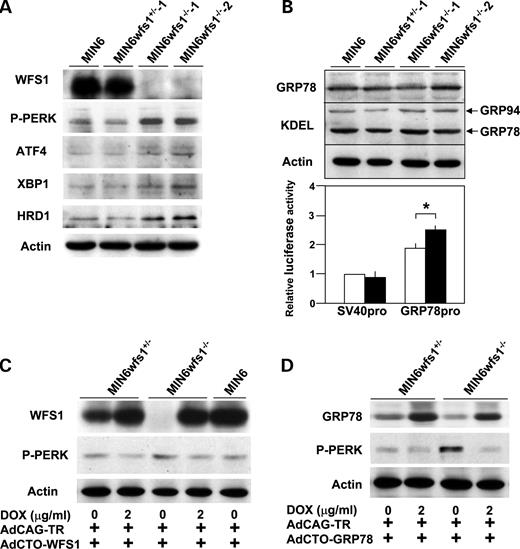
Figure 2. Increased UPR and its reversal by expression of WFS1 or GRP78 in an SV40 transformed wfs 1-deficient β-cell line (MIN6wfs1 −/− ). ( A ) Expression of UPR-related proteins in various MIN6 cell lines. MIN6, MIN6wfs1 +/− −1, MIN6wfs1 −/− −1 and MIN6wfs1 −/− −2 cells were lysed and probed with the indicated antibodies. Data shown are representative of at least three experiments with different sets of samples. ( B ) Expressions of chaperone proteins in MIN6wfs1 −/− cells. (Upper panel) Cellular lysates were probed with anti-GRP78, anti-KDEL and anti-actin (loading control) antibodies. (Lower panel) MIN6wfs1 +/− (open columns) and MIN6wfs1 −/− (closed columns) cells were transiently transfected with the pGL3-promoter plasmid containing the SV40 promoter-luciferase (SV40pro: 0.5 µg) or pGRP78pro(−172)-Luc (GRP78pro: 0.5 µg) together with the reference plasmid pTK-RL (0.05 µg) encoding Renilla luciferase. Twenty-four hours after transfection, cellular lysates were subjected to luciferase assay. The luciferase activity of the pGL3-promoter in MIN6wfs1 +/− was defined as 1. The averages of three independent experiments, each performed in duplicate, are presented. * P <0.05, n =3. ( C ) Suppression of PERK phosphorylation by WFS1 re-expression in MIN6wfs1 −/− cells. Cells were infected with AdCAG-TR expressing Tet-repressor and AdCTO-WFS1 harboring WFS1 cDNA. WFS1 expression was induced by 48 h doxycycline (DOX, 2 µg/ml) treatment. The experiment was repeated three times and similar results were obtained. ( D ) Suppression of PERK phosphorylation by GRP78 overexpression in MIN6wfs1 −/− cells. Human GRP78 expression was induced by 48 h DOX treatment. The experiment was repeated four times and similar results were obtained.
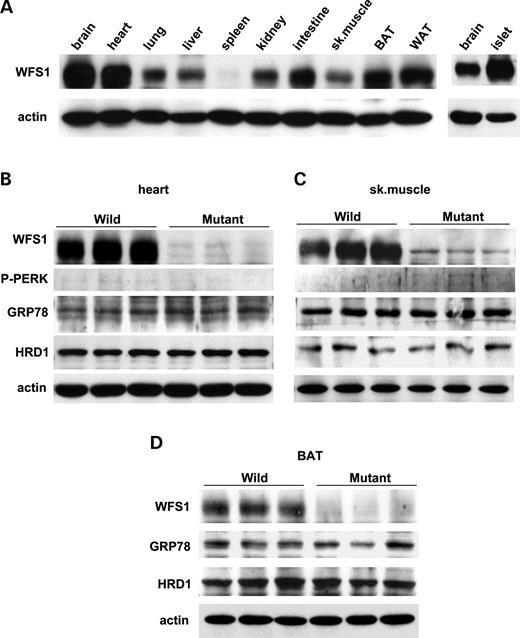
Figure 3. No UPR changes in heart, skeletal muscle or brown adipose tissue from wfs1 -deficient mice. ( A ) WFS1 protein distribution in mice. Approximately, 100 µg of protein from wild-type mouse tissues were analyzed for the presence of WFS1 protein. BAT, brown adipose tissue; WAT, white adipose tissue. ( B – D ) UPR activation was not observed in heart (B), skeletal muscle (C) or BAT (D) from wfs1 -deficient mice. The western blot data shown are representative of two experiments, each performed using three mice of each genotype.
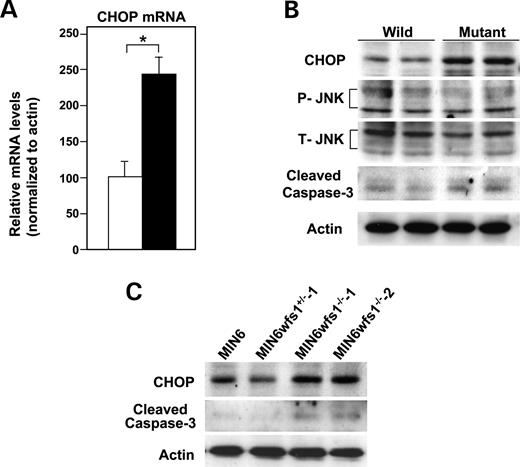
Figure 4. Activation of apoptosis signaling in wfs1 -deficient islets and MIN6 cells. ( A ) Real-time RT–PCR analysis of CHOP mRNA in wild-type (open column) and wfs1 -deficient (closed column) islets. Relative mRNA levels were obtained after normalization to actin mRNA. * P <0.05, n =6. ( B ) Western blot analysis of apoptosis signaling proteins in wfs1 -deficient islets. Lysates of islets were probed with the indicated antibodies: P-JNK, phospho-JNK; T-JNK, total-JNK. Data shown are representative of three experiments with different sets of samples. ( C ) Increased expression of CHOP and cleaved caspase-3 in wfs1 -deficient MIN6 cells. Lysates of MIN6 cell derivatives were probed with the indicated antibodies. Data shown are representative of three experiments.
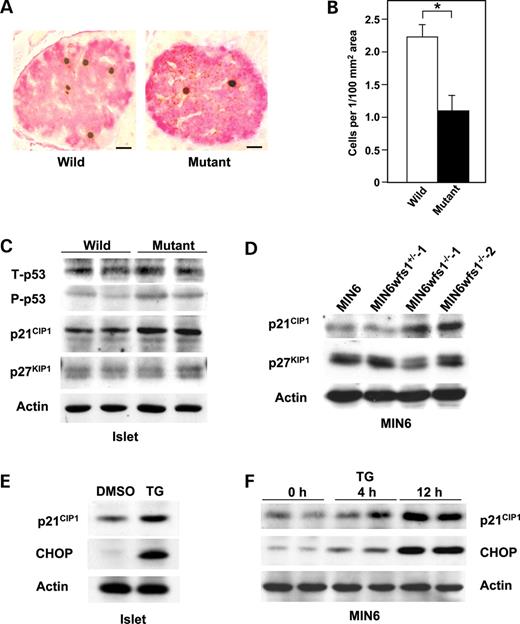
Figure 5. Impaired cell cycle progression and increased p21 CIP1 expression in wfs1 -deficient islets. ( A and B ) Impaired cell cycle progression in wfs1 -deficient β-cells. Incorporated BrdU and insulin were probed with specific antibodies (A) and BrdU positive β-cells were counted (B). Bars, 10 µ m . * P <0.05, n =4 mice per group. ( C and D ) Increased p21 CIP1 expression in wfs1 -deficient islets and MIN6 cells. Lysates of wild-type and wfs1 -deficient islets (C) or MIN6 cells (D) were probed with the indicated antibodies: T-p53, total-p53; P-p53, phospho-p53. Data shown are representative of three experiments with different sets of samples. ( E and F ) Induction of p21 CIP1 expression by TG in islets (E) and MIN6 cells (F). Wild-type islets were challenged with 0.5 µ m TG for 12 h. MIN6 cells were also treated with 0.5 µ m TG for the indicated durations. Lysates of islets or MIN6 cells were probed with the indicated antibodies. The experiment was repeated three times and similar results were obtained.
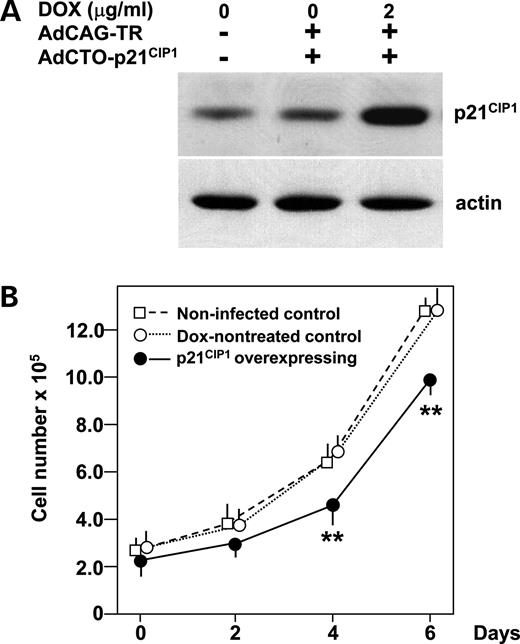
Figure 6. Decrease in MIN6 cell numbers in response to forced p21 CIP1 expression. ( A ) Forced expression of p21 CIP1 in MIN6 cells. Cells were either uninfected or infected with AdCAG-TR (m.o.i. of 30) and AdCTO-p21 CIP1 (m.o.i. of 100) harboring p21 CIP1 cDNA. Expression of p21 CIP1 was induced by 48 h DOX (2 µg/ml) treatment. MIN6 cell lysates were subjected to immunoblot analysis using anti-p21 CIP1 and actin antibodies. ( B ) Numbers of MIN6 cells overexpressing p21 CIP1 . One day after adenovirus transduction, cells were reseeded (2×10 5 per well) and divided into two groups, and, after two more days, treatment with (closed circles) or without (open circles) DOX (2 µg/ml) was commenced (day 0). Uninfected MIN6 cells (open squares) were also seeded 2 days before. Cells were then harvested on days 0, 2, 4 and 6, stained with trypan blue and counted. Data are means±S.E. for triplicate wells. ** P <0.01 against both controls. The experiment was repeated three times and similar results were obtained.
Primers used for quantitative real-time RT–PCR
| Genes | Forward | Reverse |
|---|---|---|
| ATF4 | 5′-TCCTGAACAGCGAAGTGTTG-3′ | 5′-ACCCATGAGGTTTCAAGTGC-3′ |
| GRP94 | 5′-TGATGAAGTCGACGTGGATG-3′ | 5′-TCCTGTTCACTTCAGCTTGG-3′ |
| GRP78 | 5′-GACATTTGCCCAGAAGAAA-3′ | 5′-CTCATGACATTCAGTCCAGCA-3′ |
| P58IPK | 5′-CCTTATCGGACAGTCCTTCG-3′ | 5′-TCAGAGTCCTGATTTCATCTTCA-3′ |
| EDEM | 5′-GGAAATTCATCCGAGTTCCA-3′ | 5′-GGGCCATGTACAACAATTCA-3′ |
| CHOP | 5′-CCTAGCTTGGCTGACAGAGG-3′ | 5′-CTGCTCCTTCTCCTTCATGC-3′ |
| GADD34 | 5′-CGGAGAGAAGCCAGAATCAC-3′ | 5′-CAGCAAGGAAATGGACTGTG-3′ |
| P21CIP1 | 5′-ACATCTCAGGGCCGAAAAC-3′ | 5′-CCTGACCCACAGCAGAAGAG-3′ |
| Genes | Forward | Reverse |
|---|---|---|
| ATF4 | 5′-TCCTGAACAGCGAAGTGTTG-3′ | 5′-ACCCATGAGGTTTCAAGTGC-3′ |
| GRP94 | 5′-TGATGAAGTCGACGTGGATG-3′ | 5′-TCCTGTTCACTTCAGCTTGG-3′ |
| GRP78 | 5′-GACATTTGCCCAGAAGAAA-3′ | 5′-CTCATGACATTCAGTCCAGCA-3′ |
| P58IPK | 5′-CCTTATCGGACAGTCCTTCG-3′ | 5′-TCAGAGTCCTGATTTCATCTTCA-3′ |
| EDEM | 5′-GGAAATTCATCCGAGTTCCA-3′ | 5′-GGGCCATGTACAACAATTCA-3′ |
| CHOP | 5′-CCTAGCTTGGCTGACAGAGG-3′ | 5′-CTGCTCCTTCTCCTTCATGC-3′ |
| GADD34 | 5′-CGGAGAGAAGCCAGAATCAC-3′ | 5′-CAGCAAGGAAATGGACTGTG-3′ |
| P21CIP1 | 5′-ACATCTCAGGGCCGAAAAC-3′ | 5′-CCTGACCCACAGCAGAAGAG-3′ |
Primers used for quantitative real-time RT–PCR
| Genes | Forward | Reverse |
|---|---|---|
| ATF4 | 5′-TCCTGAACAGCGAAGTGTTG-3′ | 5′-ACCCATGAGGTTTCAAGTGC-3′ |
| GRP94 | 5′-TGATGAAGTCGACGTGGATG-3′ | 5′-TCCTGTTCACTTCAGCTTGG-3′ |
| GRP78 | 5′-GACATTTGCCCAGAAGAAA-3′ | 5′-CTCATGACATTCAGTCCAGCA-3′ |
| P58IPK | 5′-CCTTATCGGACAGTCCTTCG-3′ | 5′-TCAGAGTCCTGATTTCATCTTCA-3′ |
| EDEM | 5′-GGAAATTCATCCGAGTTCCA-3′ | 5′-GGGCCATGTACAACAATTCA-3′ |
| CHOP | 5′-CCTAGCTTGGCTGACAGAGG-3′ | 5′-CTGCTCCTTCTCCTTCATGC-3′ |
| GADD34 | 5′-CGGAGAGAAGCCAGAATCAC-3′ | 5′-CAGCAAGGAAATGGACTGTG-3′ |
| P21CIP1 | 5′-ACATCTCAGGGCCGAAAAC-3′ | 5′-CCTGACCCACAGCAGAAGAG-3′ |
| Genes | Forward | Reverse |
|---|---|---|
| ATF4 | 5′-TCCTGAACAGCGAAGTGTTG-3′ | 5′-ACCCATGAGGTTTCAAGTGC-3′ |
| GRP94 | 5′-TGATGAAGTCGACGTGGATG-3′ | 5′-TCCTGTTCACTTCAGCTTGG-3′ |
| GRP78 | 5′-GACATTTGCCCAGAAGAAA-3′ | 5′-CTCATGACATTCAGTCCAGCA-3′ |
| P58IPK | 5′-CCTTATCGGACAGTCCTTCG-3′ | 5′-TCAGAGTCCTGATTTCATCTTCA-3′ |
| EDEM | 5′-GGAAATTCATCCGAGTTCCA-3′ | 5′-GGGCCATGTACAACAATTCA-3′ |
| CHOP | 5′-CCTAGCTTGGCTGACAGAGG-3′ | 5′-CTGCTCCTTCTCCTTCATGC-3′ |
| GADD34 | 5′-CGGAGAGAAGCCAGAATCAC-3′ | 5′-CAGCAAGGAAATGGACTGTG-3′ |
| P21CIP1 | 5′-ACATCTCAGGGCCGAAAAC-3′ | 5′-CCTGACCCACAGCAGAAGAG-3′ |
References
Donath, M.Y. and Halban, P.A. (
Rhodes, C.J. (
Porter, J.R. and Barrett, T.G. (
Butler, A.E., Janson, J., Bonner-Weir, S., Ritzel, R., Rizza, R.A. and Butler, P.C. (
Harding, H.P. and Ron, D. (
Wu, J. and Kaufman, R.J. (
Schroder, M. and Kaufman, R.J. (
Harding, H.P., Zhang, Y., Zeng, H., Jungries, R., Chung, P., Plesken, H., Sabatini, D.D. and Ron, D. (
Delepine, M., Nicolino, M., Barrett, T., Golamaully, M., Lathrop, G.M. and Julier, C. (
Scheuner, D., Song, B., McEwen, E., Liu, C., Laybutt, R., Gillespie, P., Saunders, T., Bonner-Weir, S. and Kaufman, R.J. (
Ladiges, W.C., Knoblaugh, S.E., Morton, J.F., Korth, M.J., Sopher, B.L., Baskin, C.R., MacAuley, A., Goodman, A.G., LeBoeuf, R.C. and Katze, M.G. (
Wolfram, D.J. and Wagener, H.P. (
Inoue, H., Tanizawa, Y., Wasson, J., Behn, P., Kalidas, K., Bernal-Mizrachi, E., Mueckler, M., Marshall, H., Donis-Keller, H., Crock, P. et al . (
Strom, T.M., Hortnagel, K., Hofmann, S., Gekeler, F., Scharfe, C., Rabl, W., Gerbitz, K.D. and Meitinger, T. (
Takeda, K., Inoue, H., Tanizawa, Y., Matsuzaki, Y., Oba, J., Watanabe, Y., Shinoda, K. and Oka, Y. (
Karasik, A., O'hara, C., Srikanta, S., Swift, M., Soeldner, J.S., Kahn, C.R. and Herskowitz, R.D. (
Ishihara, H., Takeda, S., Tamura, A., Takahashi, R., Yamaguchi, S., Takei, D., Yamada, T., Inoue, H., Soga, H., Katagiri, H. et al . (
Yamaguchi, S., Ishihara, H., Tamura, A., Yamada, T., Takahashi, R., Takei, D., Katagiri, H. and Oka, Y. (
Ueda, K., Kawano, J., Takeda, K., Yujiri, T., Tanabe, K., Anno, T., Akiyama, M., Nozaki, J., Yoshinaga, T., Koizumi, A. et al . (
Fonseca, S.G., Fukuma, M., Lipson, K.L., Nguyen, L.X., Allen, J.R., Oka, Y. and Urano, F. (
Riggs, A.C., Bernal-Mizrachi, E., Ohsugi, M., Wasson, J., Fatrai, S., Welling, C., Murray, J., Schmidt, R.E., Herrera, P.L. and Permutt, M.A. (
Kaneko, M., Ishiguro, M., Niinuma, Y., Uesugi, M. and Nomura, Y. (
Hosokawa, N., Wada, I., Hasegawa, K., Yorihuzi, T., Tremblay, L.O., Herscovics, A. and Nagata, K. (
Miyazaki, J., Araki, K., Yamato, E., Ikegami, H., Asano, T., Shibasaki, Y., Oka, Y. and Yamamura, K. (
Brewer, J.W. and Diehl, J.A. (
Georgia, S. and Bhushan, A. (
Kushner, J.A., Ciemerych, M.A., Sicinska, E., Wartschow, L.M., Teta, M., Long, S.Y., Sicinski, P. and White, M.F. (
Barone, M.V., Crozat, A., Tabaee, A., Philipson, L. and Ron, D. (
Kim, D.-G., You, K.-R., Liu, M.-J., Choi, Y.-K. and Won, Y.-S. (
Marciniak, S.J., Yun, C.Y., Oyadomari, S., Novoa, I., Zhang, Y., Jungreis, R., Nagata, K., Harding, H.P. and Ron, D. (
Hollander, M.C., Poola-Kella, S. and Fornace, A.J., Jr (
Yagi, A., Hasegawa, Y., Xiao, H., Haneda, M., Kojima, E., Nishikimi, A., Hasegawa, T., Shimokata, K. and Isobe, K. (
McBain, S.C. and Morgan, N.G. (
Sherr, C.J. and Roberts, J.M. (
Cozar-Castellano, I., Weinstock, M., Haughty, M., Velazquez-Garcia, S., Sipula, D. and Stewart, A.F. (
Yang, L., Sara, G., Carlson, S.G., McBurney, D. and Horton, W.E., Jr (
Zu, K., Bihani, T., Lin, A., Park, Y.M., Mori, K. and Ip, C. (
Uchida, T., Nakamura, T., Hashimoto, N., Matsuda, T., Kotani, K., Sakaue, H., Kido, Y., Hayashi, Y., Nakayama, K.I., White, M.F. and Kasuga, M. (
Shi, Y., Taylor, S.I., Tan, S.L. and Sonenberg, N. (
Yoshida, H., Haza, K., Yanagi, H., Yura, T. and Mori, K. (
Lee, A.S. (
Niwa, H., Yamamura, K. and Miyazaki, J. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}