



Abstract
Mutations in the human parkin gene encoding an E3 ubiquitin ligase have been associated with early-onset recessive forms of Parkinson's disease (PD). However, the molecular mechanisms by which mutations in the parkin gene cause PD are still under debate. Here, we identified and characterized the Caenorhabditis elegans parkin homolog, pdr-1. PDR-1 protein physically associates and cooperates with a conserved degradation machinery to mediate ubiquitin conjugation. Strikingly, in contrast to pdr-1 loss-of-function mutants, an in-frame deletion variant with altered solubility and intracellular localization properties is hypersensitive toward different proteotoxic stress conditions. Both endoplasmic reticulum-derived folding stress and cytosolic stress conferred by expression of mutant human α-synuclein resulted in severe developmental defects and lethality in pdr-1(lg103) mutant background. Furthermore, we show that the corresponding truncated protein PDR-1(Δaa24–247) aggregates in cell culture, but still interacts with its ubiquitylation co-enzymes. Thus, it might block the cellular degradation/detoxification machinery and therefore renders worms highly vulnerable to protein folding stress. In contrast to other complete gene knockouts or RNAi models of Parkin function, this C. elegans model recapitulates Parkin insolubility and aggregation similar to several autosomal recessive juvenile parkinsonism (AR-JP)-linked Parkin mutations. We suggest that such Parkin variants that either confer a neomorphic function or a partial loss-of-function may help to further elucidate the biological function of Parkin in vivo and the pathogenic mechanisms resulting in AR-JP. Due to high-throughput capacity of C. elegans, this model is particularly well suited to identify genetic and chemical modifiers of toxicity.
INTRODUCTION
Parkinson's disease (PD) is the second most common neurodegenerative disorder affecting about 1–2% of the population over the age of 65. PD is mainly characterized by motor dysfunctions, resulting from selective loss of dopaminergic neurons in the substantia nigra (1). A neuropathological hallmark of idiopathic PD is the formation of cytoplasmic protein inclusions called Lewy bodies. A major component of these aggregates is the presynaptic protein α-synuclein, implicated in many biological processes (2). Moreover, rare dominant mutations of α-synuclein (3–5) and genomic multiplications (6–8) have been identified to be causative for the disease. A number of transgenic animal models have been generated that overexpress α-synuclein in mice, flies (reviewed in 9) and in Caenorhabditis elegans (10).
In contrast to the rare dominant α-synuclein mutations, mutations in the parkin gene cause autosomal recessive juvenile parkinsonism (AR-JP) (11), accounting for nearly 50% of cases with autosomal recessive PD and early onset of the disease. Human Parkin consists of four known domains: an N-terminal ubiquitin-like domain (UBL) and two RING finger domains flanking a cysteine rich in-between RING finger (IBR) domain (Fig. 1A). The UBL domain has been implicated in proteasome binding (12), substrate recognition (13) and regulation of Parkin stability (14). Like many proteins with RING finger domains, Parkin acts as an E3 ubiquitin ligase in vitro (15). E3 proteins function as components of the ubiquitin/proteasome system that controls degradation of misfolded and regulatory proteins (16).
In cell culture experiments, it has been observed that expression of human parkin is up-regulated in response to unfolded protein stress (17). The accumulation of misfolded proteins in the lumen of the endoplasmic reticulum (ER) activates an intracellular signaling pathway, the unfolded protein response (UPR). This adaptive response augments ER folding capacity by transcriptional induction of ER-resident chaperones, folding catalysts and protein degradation complexes, and additionally limits further accumulation of unfolded proteins in the ER by translational attenuation (18). Unfolded proteins in the ER are retro-translocated to the cytosol and turned over by the ER-associated degradation (ERAD) pathway, a process regulated by the UPR (19). Both pathways are required for the coordinated disposal of misfolded proteins even in the absence of acute stress (20,21).
A variety of potential Parkin substrates, including the Pael-R receptor of the cytoplasmic membrane (22) and a rare, O-glycosylated form of α-synuclein (13), have already been identified (reviewed in 23). Moreover, Parkin has been shown to protect against neurotoxicity induced by diverse cellular insults including overexpression of some of its substrates (24,25). It was shown that a hotspot of familial mutations (Fig. 1B) occurs in the RING–IBR–RING domains (26) and interferes with Parkin's ubiquitylation activity, inhibiting its neuroprotective function (15). Although loss of Parkin function is considered to be causative for AR-JP, it had been suggested that some disease-linked point mutations of Parkin may retain substantial catalytic activity, and, therefore, should not be considered complete null alleles (27–31). Moreover, some of these mutations decrease the solubility of the protein, and, thus, result in Parkin variants with altered intracellular localization and/or a tendency to misfolding, aggregation and aggresome-formation (27,32–34).
Animal models generated so far, i.e. by simply deleting the parkin gene locus, did not show the expected severe neurotoxicity typical for the familial PD mutations. As a possible explanation, compensatory pathways have been suggested that may take over degradation/detoxification of neurotoxic substrates (23). Therefore, animal models recapitulating Parkin insolubility, aggregation and aggresome-formation, rather than complete gene knockouts, may thus provide an opportunity for analyzing Parkin misfunction.
In agreement with this notion, we identified the C. elegans homolog of human parkin, pdr-1, and characterized several pdr-1 mutants. In contrast to loss-of-function mutants of this gene locus, the in-frame deletion allele pdr-1(lg103) is hypersensitive for proteotoxic stress conditions. Both cytosolic stress and ER dysfunction exert specific toxicity in pdr-1(lg103) mutants. PDR-1 is involved in and regulated by the UPR, and truncated PDR-1(Δaa24–247) seems to sequester critical components of the ubiquitylation machinery. We show that this allele, when expressed in a human cell culture system, exhibits an increased tendency to form aggregates. We also found an increased tendency of α-synuclein to co-localize in these aggregates. In the worm, the corresponding C. elegans parkin mutant pdr-1(lg103) confers a highly penetrant, temperature-sensitive lethality in combination with transgenic expression of the human α-synuclein mutant A53T, but not of wild-type α-synuclein. Thus, by combining a neomorphic PDR-1/Parkin mutant similar to the aggregation-prone AR-JP variants, with an α-synuclein mutation, we have generated an animal model with high-throughput capacity that allows screening for PD modifiers.
RESULTS
C. elegans pdr-1 is the homolog of human parkin
We identified a single C. elegans homolog of human parkin [predicted open reading frame (ORF) K08E3.7] and named it pdr-1 (Parkinson's disease-related gene-1). It encodes a 386 amino acid protein, PDR-1, and shares the characteristic domain organization with human Parkin along with 28% overall sequence identity and 41% similarity (Fig. 1A and B).
To examine the expression pattern of C. elegans pdr-1, wild-type animals (N2) transgenic for different green fluorescent protein (gfp) reporter constructs (Fig. 1C) were analyzed. Expression starts in embryogenesis (Fig. 2A) and is maintained throughout development (Fig. 2B) until adulthood (Fig. 2C–H). The translational fusion gfp::pdr-1 is highly expressed in most neurons (Fig. 2B–E), where it localizes to both cell bodies and processes (Fig. 2B and D). GFP staining is cytoplasmic and in most cases excluded from the nucleus (Fig. 2C). A strong GFP signal was observed in all muscle cells (Fig. 2B and E–H), and is also detectable in other, probably all, tissues. Northern blot analyses using total RNA from each single developmental stage of C. elegans confirmed the temporal expression pattern of pdr-1 observed with gfp reporter constructs. Beginning at the larval L2 stage, pdr-1 transcript levels are up-regulated and reach a maximum in adulthood (Fig. 2I), suggesting developmental dynamics in pdr-1 function.
PDR-1 acts as an E3 ubiquitin ligase in a conserved ubiquitylation complex
To investigate the role of PDR-1 protein in C. elegans, we screened for its interaction partners using the yeast two-hybrid system. PDR-1 specifically interacts and cooperates with C. elegans UBC-2, UBC-18 and UBC-15 (Fig. 3A), homologs of the human E2 enzymes UbcH4/5, UbcH7/8 and Ubc6, respectively (35). Additionally, we identified CHN-1 (36) (Fig. 3A), the homolog of human E4 enzyme CHIP, that has been shown previously to interact with human Parkin and to enhance its E3 ligase activity (22). E4 enzymes such as CHN-1/CHIP function as ubiquitin-chain elongation factors that ensure efficient multi-ubiquitylation of substrates (37). Furthermore, we found an association of PDR-1 with RPT-2, a regulatory subunit of the C. elegans 26S proteasome (Fig. 3A). These yeast two-hybrid interactions of PDR-1 were confirmed by GST-pull-down experiments (Fig. 3B).
Self-ubiquitylation is characteristic for RING finger containing E3 ligases (38). To confirm the enzymatic activities of PDR-1, we performed an assay in which self-ubiquitylation in the absence of a specific substrate occurs on the ubiquitin ligase itself. In conjunction with the complete set of E1, E2 and E4 enzymes from C. elegans, and even in concert with human E2 enzyme UbcH7, a known binding partner of human Parkin (13), PDR-1 shows self-ubiquitylation (Fig. 3C). In this assay, only a minor fraction of PDR-1 was ubiquitylated. This was not necessarily surprising, as the self-ubiquitylation activity of human Parkin was also generally weaker than its poly-ubiquitylation activity on defined substrate proteins, and in some cases had required a more sensitive mode of detection using I125-labeled ubiquitin (15).
In summary, these biochemical data demonstrate that PDR-1 acts as an E3 ligase in a highly conserved ubiquitylation complex. We thus suggest that PDR-1 is the functional ortholog of human Parkin.
Molecular and biochemical analyses of mutant C. elegans pdr-1 alleles
To further investigate the in vivo function of pdr-1, we analyzed four different deletion mutants represented by the alleles lg103, tm598, lg101 and tm395 that were isolated from UV/trimethylpsoralen-mutagenized C. elegans deletion libraries (Fig. 4A and for overview see Fig. 1C). Cloning of the corresponding mutant pdr-1 cDNAs by polymerase chain reactions with reverse-transcribed RNA (RT–PCR) revealed that the alleles lg103 and tm598 carry in-frame deletions encoding the internally truncated PDR-1 proteins Δaa24–247 and Δaa140–263, respectively (Fig. 4A). The allele lg103 fuses exons 1 and 5, whereas the allele tm598 fuses exons 3 and 5 of pdr-1. Both these alleles delete the UPD and the first RING finger, but preserve the intact C-terminal IBR and second RING finger domain. However, the allele lg103 additionally lacks the important UBL domain which is still present in the allele tm598 (Fig. 4A). In contrast, the alleles lg101 and tm395 remove most of the pdr-1 ORF and result in out-of-frame mutations creating a premature translational stop codon (Fig. 4A). The deletion of allele lg103 was confirmed by northern blot analysis and revealed that the mutant gene is transcribed at a level comparable to wild-type. Importantly, transcription efficiency of the downstream gene K08E3.8 is unaffected by this deletion (Fig. 4B).
Surprisingly, we found by in vitro and yeast two-hybrid assays that the gene products of the in-frame deletion mutants pdr-1(lg103) and pdr-1(tm598) are still able to interact with distinct proteins of the ubiquitylation machinery (Fig. 4C and D). Truncated PDR-1(Δaa24–247), which bears the intact IBR and second RING finger domains, binds the same set of E2 and E4 enzymes as full-length PDR-1 (as shown in Fig. 3A and B). Similarly, PDR-1(Δaa140–263), which shows the same C-terminal domains, is also still able to associate with the E2 enzyme UBC-2 (Fig. 4C). However, association of truncated PDR-1(Δaa24–247) with the proteasomal subunit RPT-2 is disrupted (Fig. 4C), as this mutant protein lacks an intact UBL domain that is essential for coupling to the proteasome (12). In agreement with this, truncated PDR-1(Δaa140–263) and PDR-1(Δaa1–121Stop) which preserve an intact UBL domain are still able to associate with the proteasome (Fig. 4C).
Truncated PDR-1(Δaa24–247) encoded by the pdr-1(lg103) mutant is prone to aggregation
As pathogenic parkin point mutations have been shown to decrease the stability of the corresponding protein and/or tend to aggregate, we next tested the solubility of the C. elegans wild-type and mutant PDR-1 variants by transgenic expression in HEK293T cells (Fig. 4E). For this purpose, cells were transiently transfected with FLAG::pdr-1 constructs and extracts were prepared at different time points. All PDR-1 proteins could be extracted in a soluble form 24 h post-transfection. However, after 48 h post-transfection, only wild-type protein and mutant PDR-1 derived from alleles tm598 and tm395 could be solubilized in Triton X-100, whereas the majority of truncated PDR-1(Δaa24–247), derived from the allele lg103, was insoluble and could only be solubilized after SDS treatment.
Recently, it was shown that many of the disease-linked point mutations in parkin produce alterations in the solubility of the protein that influences its detergent extraction property and localization (27,32,34,39,40). We suggest that pdr-1 (lg103), but not the other three alleles we tested, shows a similar behavior like these PD-associated human Parkin mutants.
The pdr-1(lg103) mutant is hypersensitive to ER stress conditions
All pdr-1 mutants are viable and display no obvious morphological defects or alterations in motility, egg-laying behavior, brood size or life span under standard growth conditions. Furthermore, we could not detect any influence of the mutations on the survival of the dopaminergic neurons in the worms (Fig. 7A) as judged by the continued expression of an integrated marker that expresses gfp under the control of a dopaminergic neuron-specific promoter [dopamine transporter Pdat-1::gfp (41)].
Next, we challenged pdr-1 mutants with a variety of stressors. For this purpose, we treated worms with the reducing agents DTT, β-mercaptoethanol, as well as with tunicamycin, a specific inhibitor of N-linked glycosylation that leads to accumulation of unfolded proteins in the ER. In mammals and C. elegans, accumulation of misfolded proteins in the ER activates the unfolded protein response (UPR) that is mediated by three transducers: the protein–kinase and site-specific endoribonuclease IRE1; the eukaryotic translation initiation factor 2 kinase, PERK/PEK; and the transcriptional activator ATF6. C. elegans encodes single homologs of each stress sensor, ire-1, pek-1 and atf-6, and mutants are sensitive to elevated ER stress. An intact UPR is absolutely required for normal development as ire-1(v33) loss-of-function mutant suffers from developmental defects and a reduced brood size, whereas pek-1(ok275) and atf-6(RNAi) loss-of-function mutants are indistinguishable from wild-type animals. In conclusion, C. elegans, IRE-1 is the central regulator of the UPR, whereas PEK-1 and ATF-6 provide redundant protection against ER stress (42–44).
N2 wild-type animals were unaffected by stress treatments, whereas pdr-1(lg103) mutants grown in the presence of any of these ER stress-inducing chemicals displayed severe developmental defects and lethality at early larval stages. The majority of pdr-1(lg103) mutant animals died or arrested at, or prior to, the larval L3 stage (Fig. 5A). This result is similar to the behavior of ire-1(v33) and pek-1(ok275) mutants that are defective in the proper execution of the UPR pathway (42). Surprisingly, none of the other three analyzed pdr-1 deletion alleles showed a comparably strong phenotype (Fig. 5A).
We quantified the stress-sensitive phenotype of pdr-1 (lg103) mutants after treatment with low concentrations (1.5 µg/ml) of tunicamycin. A stress-inducible Phsp-4::gfp transcriptional reporter (43) was used in all experiments to monitor efficient induction of the UPR. C. elegans hsp-4 is a homolog of the mammalian ER chaperone BiP. At this concentration of tunicamycin, the overwhelming majority of wild-type animals matured to fertile adults, whereas most of ire-1(v33) and pek-1(ok275) mutant animals arrested during development and died, indicating increased stress sensitivity of mutants in the UPR. Almost 70% of homozygous pdr-1(lg103) animals showed ER stress hypersensitivity like ire-1 and pek-1 mutants (Fig. 5B).
As in our experiments pdr-1(lg103) behaved differently than the other C. elegans parkin alleles, it is possible that this mutation confers a neomorphic or a toxic gain-of-misfunction phenotype. In agreement with such a model is our finding that the mutant protein PDR-1(Δaa24–247) retains its capacity to interact with the E4 enzyme CHN-1/CHIP and with E2 enzymes, but fails to interact with the proteasome (Fig. 4C and D), and, moreover, shows a tendency to aggregate (Fig. 4E). To test this assumption, we first monitored effects of pdr-1 gene doses by analyzing single-heterozygous and compound-heterozygous mutants (i.e. with different mutations on both alleles). Although homozygous pdr-1(lg103) mutants showed the highest sensitivity upon ER stress, the combination of the two in-frame deletion alleles lg103 and tm598 also resulted in a significantly increased sensitivity. Notably, the presence of one wild-type copy of pdr-1, either genomic in a pdr-1(lg103)/+ heterozygote or from a transgenic array, was sufficient to restore viability and fertility (Fig. 5B and C). In conclusion, like the majority of familial parkin mutants in AR-JP, pdr-1(lg103) is recessive.
PDR-1 is involved in, and regulated by, the UPR
Next, we tested the genetic interactions between pdr-1 mutants and the UPR mutants ire-1(v33), pek-1(ok275) and atf-6(ok551). To do this, we counted the progeny of the corresponding single and double mutants (Fig. 6A). pdr-1 mutants have a brood size similar to wild-type, whereas ire-1(v33) single mutants have significantly less progeny. Strikingly, ire-1(v33);pdr-1(lg103) and ire-1(v33);pdr-1 (tm598) double mutants showed a dramatically reduced brood size with respect to each single mutant. As already observed in the tunicamycin stress assay, the allele lg103 confers a stronger phenotype as the second in-frame deletion tm598. Notably, complete loss of pdr-1 function in the ire-1 mutant background, i.e. in the double mutant ire-1(v33);pdr-1 (tm395), had no effect on the brood size. In contrast, both pek-1(ok275) and atf-6(ok551) single mutants, as well as pek-1(ok275);pdr-1(lg103) or atf-6(ok551);pdr-1(lg103) double mutants had brood sizes similar to wild-type. Previous experiments suggested that ire-1(v33) represents a null allele. Therefore, the most simple explanation for the strong synergistic effect of the ire-1(v33);pdr-1(lg103) and ire-1 (v33);pdr-1(tm598) double mutants is that both genes act in parallel pathways of the UPR.
To position pdr-1 functionally, we analyzed pdr-1 transcript levels in the background of UPR mutants. The pdr-1 transcription in pdr-1(lg103) animals is not affected, arguing against an auto-regulatory role on the transcript level. However, in pdr-1(lg103) mutants the UPR remained un-induced, but was still inducible by exogenous stress, as judged by hsp-4 expression in vivo and by northern blot analyses (Fig. 6B). Transcript levels of pdr-1 were reduced in ire-1(v33) and pek-1(ok275) loss-of-function alleles, and were as strongly up-regulated in the constitutive active atf-6(ok551) mutant background (Fig. 6B). We conclude that parkin expression is controlled by all three regulators of the unfolded protein response, and may therefore be positioned downstream of the UPR transducers IRE-1, PEK-1 and ATF-6.
Overexpression of α-synuclein A53T mutation in pdr-1(lg103) mutants leads to developmental arrest and lethality
Accumulation of α-synuclein in Lewy bodies is one of the hallmarks of sporadic PD and of its hereditary forms caused by mutations in the α-synuclein gene. We wanted to test whether a genetic link between pdr-1 dysfunction and α-synuclein aggregation exists in C. elegans. As the C. elegans genome does not encode an obvious homolog of α-synuclein, we expressed genomically integrated copies (to enhance expression levels) of human wild-type and mutant α-synuclein from a pan-neuronal promoter [Paex-3::α-synuclein, gift of G. Wong, Kuopio (10)] in pdr-1 mutant background. Recently, a neurotoxic effect after overexpression of α-synuclein in the eight dopaminergic neurons of C. elegans had been reported (45). In wild-type background, we did not observe any morphological phenotype on overexpressing wild-type or mutant α-synuclein. In contrast, the α-synuclein A53T mutant caused developmental defects and a temperature-sensitive lethal phenotype in pdr-1(lg103), whereas wild-type α-synuclein was inconspicuous in this genetic background (Fig. 7A). At 15°C, 15% of pdr-1(lg103) mutants expressing α-synuclein A53T arrested at early larval stages (Fig. 7B). At 20°C, this lethal phenotype became fully penetrant (Fig. 7C). In contrast, even at 25°C expression of α-synuclein A53T did not cause a phenotype in other pdr-1 alleles, and expression of wild-type α-synuclein was inconspicuous in any pdr-1 mutant background. In none of the pdr-1 mutants we observed death of dopaminergic neurons, as judged from the gfp expression pattern of an integrated Pdat-1::gfp marker (Fig. 7A).
Cytotoxicity is dependent on α-synuclein A53T and pdr-1(lg103) expression level
The incidence of PD increases exponentially with age in humans. The lack of a phenotype upon expression of α-synuclein in C. elegans may be the consequence of the short lifespan (generally less than 20 days) of these animals. The increase of transgenic α-synuclein expression could potentially phenocopy the situation seen in old patients. Therefore, we used genetic tools to monitor the consequences of different levels of α-synuclein accumulation in various mutant backgrounds. For this purpose, we crossed the pdr-1 mutants into strains that expressed α-synuclein wild-type or A53T mutation from varying genomic copies (Fig. 8A). Several results from these experiments are noteworthy. First, only strong expression of α-synuclein A53T (two copies of the transgene) resulted in a strong phenotype in pdr-1 (lg103). Secondly, similar to the results we had obtained after exposing the animals to ER stress, only a homozygous pdr-1(lg103) mutant background or, to some extent, a trans-heterozygous pdr-1(lg103)/pdr-1(tm598) genetic background resulted in a phenotype in α-synuclein A53T expressing worms. Thirdly, a single copy of the wild-type pdr-1 or of the alleles lg101 or tm395 was sufficient to prevent the establishment of a phenotype, even in animals that expressed two copies of α-synuclein A53T (Fig. 8A).
Our previous experiments have suggested that pdr-1(lg103) might cause a neomorphic phenotype (see Figs 4–7). If this were the case, then further increasing the expression level of this mutant should result in an aggravation of the α-synuclein lethal phenotype. To test this hypothesis, we analyzed the consequences of α-synuclein A53T expression in the pdr-1 (lg103);atf-6(ok551) double mutant background (Fig. 8B), as we have shown that the atf-6(ok551) mutant enhances the transcription rate of pdr-1. Lethality/arrest was strongly enhanced already at 15°C with respect to the pdr-1(lg103) single mutant. We conclude that lethality results from the expression of mutant α-synuclein in the sensitized background of recessive toxic gain-of-function alleles of the C. elegans parkin pdr-1. As α-synuclein was expressed from a pan-neuronal promotor, lethality seems to be caused by a neuron-specific toxicity.
Blocking the UPR is not sufficient for causing α-synuclein-mediated toxicity
The aggravation of a mutant α-synuclein phenotype by pdr-1 (lg103) could either be caused by blocking the ER stress response or by an effect of the mutant protein that does not involve its role in the UPR. To distinguish between these two possibilities, we analyzed α-synuclein-expressing animals, in which individual genes of the UPR had been mutated, as well as animals treated with exogenous ER stress. Expression of neither wild-type α-synuclein nor the A53T variant caused a detectable phenotype in the ire-1 (v33) mutant (Fig. 9A). Furthermore, animals expressing α-synuclein are not sensitive to tunicamycin treatment (Fig. 9B). Consistent with the latter finding, α-synuclein-expressing strains did not induce the UPR, as monitored by northern and hsp-4::gfp expression analyses in vivo (data not shown). We conclude from these results that the phenotype caused by the combination of α-synuclein A53T expression and pdr-1 toxic gain-of-function mutations is not mediated by the impairment of an ER stress pathway. This conclusion is further substantiated by the inconspicuous behavior of pdr-1 mutants and α-synuclein-expressing strains in assays testing increased oxidative and heat stress. Unlike mev-1 (kn1) (encoding a mutant version of a cytochrome b subunit) or ire-1(v33) strains, respectively, pdr-1 mutants and α-synuclein strains were not sensitive against paraquat and short heat-shock treatment (2 h 35°C) and showed no discernable phenotype (Fig. 9C and D).
Co-expression of α-synuclein A53T and PDR-1 variants enhances aggregate formation
To understand the reason for the synergistic effect of pdr-1 (lg103) and α-synuclein A53T, we co-expressed C. elegans Parkin variants and human α-synuclein in HEK293T cells and monitored the localization of the proteins by antibody stainings. In agreement with in vitro data (see Fig. 4E), we found that only PDR-1(Δaa242–247) showed altered localization and was prone to aggregation in human cell culture, whereas wild-type protein and the other PDR-1/Parkin derivatives showed uniform cellular distribution (Fig. 10A). These data suggest that the C-terminal IBR and RING2 domains in combination with the deletion of the UBL domain result in PDR-1/Parkin mislocalization and aggregation. Co-transfection of α-synuclein wild-type did not alter the localization of any of the PDR-1 variants. In all cases, α-synuclein wild-type was uniformly distributed within the cells. In contrast, combined expression of α-synuclein A53T with PDR-1 wild-type, or either of the two in-frame PDR-1 deletion variants Δaa24–247 or Δaa140–263, resulted in aggregation of both proteins. However, expression of α-synuclein A53T together with the C-terminal PDR-1 deletion amino acids 1–199Stop did not affect localization and thus did not result in aggregate-like structures. These data indicate that the cysteine-rich C-terminus of PDR-1 mediates α-synuclein A53T-dependent aggregation.
In addition, the solubility of the PDR-1 derivatives was tested in cellular extracts. Similar to human Parkin in a previous report (27), C. elegans PDR-1 in an α-synuclein-expressing background is observed both in the Triton X-soluble and Triton X-insoluble fractions (Fig. 10B). The shift of PDR-1 wild-type and Δaa140–263 to the insoluble fraction in an α-synuclein A53T background correlates with its aggregation (compare Fig. 10A). No A53T-dependent increase of the insoluble fraction was observed for PDR-1(Δaa24–247) that already forms aggresome-like structures without co-expression of α-synuclein (compare Figs 4E and 10B).
DISCUSSION
Many different mutations in human parkin have been associated with particularly severe recessive forms of PD resulting in the clinical and pathological heterogeneity (26). However, the biochemical mechanisms leading to AR-JP are poorly understood. Although complete loss of Parkin function is considered to be causative for disease, at least some disease-linked point mutants of Parkin have been described to retain substantial biochemical activity (27–31) and additionally lead to Parkin protein aggregation and aggresome formation (28,29,32–34,39,40,46). In contrast, Parkin animal models generated so far are exclusively based on complete knockout strategies, although there is growing evidence that the disease-linked mutations may alter several properties of Parkin function, not all of them can be phenocopied by abolishing the expression of this E3 ligase (27).
This may explain the weak phenotypes observed in several mouse knockout strains of parkin (reviewed in 3), compared with the severe cellular effects in humans. Interestingly, none of the proposed Parkin substrates are stabilized in parkin knockout mice (47–49), implying that either the bona fide substrates have not been identified yet, or that the protection/detoxification mechanism to which Parkin contributes does not depend on the degradation of toxic substrates. Alternatively, at least partially redundant pathways for removal of Parkin substrates might exist. Consistently, the Parkin substrate synphilin-1 has been shown to be ubiquitylated by other E3 ligases (50–53) that might compensate for Parkin loss-of-function. In contrast, a protein exerting a partial loss-of-function or an altered localization or regulation (i.e. caused by a loss of only one functional domain) might be more deleterious to the cell, in particular, under stress conditions. Therefore, animal models that are generated to further study the disease mechanism and to screen for genetic of pharmacological modification might be based on parkin neomorphic or partial loss-of-function mutants similar to the disease-linked alleles rather than on knocking out/down Parkin activity. We, for the first time, describe such a model in this study.
Like other RING finger containing proteins, Parkin is an E3 ubiquitin ligase which targets several substrates for degradation, including, as proposed, α-synuclein (13). Here we show that the C. elegans protein PDR-1 is the ortholog of human Parkin, as it interacts with enzymes of the ubiquitin/proteasome pathway and mediates E3 activity in a fully synthetic in vitro assay (Fig. 3C). Like human Parkin (13,22,46), PDR-1 specifically cooperates with E2 and E4 enzymes involved in cytosolic protein stress response and the ERAD pathway (Fig. 3A and B). Our data are consistent with Parkin/PDR-1 having several roles in the removal of proteotoxic stress: it contributes to the UPR and participates in the cytosolic stress response. We have analyzed and compared two PDR-1/Parkin loss-of-function alleles and two in-frame deletions encoding truncated proteins in biochemical, genetic and pharmacological tests, and showed that only the toxic gain-of-misfunction mutant pdr-1(lg103) is hypersensitive to distinct proteotoxic stress conditions.
PDR-1 is part of the UPR pathway
UPR is an intracellular signaling pathway that mediates an adaptation to ER stress at both transcriptional and translational levels. It augments folding and degradation capacity and also acts by reducing the protein load in the ER (18). Treatment with tunicamycin, an inhibitor of N-linked glycosylation, results in high amounts of unfolded proteins in the ER lumen and thus is a potent inducer of the UPR pathway.
We found that particularly one parkin allele, pdr-1(lg103), an in-frame deletion resulting in a PDR-1 protein without functional UBL and RING1 domains, is sensitive toward tunicamycin treatment. These animals specifically suffer from severe developmental defects and lethality (Fig. 5), but they are, at the same time, not more sensitive than wild-type to heat or oxidative stress (Fig. 9C and D). The hypersensitivity of pdr-1(lg103) animals against ER stress is similar to the phenotype of mutants defective in the UPR, which is required for normal development in C. elegans. Tunicamycin-treated mutants, or non-treated double mutants of the pathway, including ire-1;pek-1, xbp-1;pek-1 or xbp-1;atf-6(RNAi) animals, typically arrest at the L2/L3 larval stages due to the degeneration of the intestine (42). At L2, the C. elegans intestine induces high-level synthesis of secretory proteins which in the absence of proper UPR function make the animals more susceptible toward ER stress (42). Here we show that PDR-1 contributes to this anti-stress response, and is consequently up-regulated in the L2/L3 larval stages (Fig. 2I).
Our results indicate that specifically the in-frame deletions pdr-1(lg103) and pdr-1(tm598) genetically interact with ire-1 rather with pek-1 or atf-6, as a synergistic effect was only observed in the ire-1(v33);pdr-1(lg103) and ire-1 (v33);pdr-1(tm598) double mutants (Fig. 6A). This suggests that pdr-1 may act to some extent in parallel to ire-1 signaling, perhaps by directly contributing to either the pek-1 or atf-6 pathway. IRE-1 and PEK-1 pathways have indeed been shown to provide redundant protection against ER stress (42). Moreover, we found that transcription of pdr-1 is controlled by all three signaling pathways IRE-1, PEK-1 and ATF-6 (Fig. 6B) Therefore, PDR-1 can be positioned downstream of the UPR transducers. Unfolded proteins in the ER are retro-translocated to the cytosol and turned over by the ERAD pathway which function is regulated by the UPR (19). Mutations in ERAD genes and in the recently identified C. elegans abu gene family (activated in blocked UPR) also result in synthetic lethality in combination with defective UPR mutants, but are, in contrast to pdr-1, themselves activators of the UPR (20,21). Taken together, these data suggest a general role for PDR-1/Parkin in the UPR pathway.
Interestingly, there are several indications for a role of ER stress and the UPR in the pathophysiology of PD (54,55). For example, it was shown that some PD mimetics like 6-OHDA, MPP+ and rotenone specifically induce ER stress and activate the UPR in cultured neuronal cells (56). The biochemistry of stress induction is most likely very similar in C. elegans, as we could recently demonstrate the susceptibility of worms to MPP+ treatment and the amelioration of neurotoxicity by anti-PD drugs (57).
Overexpression of the α-synuclein A53T variant is toxic in the background of pdr-1(lg103)
Accumulation of the cytosolic protein α-synuclein in Lewy bodies is a hallmark of PD, and mutations result in autosomal dominant familial forms. The A53T mutation enhances aggregation of α-synuclein by accelerated fibril formation. This in turn impairs the proteolytic system and increases the sensitivity of cells to proteasome inhibition (reviewed in 58). Here we demonstrated that expression of human mutant α-synuclein (A53T), but not of wild-type (10), also exerts a cytotoxic effect in C. elegans pdr-1(lg103) animals that results in developmental arrest and lethality (Fig. 7). This phenotype is similar to the ER stress-induced one and is dependent on temperature and on the gene dose of the α-synuclein A53T (Figs 7B–D and 8A). The fact that this phenotype in C. elegans already arises on the second to third day after fertilization is remarkable, given that α-synuclein aggregation in mice is only toxic after months (59). Probably due to the short lifespan of the animals (less than 20 days), this toxicity was only observed in animals that combined expression of both mutant α-synuclein and the aggregation-prone Parkin encoded by pdr-1(lg103). Our results revealed neither increased mRNA nor elevated protein levels of chaperones in pdr-1(lg103) animals or worms overexpressing α-synuclein, that would have indicated an elevated stress level as a consequence of this mutation/expression (Fig. 6B and data not shown).
As α-synuclein is a cytosolic protein and accumulates in C. elegans neurons, both in the cytoplasm and axonal processes (data not shown), toxicity most likely resembles a cytosolic mechanism. Consistently, this effect is independent of the ER stress response and the UPR signaling pathway (Fig. 9A and B). Expression of mutant α-synuclein in wild-type worms treated with tunicamycin or in ire-1-deficient worms did not impair development or viability. Moreover, in α-synuclein-expressing animals the ER stress marker hsp-4/BiP was not induced (data not shown). Thus, pdr-1(lg103) exacerbates mutant α-synuclein-induced toxicity in an UPR-independent way.
Our PDR-1 mutants, similar to the parkin mutants tested recently (27,40), have differential solubility properties in transfected cells. PDR-1(Δaa242–247) is unique because it is already mostly detergent-insoluble and forms intracellular, aggresome-like structures in the absence of transgenic co-expression of α-synuclein. Aggregate formation of PDR-1 derivatives containing the C-terminal IBR and RING2 domains is induced by α-synuclein A53T co-expression. The second RING finger had been implicated in substrate binding of synphilin-1 previously (27). One could argue that although these variants are capable of binding substrates, at least PDR-1(Δaa242–247) will be unable to function in the ubiquitylation process due to the truncated UBL domain, resulting in the accumulation of both PDR-1 and misfolded α-synuclein. In C. elegans neurons, this combination is cytotoxic and causes the death of the animals. Moreover, aggregation is dependent on the PDR-1 C-terminus, because in PDR-1(aa1–199Stop) even α-synuclein A53T is uniformly distributed throughout the cell.
Misfolding, not loss of expression, of Parkin may be the prime cause of neurotoxicity
Similar to complete loss of fly (60,61) or mouse Parkin (47,48,62,63), we showed here that deletion mutants of the C. elegans parkin ortholog pdr-1 are viable and do not display obvious defects in dopaminergic neurons (Fig. 7A). Furthermore, C. elegans parkin mutants are not sensitive to various stress conditions (Figs 5A and B, 7B–D and 9C and D). In contrast, homozygous pdr-1(lg103) in-frame deletion mutants are particularly hypersensitive to ER-derived and cytosolic protein folding stress (Figs 5 and 7). Although the other in-frame deletion mutant pdr-1(tm598) did not show increased stress sensitivity that was detectable in our assay, it can serve as a sensitized background. pdr-1(lg103)/pdr-1 (tm598) compound heterozygotes showed an augmented susceptibility to tunicamycin treatment and α-synuclein expression (Figs 5B and 8A). Moreover, in contrast to the pdr-1 loss-of-function allele tm395, both pdr-1 in-frame deletion alleles, lg103 and tm598, show a clear synergistic effect on the brood size of ire-1(v33) loss-of-function mutants (Fig. 6A). Taken together, mutations that eliminate or reduce pdr-1 expression obviously do not exert such effects. The observation that the increased transcription of pdr-1(lg103) in the atf-6(ok551) hypermorphic background exacerbates the α-synuclein A53T conferred cytotoxicity (Fig. 8B) provides further support for the neomorphic nature of the lg103 mutation. We therefore conclude that small domain-specific in-frame deletions of pdr-1 confer a different quality than a complete gene knockout.
Interestingly, allele lg103 confers a stronger phenotype than allele tm598. Both pdr-1 in-frame deletion variants lack the first RING domain, the difference being that in pdr-1(lg103) the UBL domain is also missing whereas it remains intact in pdr-1(tm598) (Fig. 4A). It was recently shown that the UBL domain regulates the stability of the Parkin protein (14). In line with these data, we obtained higher amounts of the corresponding truncated PDR-1(Δaa24–247) protein from recombinant expression in SF9 insect cells compared with full-length PDR-1 (data not shown). Although both PDR-1 (Δaa24–247) and also PDR-1(Δaa140–263) still bind enzymes of the ubiquitylation machinery, interaction of PDR-1(Δaa24–247) with the proteasome is abrogated (Fig. 4C and D). Moreover, only this allele expressed mutant Parkin that became insoluble within 48 h after expression in human cell culture (Fig. 4E). Thus, an increased intracellular concentration and/or a misregulation of PDR-1 (Δaa24–247) combined with the residual binding activity may confer a more severe phenotype.
As we showed that PDR-1(Δaa24–247) still binds to essential components of the ubiquitylation system, including CHIP/CHN-1 (Fig. 4C and D), it is possible that this mutant (and other PD-related parkin variants with similar capacities) sequesters and inactivates critical components of the protein degradation machinery. Recently, we have shown that CHN-1 is expressed in the cytosol and binds to chaperones (36), similar to its human ortholog CHIP, which is involved in the chaperone/Parkin-mediated quality control of the ER protein Pael-R (22). Thus, CHN-1 might assist in regulating the cellular balance between folding and degradation and its titration could lead to a dramatic change in the folding capacity of the cytosol. Thus, it is tempting to speculate that PDR-1 (Δaa24–247) might sequester critical components of the cytosolic/ERAD ubiquitylation machinery and its associated chaperones, rendering these animals sensitive toward proteotoxic stress. pdr-1(lg103) can tolerate a certain threshold of aggregation-prone proteins, but high cellular levels, together with an impaired detoxification system, cause developmental defects. The recent findings indicate that overexpression of torsin protects from α-synuclein-induced toxicity is fully consistent with this model, as torsin has been suggested to manage protein folding (45).
In summary, these data suggest that studying parkin alleles, either neomorphic (gain-of-misfunction) alleles or partial loss-of-function alleles that have retained some properties of the enzyme (like substrate binding), may provide further insights into the biological role of Parkin. It would be interesting in the future to adopt similar strategies in other model organisms to help elucidating the molecular and cellular mechanisms resulting in AR-JP. Moreover, the powerful genetics of C. elegans can now be exploited to identify modifiers of parkin/α-synuclein-induced proteotoxicity as well as to study the effects of specific human disease-linked parkin mutations.
MATERIALS AND METHODS
Strains
Worms were handled according to the standard procedures as previously described (64). The mutations and integrated arrays used in this study are listed by chromosomes (LG) as follows. LGII: ire-1(v33); LGIII: pdr-1:lg101, lg103, tm395, tm598;mev-1(kn1); LGIV: WG8 Is[aex-3::α-syn(A53T); Pdat-1::gfp]; LGV: zcIs4[Phsp-4::gfp], Is[Pdat-1::gfp;rol-6]; LGX: pek-1(ok275), atf-6(ok551); Unmapped: WG3 Is[aex-3::α-syn(WT);Pdat-1::gfp]. N2 (Bristol) was used as the C. elegans wild-type strain, VT847 as the C. briggsae and EM464 as the C. remanei strain.
Isolation and characterization of pdr-1 deletion mutants
Nested PCR was used to screen an UV/TMP mutagenized C. elegans library with pdr-1-specific primers and isolated two pdr-1 mutant deletion alleles, lg103 and lg101, and received two additional deletion alleles tm598 and tm395 provided by Dr Shohei Mitani. Deletion breakpoints (cosmid K08E3 coordinates): lg103, 30885/30886–32017/32018; lg101, 31312/31313–33059/33060; tm598, 31365/31366–32062/32063; tm395, 31601/31602–32081/32082. The mutants were backcrossed several times with N2 wild-type animals. Twenty-five ng/µl of the rescuing subclone was co-injected with 25 ng/µl marker sel-12::gfp into pdr-1(lg103) mutant animals. Six independent transgenic lines [pdr-1(lg103); byEx417-422[pdr-1(Rescue);sel-12::gfp]] were analyzed for rescue on plates containing 2.5 µg/ml tunicamycin.
Plasmids
To generate pdr-1 GFP reporter constructs, a 650-bp promoter region immediately 5′ of the predicted initiation ATG codon of pdr-1 was amplified by PCR and ligated in-frame with GFP in the plasmid pPD117.01 (gift of Andy Fire). The translational fusion construct, Ppdr-1::gfp::pdr-1, was engineered by in-frame ligation of an 8.5-kb fragment, containing the complete genomic region of pdr-1, into the promoter construct Ppdr-1::gfp. To generate the rescuing subclone, a 13 707-bp EcoRV fragment of cosmid K08E3 carrying the pdr-1/K08E3.8 operon was subcloned into vector pCRScript-AmpSK(+) (Stratagene) and a 4-bp insertion was introduced into exon 3 of the downstream gene K08E3.8, causing a frame shift at nucleotide 192 of the coding sequence. To verify the yeast two-hybrid results, full-length cDNAs of pdr-1, pdr-1(lg103), pdr-1(tm598), pdr-1(lg101), ubc-2, ubc-6, ubc-7, ubc-14, ubc-15, ubc-18, chn-1 and rpt-2 were cloned into vectors pGBKT7 and pGADT7 (Clontech). pdr-1 and pdr-1(lg103) ORFs were cloned into vector pCite-4a(+) (Novagen) for in vitro translation. To generate GST-fusion proteins, ubc-2 and ubc-18 ORFs were cloned into vector pGSTparallel-3, pdr-1 ORF was cloned into a modified Baculovirus vector pAcUW51 (Pharmingen) and chn-1 ORF was cloned into vector pGEX4T1 (Pharmacia). For in vitro ubiquitylation assays, chn-1 ORF was cloned into vector pET21a(+) (Novagen). pET21a-UbcH7 (gift of Martin Scheffner) was used to express human E2 enzyme UbcH7 in Escherichia coli. To transfect human cells with pdr-1, full-length cDNAs of wild-type pdr-1 and truncated versions of the mutants lg103, tm598 and tm395 were cloned into a modified pcDNA3.1-FLAG (Invitrogen).
RNA isolation and northern blot analyses
Total RNA was isolated from mixed or a specific staged worm plates and prepared with an RNAeasy kit according to the manufacturer's instructions (Qiagen). Five micrograms of total RNA was loaded per lane and blots were performed following standard procedures. Probes were labeled with α32P dCTP using the Megaprime labeling kit according to the manufacturer's instructions (Amersham). ama-1- and act-1-specific probes were used as controls to adjust for equal loading.
RT–PCR
Single-strand cDNA synthesis was carried out using oligo-dT primer and reverse transcriptase with total RNA samples of the respective strains. All cDNAs were isolated by PCR on single-strand cDNA, using combinations of oligo-dT and gene-specific primers. Designed species-specific oligonucleotides were then used to amplify the respective pdr-1 ORFs from related species of the genus Caenorhabditis.
GFP in vivo expression studies
Both plasmids, the promoter construct Ppdr-1::gfp or the translational fusion Ppdr-1::gfp::pdr-1, were microinjected at a concentration of 25 ng/µl together with pRF4 at 75 ng/µl as an injection marker into N2 wild-type animals. Five to six independent lines transgenic for either the promoter construct (N2 byEx3187-3191[Ppdr-1::gfp;rol-6]) or the translational fusion (N2 byEx411-416[Ppdr-1::gfp::pdr-1;rol-6]), respectively, were used for expression pattern analysis.
Yeast two-hybrid assays
Protein interaction studies were performed using the MATCHMAKER GAL4 Two-Hybrid System 3 according to the manufacturer's instructions (Clontech). Full-length PDR-1 was fused to GAL4 DNA-binding domain (pGBKT7) and transformed into yeast AH109. C. elegans GAL4 activation domain libraries (gift of Robert Barstead) were used as prey. Approximately 150 000 transformants were screened and about 50 independent clones were identified as true positives, including several isolates of distinct ORFs. Protein interaction studies were carried out under high stringency conditions according to the manufacturer's instructions.
Expression and purification of proteins
Recombinant Baculovirus was generated using the Baculo Gold System as described by the manufacturer (Pharmingen). For protein production, 20 ml of infected SF9 cells (3×106 cells/ml) were grown for 2 days. Cells were lysed in twice the volume of the cell pellet in buffer A [10 mm Tris, pH 8; 10 mm DTT or 10 mm β-mercaptoethanol+complete protease inhibitors (Boehringer)] using a dounce homogenizer. BL21(pRIL) E. coli cells were used to produce recombinant E2 and E4 enzymes from the described clones and lysed in buffer A. For purification of GST-fusion proteins (GST::UBC-2/GST::UBC-18/GST::CHN-1/GST-mycPDR-1) cleared lysates were allowed to bind to 500 µg of glutathione-Sepharose beads (Pharmacia). After extensive washing in buffer A with 0.1% Triton X-100 and 500 mm NaCl, GST-fusion proteins were eluted with 10 mm glutathione.
GST-pull-down experiments
PDR-1 and PDR-1(Δaa24–247) mutant versions were radioactively labeled with 35S methionine/cysteine by in vitro translation using the respective plasmids according to the manufacturer's instructions (TNT Coupled Reticulocyte Lysate System, Promega). Lysates were incubated overnight at 4°C on the loaded glutathione-Sepharose beads and washed at least five times in buffer A with 0.1% Triton X-100 and 150 mm NaCl. Reactions were separated by SDS–PAGE and visualized by Coomassie blue staining and autoradiography.
In vitro ubiquitylation assay
Reactions were done as previously described (36). Purified rabbit E1 (Affiniti), purified GST-UBC-2 as well as UbcH7 and CHN-1 crude E. coli cell extracts were used for self-ubiquitylation reactions of purified GST::myc::PDR-1. Reactions were separated by SDS gel electrophoresis followed by western blotting using 9E10 anti-myc (Boehringer) antibody.
Expression of mutant PDR-1 in HEK293T cells
HEK293T cells were cultured in Dulbecco's modified Eagle's medium with Glutmax (PAA Laboratories GmbH) supplemented with 10% fetal calf serum. Cells were transfected using Lipofectamine 2000 Reagent (Invitrogen) according to the supplier's instructions. Cells were harvested 24 or 48 h post-transfection and lysed in lysis buffer (10 mm Tris–HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, containing 1% Triton X-100) with proteinase inhibitors (Sigma) on ice for 15 min. Cell lysates were centrifuged at 50 000g for 1 h at 4°C. Supernatants were saved as the Triton X-100 soluble fractions (S) and pellets were washed once with lysis buffer containing Triton X-100 before re-extraction with SDS buffer (lysis buffer containing 1% SDS in place of Triton X-100). After centrifugation at 50 000g for 1 h at 4°C supernatants were saved as the SDS fractions (P). Equivalent amounts of proteins among different S and P fractions were resolved by SDS–PAGE and analyzed by western blotting procedures using anti-FLAG and anti-β-actin antibodies (Sigma).
Immunocytochemistry with FLAG-tagged PDR-1
HEK293T were cultivated on 4-well chamber slides (Nunc) and transfected using Lipofectamine 2000 according to the manufacturer's instruction (Invitrogen). After 48 h, transfected cells were washed once with PBS and fixed with cold methanol for 10 min and acetone for 5 min at −20°C. Fixed cells were washed twice with PBS and blocked with 1% BSA for 30 min. Cells were then incubated with the primary rabbit anti-FLAG antibody (1:200, Sigma) and the monoclonal anti-Synuclein antibody (1:500, Biosource). The secondary antibody against mouse was labeled with Cy3 (1:800, Jackson Immunoresearch) and the anti-rabbit antibody was labeled with Alexa 488 (1:800, Invitrogen). Single-optic sections were recorded using Zeiss Axiovision microscope.
Drug treatments
Adult worms were allowed to lay eggs for 3 h at 20°C on NGM agar plates containing varying concentrations of DTT, β-mercaptoethanol or tunicamycin (Calbiochem). Eggs were counted and progenies were studied 3 days later (42). Synchronized L1 worms were treated with 2 mm paraquat (Sigma) solution and survival at 20°C was studied 3 days later. Mean values were calculated from different experimental groups of 3–10 independent assays, each.
Heat-shock experiments
Synchronized L2 larvae grown at 20°C were heat stressed for 2 h at 35°C and afterwards further maintained at 20°C. Development and survival was scored 2 days later. Mean values were calculated from different experimental groups of six independent assays.
Software and microscopy
Quantitative evaluation of northern blots was performed using ImageQuant 5.0 software (Molecular Dynamics). The amino acid alignment was generated using Vector NTI version 6.0 (InforMax). Pictures of GFP expression were taken with an AxioPlan 2 Microscope (Zeiss) using the AxioVision 3.0 software.
ACKNOWLEDGEMENTS
We thank Bianca Sperl for excellent technical assistance, Philipp Kahle, Stefan Eimer and the members of the Baumeister lab for helpful discussions and critical reading of the manuscript, Christian Haass for support, Claudia Rudolph (EleGene) for isolating pdr-1(lg103) and pdr-1(lg101), Julia Sämann for help with the yeast experiments, Shohei Mitani for providing pdr-1(tm598) and pdr-1(tm395), Garry Wong for integrated α-synuclein strains, Randal Kaufmann, David Ron, Randy Blakely, Andy Fire, Martin Scheffner and Robert Barstead for strains, plasmids and cDNA libraries. Some strains used in this study were obtained from the C. elegans Genetics Center, St Louis, which is funded by the NIH. This work was supported by grants from the European Community (through the Integrated Project APOPIS), the Friedrich-Baur Stiftung, the Fonds der Chemischen Industrie to R.B., and the Deutsche Forschungsgemeinschaft to R.B. (SFB596) and to T.H. (SFB 444).
Conflict of Interest statement. All authors have no conflict of interest to declare.
Present address: Centre for Molecular Neurobiology (ZMNH), University of Hamburg, Germany.
![Figure 1.C. elegans PDR-1 is conserved in evolution. (A) Schematic comparison of human Parkin and C. elegans homolog PDR-1. The respective domains are boxed: UBL: ubiquitin-like domain; UPD: unique Parkin domain; RING: C3HC4 RING finger domains; IBR: C6HC in-between RING finger domain. Identity and similarity values for each domain are shown. (B) PDR-1/Parkin protein alignment. Cloning and sequencing of the C. elegans pdr-1 cDNA revealed an additional coding exon (exon 4) not recognized by gene predictions. Additionally, the cDNAs of pdr-1 from the related nematode species C. briggsae and C. remanei were cloned. Length of PDR-1/Parkin proteins: C. elegans: 386 amino acids; C. briggsae: 385 amino acids; C. remanei: 387 amino acids; D. melanogaster: 468 amino acids; M. musculus: 464 amino acids; R. norvegicus: 465 amino acids; B. taurus: 465 amino acids; H. sapiens: 465 amino acids. Black shading indicates sequence identity, gray shading sequence similarity. Asterisks indicate positions of familial PD missense mutations in human Parkin (26). (C) Genomic organization and gene structure of pdr-1. Coding exons are depicted as boxes, introns as lines. pdr-1, together with its downstream gene K08E3.8 forms an operon (depicted by an arrow) driven by a single promoter, whereas the upstream gene cyk-4 (ORF K08E3.6) is located in a head-to-head orientation. pdr-1 is trans-spliced to splice leader 1 (SL1), the downstream gene K08E3.8 is trans-spliced to SL2. As both genes are co-transcriptionally regulated, K08E3.8 served as an internal control in this study. Shown are promoter construct (Ppdr-1::gfp), translational gfp fusion (Ppdr-1::gfp::pdr-1) and the rescuing subclone [Rescue pdr-1(wt)]. Lines represent the genomic regions cloned into the respective constructs. The asterisk indicates the position of an engineered frame-shift mutation in the downstream gene of the rescuing clone. The position and extent of the four analyzed pdr-1 deletions alleles (lg103, tm598, lg101 and tm395) are depicted by lines.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/22/10.1093/hmg/ddi371/2/m_ddi37101.jpeg?Expires=1716359187&Signature=H6l8j2MkSwHaBktTV8wBQ1sYgvfpHfwYHufSBS2RJoN15KkBXrxnuVtkopinKrF6qPFjv-neZN-odx4Uhrdz2Bv7MJjKlmqZXOfz03JtksqnKfiwNYe6Xdo~aE1zpo5Y9f4-BRYLkNj3-iCpTVtlA2xGiv7x6JOq4McwDWMYYfMfQfHGtF4luACBFwDwUFcn~XJF38T8fR6KALKiKbySH~fstRzfLdg0W~tJ~eUgDMXNxhv0PPjyAvkXhU6y0jy00nV3GwXEFNccScaCTejitCZSLwng5cDkeLTB0Dil2FjGChGurwgd8RuB9dF0a2MD1lokP~2irTlg7TXH5Iop2w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 1.C. elegans PDR-1 is conserved in evolution. (A) Schematic comparison of human Parkin and C. elegans homolog PDR-1. The respective domains are boxed: UBL: ubiquitin-like domain; UPD: unique Parkin domain; RING: C3HC4 RING finger domains; IBR: C6HC in-between RING finger domain. Identity and similarity values for each domain are shown. (B) PDR-1/Parkin protein alignment. Cloning and sequencing of the C. elegans pdr-1 cDNA revealed an additional coding exon (exon 4) not recognized by gene predictions. Additionally, the cDNAs of pdr-1 from the related nematode species C. briggsae and C. remanei were cloned. Length of PDR-1/Parkin proteins: C. elegans: 386 amino acids; C. briggsae: 385 amino acids; C. remanei: 387 amino acids; D. melanogaster: 468 amino acids; M. musculus: 464 amino acids; R. norvegicus: 465 amino acids; B. taurus: 465 amino acids; H. sapiens: 465 amino acids. Black shading indicates sequence identity, gray shading sequence similarity. Asterisks indicate positions of familial PD missense mutations in human Parkin (26). (C) Genomic organization and gene structure of pdr-1. Coding exons are depicted as boxes, introns as lines. pdr-1, together with its downstream gene K08E3.8 forms an operon (depicted by an arrow) driven by a single promoter, whereas the upstream gene cyk-4 (ORF K08E3.6) is located in a head-to-head orientation. pdr-1 is trans-spliced to splice leader 1 (SL1), the downstream gene K08E3.8 is trans-spliced to SL2. As both genes are co-transcriptionally regulated, K08E3.8 served as an internal control in this study. Shown are promoter construct (Ppdr-1::gfp), translational gfp fusion (Ppdr-1::gfp::pdr-1) and the rescuing subclone [Rescue pdr-1(wt)]. Lines represent the genomic regions cloned into the respective constructs. The asterisk indicates the position of an engineered frame-shift mutation in the downstream gene of the rescuing clone. The position and extent of the four analyzed pdr-1 deletions alleles (lg103, tm598, lg101 and tm395) are depicted by lines.
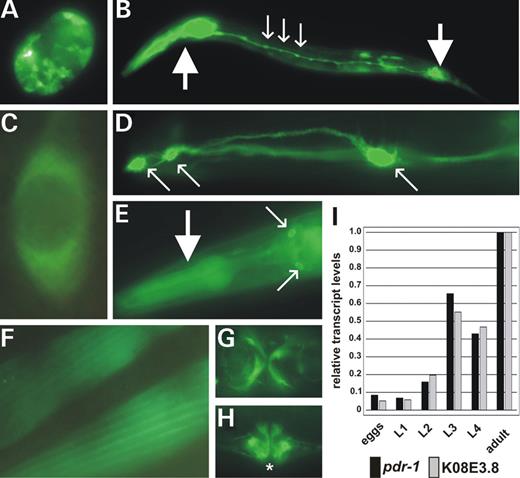
Figure 2.pdr-1 is ubiquitously expressed and developmentally regulated. (A–H) Transgenic expression of different gfp reporter constructs in N2 wild-type animals (for constructs, see Fig. 1C). In all the 11 independent transgenic lines analyzed, gfp expression patterns, although mosaic, were almost identical. (A) Embryo expressing gfp in almost all cells. (B) L2 larval gfp expression in pharyngeal and anal muscles (big arrows) as well as in neurons of the ventral nerve cord (small arrows). (C) Cytoplasmatic localization of the translational fusion GFP::PDR-1 in a neuron. (D) GFP::PDR-1 localization in cell bodies (small arrows) and processes of head neurons. (E) Pharyngeal muscles (big arrow) and neurons of the head (small arrows). (F) Body-wall muscles. (G) Vulval muscles (ventral view). (H) Vulval muscles (lateral view). The vulva opening is marked by an asterisk. (I) Northern blot analyses show co-transcriptional regulation of pdr-1 and K08E3.8 during all developmental stages, from embryogenesis (eggs) throughout larval stages (L1–L4) until adulthood (adult). pdr-1 and K08E3.8 transcript levels are up-regulated beginning in L2 and strongly increasing in L3. All transcription levels indicated are relative to young adult levels and were adjusted for equal loading with the corresponding ama-1 (large subunit of RNA polymerase II) level.
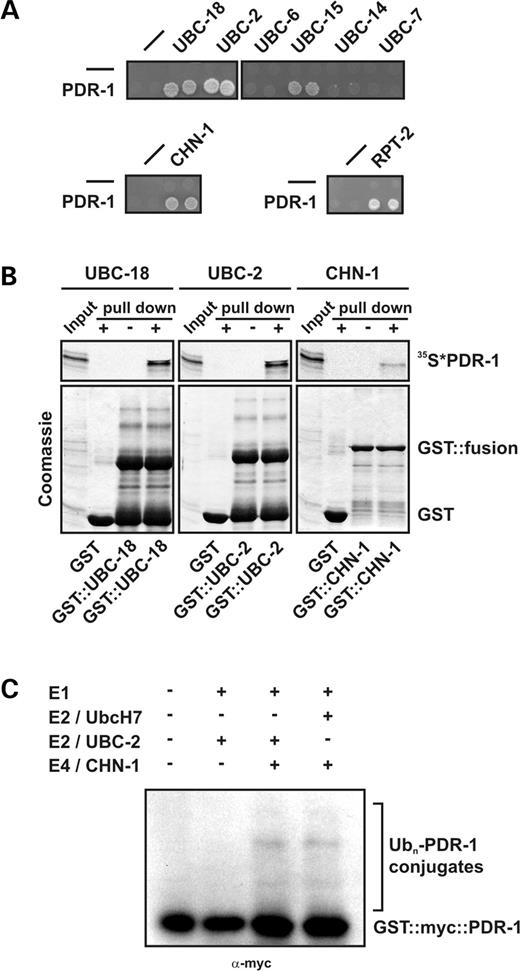
Figure 3. PDR-1 physically associates with a conserved C. elegans degradation machinery and mediates E3 ubiquitin ligase activity. (A) GAL4 yeast two-hybrid interactions studies of C. elegans PDR-1. Growth analysis of yeast cells expressing the indicated combinations of control (−), PDR-1 and its associated proteins, to test for interaction-dependent activation of HIS3 and ADE2 genes. Equal concentrations of the respective yeast strains were spotted in doublets on appropriate selective medium plates. PDR-1 specifically interacts with C. elegans E2 enzymes UBC-18/UbcH7/8, UBC-2/Ubch4/5 and UBC-15/Ubc6, but not with UBC-6, UBC-7 or UBC-14, homologs of S. cerevisiae Ubc7. In addition, PDR-1 associates with the E4 enzyme CHN-1/CHIP and with the proteasomal subunit RPT-2/P26s4. (B) PDR-1 GST-pull-down experiments with recombinant proteins analyzed by Coomassie blue staining followed by autoradiography. Immobilized GST-fused UBC-18/UbcH7/8, UBC-2/UbcH4/5 and CHN-1/CHIP are able to bind and pull down in vitro translated PDR-1, labeled with 35S methionine/cysteine. GST alone was used as a negative control. (C) In vitro self-ubiquitylation of PDR-1. The entire ubiquitylation system was reconstituted using purified proteins. Baculoviral GST::myc::PDR-1 was incubated with the combination of ubiquitin, E1 and the C. elegans E2 enzyme UBC-2 or human E2 UbcH7, as well as the E4 enzyme CHN-1, as indicated, and analyzed by western blotting using anti-myc antibody. Efficient self-ubiquitylation of PDR-1 requires E1, E2 (C. elegans UBC-2 or human UbcH7) and the E4 enzyme CHN-1.
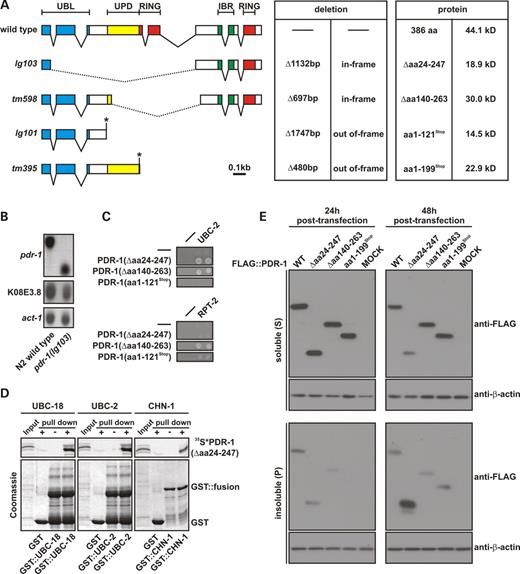
Figure 4. Molecular and biochemical analyses of pdr-1 mutants. (A) Detailed view of the pdr-1 deletions and the corresponding mutant gene products. Coding exons are indicated as boxes, introns as lines. Encoded domains are color-boxed: blue: ubiquitin-like domain (UBL); yellow: unique Parkin domain (UPD); red: C3HC4 RING finger domains (RING); green: C6HC in-between RING finger domain (IBR). The extent and the nature of the deletions as well as the resulting mutant proteins are listed. In-frame deletions are indicated by dotted lines and early stops generated by out-of-frame deletions are marked by asterisks. For exact deletion breakpoints, see Materials and Methods. (B) Northern blot analysis of pdr-1 transcripts from wild-type and pdr-1(lg103) mutants. pdr-1(lg103) produces a truncated transcript at levels comparable to wild-type pdr-1. Also the deletion pdr-1(lg103) does not affect co-transcription of the downstream gene K08E3.8. An act-1/actin-specific probe confirms slightly unequal loading (also see Fig. 5B for normalized data). (C) GAL4 yeast two-hybrid interactions studies of the gene products from pdr-1 in-frame deletion alleles lg103 and tm598 (as described for wild-type PDR-1 in Fig. 2A). Both truncated PDR-1 (Δaa24–247) and PDR-1(Δaa140–263) still associate with enzymes of the ubiquitylation machinery (UBC-2/UbcH4/5), but only PDR-1(Δaa140–263) and PDR-1(aa1–121Stop) are able to couple to the proteasome, whereas interaction of PDR-1(Δaa24–247) with RPT-2 is abrogated. (D) PDR-1(Δaa24–247) GST-pull-down experiments with recombinant proteins (as described for wild-type PDR-1 in Fig. 2B). Immobilized GST-fused UBC-18/UbcH7/8, UBC-2/UbcH4/5 and CHN-1/CHIP are able to bind and pull down in vitro translated truncated PDR-1(Δaa24–247), labeled with 35S methionine/cysteine. (E) Biochemical distribution of wild-type and mutant PDR-1 proteins in transfected human cells. Shown are representative anti-FLAG immunoblots of cell extracts sequentially prepared 48 h post-transfection with Triton X-100 (S) and SDS (P) from HEK293T cells transiently transfected either with FLAG-tagged wild-type PDR-1 or various PDR-1 mutants (as indicated). The corresponding anti-β-actin immunoblots demonstrate equal loading. The experiments were performed in triplicates and repeated at least three times with similar results. After 48 h post-transfection, only truncated PDR-1(Δaa24–247) was predominantly found in the insoluble fraction.
![Figure 5. The in-frame deletion pdr-1(lg103) causes hypersensitivity to ER stress. (A) pdr-1(lg103) mutants are specifically hypersensitive to ER stress. Photos show ER-stressed worms, treated with 1.5 µg/ml tunicamycin, after 3 days growth from synchronized eggs at 20°C. Most of the N2 wild-type animals reach adulthood, whereas most of the ire-1(v33) and pek-1(ok275) mutant animals either arrest or die. pdr-1(lg103) animals, in contrast to other pdr-1 deletion alleles, show the same ER stress hypersensitivity as mutants of the UPR. All untreated pdr-1 control mutants showed no lethality/arrest. Scale bar: 0.5 mm. (B and C) Quantitative analyses of survival/arrest (as described in Fig. 4A). Shown are mean values±SEM of dead/arrested worms. The total numbers of animals analyzed from the indicated strains is listed above each column (n). (B) Only homozygous pdr-1(lg103) mutants are hypersensitive to tunicamycin, but compound heterozygous pdr-1(lg103)/pdr-1(tm598) animals also show a significantly increased sensitivity compared with the respective heterozygous alleles. Heterozygous and compounds were obtained by crossing the respective pdr-1 mutants either with wild-type or homozygous pdr-1(lg103) males. (C) Rescue of the pdr-1(lg103) tunicamycin-sensitive phenotype. Wild-type copies of pdr-1, expressed from independent transgenic arrays in the mutant background pdr-1(lg103);byEx417-422[Rescue pdr-1(wt)] significantly restored survival, even at higher tunicamycin concentration (2.5 µg/ml).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/22/10.1093/hmg/ddi371/2/m_ddi37105.jpeg?Expires=1716359187&Signature=hmtRcEoat~wqFP0VSiimd6RHxiz6fj9u0kX2DpcnHNdTkwUF5K0zut3BepT0ekXE-0fdfsRqwdhocqkOk14GmjHQOIU426ycVvPIOncNEanGMxUwo2GhbtmTlFGerow6k6f8RDBd10~EBazgr5qF-V~Gh-M3vDHqoWBQJXmKbVtDohX7ycpmgvT~00rDQOr26uPs-BxnSLixf3w3r4pp1N6lLV7xuE6iwECtcpyHZAIxd~asxb4HSuah1rFPMNgvWNWYKFPFefkDn6SR0A-4r1NathmlACfXHsvH6JH-dzs0dBY6IeuB~KGtQMIJyI0i1zlFgTjB1of78efhZOV71w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 5. The in-frame deletion pdr-1(lg103) causes hypersensitivity to ER stress. (A) pdr-1(lg103) mutants are specifically hypersensitive to ER stress. Photos show ER-stressed worms, treated with 1.5 µg/ml tunicamycin, after 3 days growth from synchronized eggs at 20°C. Most of the N2 wild-type animals reach adulthood, whereas most of the ire-1(v33) and pek-1(ok275) mutant animals either arrest or die. pdr-1(lg103) animals, in contrast to other pdr-1 deletion alleles, show the same ER stress hypersensitivity as mutants of the UPR. All untreated pdr-1 control mutants showed no lethality/arrest. Scale bar: 0.5 mm. (B and C) Quantitative analyses of survival/arrest (as described in Fig. 4A). Shown are mean values±SEM of dead/arrested worms. The total numbers of animals analyzed from the indicated strains is listed above each column (n). (B) Only homozygous pdr-1(lg103) mutants are hypersensitive to tunicamycin, but compound heterozygous pdr-1(lg103)/pdr-1(tm598) animals also show a significantly increased sensitivity compared with the respective heterozygous alleles. Heterozygous and compounds were obtained by crossing the respective pdr-1 mutants either with wild-type or homozygous pdr-1(lg103) males. (C) Rescue of the pdr-1(lg103) tunicamycin-sensitive phenotype. Wild-type copies of pdr-1, expressed from independent transgenic arrays in the mutant background pdr-1(lg103);byEx417-422[Rescue pdr-1(wt)] significantly restored survival, even at higher tunicamycin concentration (2.5 µg/ml).
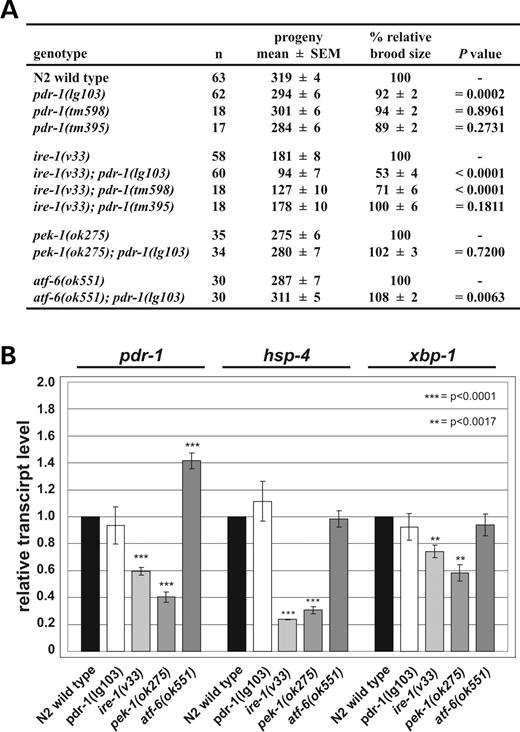
Figure 6. PDR-1 is involved in and regulated by the UPR. (A) pdr-1(lg103) and pdr-1(tm598) genetically interact with ire-1(v33). The table depicts the absolute (mean±SEM) and relative (% ±SEM) brood sizes of pdr-1 mutants as well as of ire-1(v33), pek-1(ok275) and atf-6(ok551), single and double mutants at 20°C. The total numbers of animals scored are listed (n). pdr-1 mutations show only minor effects on brood size, as compared with wild-type. ire-1(v33);pdr-1(lg103) and ire-1(v33);pdr-1(tm598) double mutants show a dramatically reduced brood size, compared with the respective single mutants, most likely as a result of a synergistic effect of both single mutations. (B) pdr-1 transcription is regulated by the UPR. Northern blot analyses of total RNA from mutants of the UPR show reduced transcript levels of pdr-1 in ire-1(v33) and pek-1(ok275) mutants, but elevated levels in atf-6 (ok551) mutants, relative to wild-type. ER stress markers hsp-4 and xbp-1, the C. elegans homologs of mammalian transcription factor XBP1, were used as control genes. Both showed a reduction of their transcriptional rate in ire-1(v33) and pek-1(ok275) mutants, whereas levels in atf-6(ok551) and pdr-1(lg103) mutants were equal to wild-type. pdr-1 transcription levels are unaffected in pdr-1(lg103) mutants compared with N2 wild-type. Wild-type levels of each transcript were set to 1.0-fold induction. An act-1/actin-specific probe was used to adjust for equal loading. The mean values of relative transcript level±SEM of 3–14 independent quantifications are shown.
![Figure 7. Combining overexpression of human α-synuclein A53T with pdr-1 (lg103) results in developmental arrest and lethality. (A) Photos show pdr-1 (lg103) mutant animals overexpressing α-synuclein WT or A53T mutation from integrated arrays. Worms were analyzed after growth from synchronized eggs at 20°C. Ectopic expression of α-synuclein WT in pdr-1(lg103) mutant background showed no effect on survival/development of the animals. In contrast, expression of α-synuclein A53T in pdr-1(lg103) mutant background resulted in a dramatic lethality/arrest. The enlarged sector shows a 3-fold magnified view on pdr-1 (lg103);Is[α-synuclein A53T] arrested at early larval stages. Expression of α-synuclein in pdr-1 mutant background did not accelerate or enhance loss of dopaminergic neurons as judged by integrated co-injection marker Pdat-1::gfp. Scale bar: 0.1 mm. (B–D) Survival of pdr-1 mutants overexpressing α-synuclein WT and A53T mutation is temperature-dependent. The Y-axis depicts the percentage of dead/arrested animals after growth from synchronized eggs at different temperatures. Shown are mean values of 10–15 independent experimental groups±SEM, the total numbers of animals analyzed is listed above each column (n). (B) Only α-synuclein A53T overexpressed in specifically pdr-1(lg103) animals results in weak but significant lethality/arrest at 15°C. (C) α-synuclein A53T-induced lethality/arrest in pdr-1(lg103) is dramatically enhanced at 20°C. (D) Increasing temperature to 25°C did not induce arrest/lethality in other pdr-1 alleles overexpressing mutant α-synuclein.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/14/22/10.1093/hmg/ddi371/2/m_ddi37107.jpeg?Expires=1716359187&Signature=3BlxIW4ki4sah9HD2rPHebKkSnONhcJwlYgiXftfLyn7sTM5~NcTYH~WDDU3ZfnQ0hgOyH9ECVIB~Qld1vZwWz0rd5Xiiuk3rxHZVPPUh93c6OSSI1v~JlP3KU1fAzn~PE0PRiTw06VeVCwDDXVh~Gb1Hvf1nnCTMABdTYGtmqVM9nXtE2~s7Q6jWGh5QxBwEvcISw8EHnJK2HE5pR0NG21AsfHzcoPRp1gpkEzqDd6m2qKBPA76hpXurYSBi6USBB0Cs7ruLwHGkwF0B-z0TFN0~yrzQH47oU6LZtYayZU4fXaV17hvzGhJ4j4lMbPSC824i-erl6lSI-Pul5J7ew__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 7. Combining overexpression of human α-synuclein A53T with pdr-1 (lg103) results in developmental arrest and lethality. (A) Photos show pdr-1 (lg103) mutant animals overexpressing α-synuclein WT or A53T mutation from integrated arrays. Worms were analyzed after growth from synchronized eggs at 20°C. Ectopic expression of α-synuclein WT in pdr-1(lg103) mutant background showed no effect on survival/development of the animals. In contrast, expression of α-synuclein A53T in pdr-1(lg103) mutant background resulted in a dramatic lethality/arrest. The enlarged sector shows a 3-fold magnified view on pdr-1 (lg103);Is[α-synuclein A53T] arrested at early larval stages. Expression of α-synuclein in pdr-1 mutant background did not accelerate or enhance loss of dopaminergic neurons as judged by integrated co-injection marker Pdat-1::gfp. Scale bar: 0.1 mm. (B–D) Survival of pdr-1 mutants overexpressing α-synuclein WT and A53T mutation is temperature-dependent. The Y-axis depicts the percentage of dead/arrested animals after growth from synchronized eggs at different temperatures. Shown are mean values of 10–15 independent experimental groups±SEM, the total numbers of animals analyzed is listed above each column (n). (B) Only α-synuclein A53T overexpressed in specifically pdr-1(lg103) animals results in weak but significant lethality/arrest at 15°C. (C) α-synuclein A53T-induced lethality/arrest in pdr-1(lg103) is dramatically enhanced at 20°C. (D) Increasing temperature to 25°C did not induce arrest/lethality in other pdr-1 alleles overexpressing mutant α-synuclein.
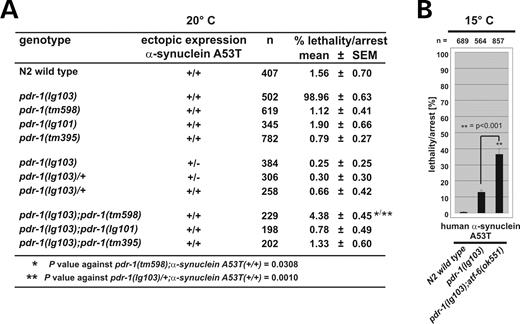
Figure 8. Dosage-dependent cytotoxicity of pdr-1(lg103) and α-synuclein A53T. Survival/arrest analyses of heterozygous mutants and the hypermorphic atf-6 background (as described in Fig. 6). (A) At 20°C, only α-synuclein A53T expressed from both integrated copies confers cytotoxicity in specifically homozygous pdr-1(lg103) mutant background, and, although milder, in pdr-1(lg103)/pdr-1(tm598) compound heterozygous mutants carrying both in-frame deletions. (B) pdr-1(lg103);atf-6(ok551) double mutants overexpressing α-synuclein A53T mutation show a significantly enhanced lethality/developmental arrest already at 15°C.
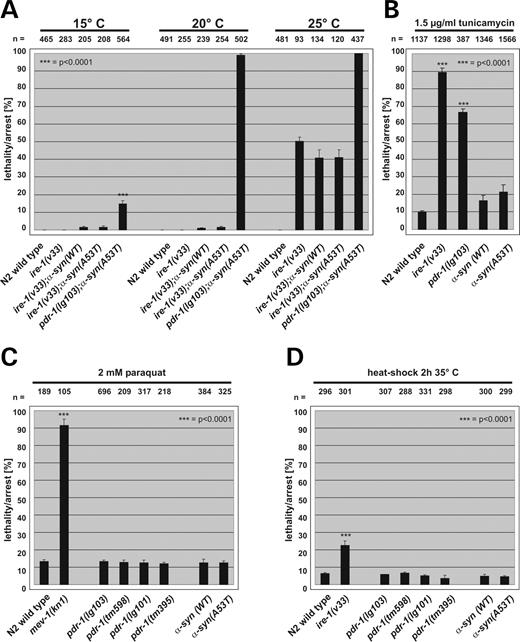
Figure 9. Cytotoxicity of α-synuclein A53T expression is independent of ER, oxidative and heat stress pathways. The Y-axis depicts the percentage of dead/arrested animals. Shown are mean values±SEM, the total numbers of animals analyzed is listed above each column (n). (A) Overexpression of α-synuclein WT and A53T in ire-1(v33) mutant background did not affect survival at any temperature tested. Survival was tested 3 days after synchronization of eggs at the temperatures depicted. (B) Exogenous ER stress had no effect on viability and development of mutants overexpressing human α-synuclein WT or A53T. Plates contained 1.5 µg/ml tunicamycin. (C) mev-1(kn1) animals used as controls arrest early in development when treated with paraquat. In contrast, oxidative stress did not affect viability and development of pdr-1 mutants or animals expressing human α-synuclein WT or A53T. Survival was analyzed after 3 days exposure of synchronized L1 larvae to 2 mm paraquat at 20°C. (D) Heat-shock treatment of pdr-1 mutants and worms overexpressing human α-synuclein WT or A53T mutation for 2 h at 35°C. Heat stress on synchronized L2 larvae had no effect on development or survival of mutant worms.
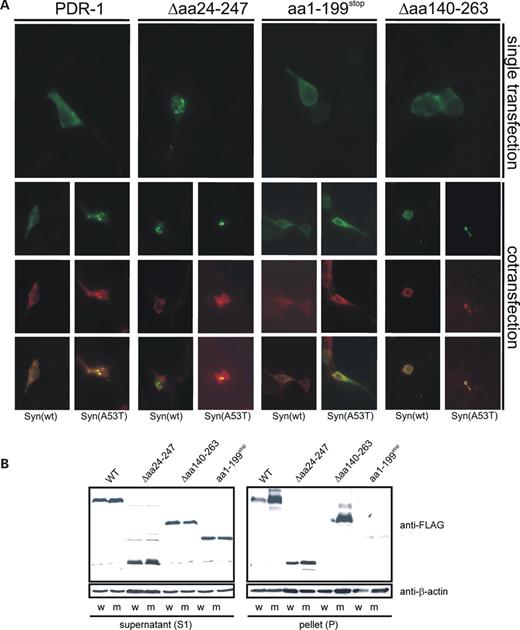
Figure 10. α-synuclein A53T, but not wild-type α-synuclein, enhances aggregation of the transgenically expressed PDR-1 in HEK293T cells. (A) The full-length or truncated forms of C. elegans PDR-1 were expressed in HEK293T cells alone or in combination with α-synuclein WT or α-synuclein A53T. All PDR-1 constructs were fused to FLAG epitope tag represented by green labeling, α-synuclein expression is shown in red. After single transfection only PDR-1(Δaa24–247), corresponding to pdr-1(lg103), formed subcellular aggregates in HEK293T cells. Both wild-type and mutant A53T α-synuclein co-localizes with the aggregates. In contrast, expression of α-synuclein A53T resulted in enhanced aggregation of both wild-type PDR-1 and PDR-1(Δaa140–263) (corresponding to allele tm598), whereas PDR-1 variants expression only UBL and UPD domains were not affected. (B) The aggregation of PDR-1 variants were investigated biochemically after co-expression in HEK293 with α-synuclein wild-type (w) or the α-synuclein A53T mutant (m) as described for Figure 4. The formation of 1% Triton X-100 resistant aggregates of the full-length and in-frame deleted Δaa140–263 variants of PDR-1 was significantly enhanced after co-expression of α-synuclein A53T but not of α-synuclein wild-type. The aggregation of the in-frame deleted variant Δaa24–247 of PDR-1 was only slightly increased after co-expression of α-synuclein A53T compared with the wild-type variant of α-synuclein. No aggregation of α-synuclein A53T or α-synuclein wild-type was observed after co-transfection with PDR-1(aa1–199Stop), suggesting that this allele represents a strong loss-of-function or null allele.
References
Riess, O., Berg, D., Kruger, R. and Schulz, J.B. (
Lykkebo, S. and Jensen, P.H. (
Polymeropoulos, M.H., Lavedan, C., Leroy, E., Ide, S.E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R. et al. (
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., Przuntek, H., Epplen, J.T., Schols, L. and Riess, O. (
Zarranz, J.J., Alegre, J., Gomez-Esteban, J.C., Lezcano, E., Ros, R., Ampuero, I., Vidal, L., Hoenicka, J., Rodriguez, O., Atares, B. et al. (
Chartier-Harlin, M.C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., Levecque, C., Larvor, L., Andrieux, J., Hulihan, M. et al. (
Singleton, A.B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., Nussbaum, R. et al. (
Ibanez, P., Bonnet, A.M., Debarges, B., Lohmann, E., Tison, F., Pollak, P., Agid, Y., Durr, A. and Brice, A. (
Maries, E., Dass, B., Collier, T.J., Kordower, J.H. and Steece-Collier, K. (
Lakso, M., Vartiainen, S., Moilanen, A.M., Sirvio, J., Thomas, J.H., Nass, R., Blakely, R.D. and Wong, G. (
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y. and Shimizu, N. (
Sakata, E., Yamaguchi, Y., Kurimoto, E., Kikuchi, J., Yokoyama, S., Yamada, S., Kawahara, H., Yokosawa, H., Hattori, N., Mizuno, Y. et al. (
Shimura, H., Schlossmacher, M.G., Hattori, N., Frosch, M.P., Trockenbacher, A., Schneider, R., Mizuno, Y., Kosik, K.S. and Selkoe, D.J. (
Finney, N., Walther, F., Mantel, P.Y., Stauffer, D., Rovelli, G. and Dev, K.K. (
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., Shimizu, N., Iwai, K., Chiba, T., Tanaka, K. et al. (
Imai, Y., Soda, M. and Takahashi, R. (
Rutkowski, D.T. and Kaufman, R.J. (
Kostova, Z. and Wolf, D.H. (
Friedlander, R., Jarosch, E., Urban, J., Volkwein, C. and Sommer, T. (
Travers, K.J., Patil, C.K., Wodicka, L., Lockhart, D.J., Weissman, J.S. and Walter, P. (
Imai, Y., Soda, M., Hatakeyama, S., Akagi, T., Hashikawa, T., Nakayama, K.I. and Takahashi, R. (
Kahle, P.J. and Haass, C. (
Petrucelli, L., O'Farrell, C., Lockhart, P.J., Baptista, M., Kehoe, K., Vink, L., Choi, P., Wolozin, B., Farrer, M., Hardy, J. et al. (
Yang, Y., Nishimura, I., Imai, Y., Takahashi, R. and Lu, B. (
Mata, I.F., Lockhart, P.J. and Farrer, M.J. (
Sriram, S.R., Li, X., Ko, H.S., Chung, K.K., Wong, E., Lim, K.L., Dawson, V.L. and Dawson, T.M. (
Imai, Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y. and Takahashi, R. (
Chung, K.K., Zhang, Y., Lim, K.L., Tanaka, Y., Huang, H., Gao, J., Ross, C.A., Dawson, V.L. and Dawson, T.M. (
Corti, O., Hampe, C., Koutnikova, H., Darios, F., Jacquier, S., Prigent, A., Robinson, J.C., Pradier, L., Ruberg, M., Mirande, M. et al. (
Hyun, D.H., Lee, M., Hattori, N., Kubo, S., Mizuno, Y., Halliwell, B. and Jenner, P. (
Gu, W.J., Corti, O., Araujo, F., Hampe, C., Jacquier, S., Lucking, C.B., Abbas, N., Duyckaerts, C., Rooney, T., Pradier, L. et al. (
Winklhofer, K.F., Henn, I.H., Kay-Jackson, P.C., Heller, U. and Tatzelt, J. (
Henn, I.H., Gostner, J.M., Lackner, P., Tatzelt, J. and Winklhofer, K.F. (
Jones, D., Crowe, E., Stevens, T.A. and Candido, E.P. (
Hoppe, T., Cassata, G., Barral, J.M., Springer, W., Hutagalung, A.H., Epstein, H.F. and Baumeister, R. (
Hoppe, T. (
Lorick, K.L., Jensen, J.P., Fang, S., Ong, A.M., Hatakeyama, S. and Weissman, A.M. (
Cookson, M.R., Lockhart, P.J., McLendon, C., O'Farrell, C., Schlossmacher, M. and Farrer, M.J. (
Wang, C., Tan, J.M., Ho, M.W., Zaiden, N., Wong, S.H., Chew, C.L., Eng, P.W., Lim, T.M., Dawson, T.M. and Lim, K.L. (
Nass, R., Hall, D.H., Miller, D.M., III and Blakely, R.D. (
Shen, X., Ellis, R.E., Lee, K., Liu, C.Y., Yang, K., Solomon, A., Yoshida, H., Morimoto, R., Kurnit, D.M., Mori, K. et al. (
Calfon, M., Zeng, H., Urano, F., Till, J.H., Hubbard, S.R., Harding, H.P., Clark, S.G. and Ron, D. (
Urano, F., Calfon, M., Yoneda, T., Yun, C., Kiraly, M., Clark, S.G. and Ron, D. (
Cao, S., Gelwix, C.C., Caldwell, K.A. and Caldwell, G.A. (
Zhang, Y., Gao, J., Chung, K.K., Huang, H., Dawson, V.L. and Dawson, T.M. (
Palacino, J.J., Sagi, D., Goldberg, M.S., Krauss, S., Motz, C., Wacker, M., Klose, J. and Shen, J. (
Goldberg, M.S., Fleming, S.M., Palacino, J.J., Cepeda, C., Lam, H.A., Bhatnagar, A., Meloni, E.G., Wu, N., Ackerson, L.C., Klapstein, G.J. et al. (
Lorenzetti, D., Antalffy, B., Vogel, H., Noveroske, J., Armstrong, D. and Justice, M. (
Lim, K.L., Chew, K.C., Tan, J.M., Wang, C., Chung, K.K., Zhang, Y., Tanaka, Y., Smith, W., Engelender, S., Ross, C.A. et al. (
Liani, E., Eyal, A., Avraham, E., Shemer, R., Szargel, R., Berg, D., Bornemann, A., Riess, O., Ross, C.A., Rott, R. et al. (
Nagano, Y., Yamashita, H., Takahashi, T., Kishida, S., Nakamura, T., Iseki, E., Hattori, N., Mizuno, Y., Kikuchi, A. and Matsumoto, M. (
Ito, T., Niwa, J., Hishikawa, N., Ishigaki, S., Doyu, M. and Sobue, G. (
Sherman, M.Y. and Goldberg, A.L. (
Forman, M.S., Lee, V.M. and Trojanowski, J.Q. (
Ryu, E.J., Harding, H.P., Angelastro, J.M., Vitolo, O.V., Ron, D. and Greene, L.A. (
Braungart, E., Gerlach, M., Riederer, P., Baumeister, R. and Höner, M. (
Eriksen, J.L., Dawson, T.M., Dickson, D.W. and Petrucelli, L. (
Giasson, B.I., Duda, J.E., Quinn, S.M., Zhang, B., Trojanowski, J.Q. and Lee, V.M. (
Greene, J.C., Whitworth, A.J., Kuo, I., Andrews, L.A., Feany, M.B. and Pallanck, L.J. (
Pesah, Y., Pham, T., Burgess, H., Middlebrooks, B., Verstreken, P., Zhou, Y., Harding, M., Bellen, H. and Mardon, G. (
Itier, J.M., Ibanez, P., Mena, M.A., Abbas, N., Cohen-Salmon, C., Bohme, G.A., Laville, M., Pratt, J., Corti, O., Pradier, L. et al. (
Von Coelln, R., Thomas, B., Savitt, J.M., Lim, K.L., Sasaki, M., Hess, E.J., Dawson, V.L. and Dawson, T.M. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}