




Abstract
Coronary artery disease (CAD), including its most serious complication myocardial infraction (MI), is the leading cause of death in the US and developed countries. We recently discovered that a seven-amino acid deletion in MEF2A, a transcription factor with a high level of expression in the endothelium of coronary arteries, co-segregates with CAD/MI in one family, and it suppresses transcription activation activity of MEF2A by a dominant-negative mechanism. In this study, we used single-strand conformation polymorphism and DNA sequence analyses to identify mutations in MEF2A in 207 independent CAD/MI patients and 191 controls with normal angiograms. We identified three novel mutations in exon 7 of MEF2A in four of 207 CAD/MI patients (1.93%). No mutations were detected in the 191 controls. The mutations identified here include N263S identified in two independent CAD patients, P279L in one patient and his father with the diagnosis of CAD and G283D in one patient. These mutations are clustered within or close to the major transcriptional activation domain of MEF2A. They significantly reduce the transcriptional activation activity of MEF2A and act by a loss-of-function mechanism. The gene carriers with loss-of-function mutations appear to be associated with less severe CAD. These results suggest that CAD/MI can result from a spectrum of MEF2A transcription dysfunctions ranging from loss-of-function to dominant-negative suppression and that a significant percent of the CAD/MI population (1.93%) may carry mutations in MEF2A, although further definition of the prevalence of MEF2A mutations is warranted.
INTRODUCTION
Coronary artery disease (CAD) is the most common form of heart disease. It is attributable to atherosclerotic plaque buildup in the walls of the epicardial coronary arteries (1,2). The atheroma accumulation can limit blood flow to the myocardium, resulting in symptoms of ischemia. In some patients, inflammation at the site of atheroma leads to plaque fissure, rupture or erosion. These events can result in sudden coronary thrombosis and myocardial infarction (MI) (1–3). CAD/MI claims more lives each year than the other five leading causes of death combined (cancer, respiratory diseases, accidents, diabetes mellitus and pneumonia) (4,5). It accounts for one of every five deaths, and overall, it is estimated to affect more than 13 million Americans. Several risk factors have been identified for CAD and MI, including family history, male gender, high blood cholesterol and other lipids, smoking, physical inactivity, overweight and obesity, hypertension and diabetes (6–9). A family history of CAD is one of the most significant risk factors for the disease. To date, several large-scale genome-wide scans using hundreds of sibpairs have been reported to identify chromosomal regions implicated in complex CAD or MI traits. We have recently mapped a genetic susceptibility locus for MI to chromosome 1p34–36 region by analyzing MI as a complex trait in 428 nuclear families with premature CAD and MI (10). Another susceptibility locus for MI was identified in the chromosome 14q region (11). Five CAD susceptibility loci have been identified on chromosome 2q21.1–22 (12), 2q36 (13), 3q13 (14), 16p13–pter (15) and Xq23–26 (12). The specific genes at all these loci, however, remain to be identified or cloned.
An effective approach for identifying a specific gene for CAD and MI appears to be a genome-wide screen in large, extended families with multiple affected family members. Focusing on a large CAD/MI family with autosomal dominant inheritance, we recently demonstrated genetic linkage of CAD/MI with a DNA polymorphism in chromosome 15q26 region, D15S120 (we designated this locus as adCAD1) (16). MEF2A is a gene that encodes a transcription factor and is located in chromosomal 15q26 region (17–19). We subsequently identified a seven-amino acid deletion of MEF2A that co-segregates with CAD/MI in the family. MEF2A was initially identified as a muscle-specific gene, but was later shown to be highly expressed in the endothelium of coronary arteries (16). MEF2A is a member of the myocyte enhancer factor-2 (MEF2) family of transcription factors, and other members include MEF2B, MEF2C and MEF2D (17–19). The MEF2 factors act as a homo- or heterodimer, and each of these proteins binds to the MEF2 target DNA sequence (the A–T rich sequence) present in the regulatory regions of many, if not all, muscle-specific genes, and controls transcription (17,18).
The previously identified seven-amino acid deletion of MEF2A is a functional mutation that disrupts the transcriptional activation activity of MEF2A by a dominant-negative mechanism (16). Here, we report identification and functional characterization of three new MEF2A mutations in four of 207 patients with CAD. We discovered that all three missense mutations are loss-of-function mutations, suggesting that MEF2A mutations can cause CAD/MI by multiple molecular mechanisms. These results strongly support the premise that MEF2A is adCAD1. Genotype–phenotype correlation studies suggest that individuals with the dominant-negative mutation may display the more severe form of CAD with a high incidence of MI than those carrying loss-of-function mutations. We also provide preliminary data to estimate the frequency of MEF2A mutations in the patient population with CAD and MI.
RESULTS
Identification of three novel MEF2A mutations associated with CAD
We have previously determined the genomic structure of MEF2A, and reported the PCR primers for successfully amplifying all 11 exons of MEF2A for mutations analysis (16). Single-strand conformation polymorphism (SSCP) and DNA sequence analyses were used to screen for mutations in MEF2A. We screened 191 controls and 207 independent patients with unequivocal diagnosis of CAD based on cardiac angiography and/or development of MI (Table 1). We selected patients with onset of CAD ≤55 years of age in males and ≤60 years of age in females. The control individuals were ≥55 years of age with coronary angiography documenting lack of any significant coronary arterial narrowings. The CAD cases and controls are matched for ethnicity, age and body mass index (P>0.05, Table 1). The CAD population has significantly higher rates of males and individuals with a history of diabetes, hypertension and smoking (P<0.01, Table 1), all of which are established risk factors for CAD.
The entire coding region of MEF2A and the intron–exon boundaries were screened for mutations in these 207 patients and 191 controls. MEF2A mutations were identified in four Caucasians of the 207 CAD patients (Figs 1–3; Table 1), accounting for 1.93% of this patient population. No mutations were identified in the 191 controls. MEF2A mutations identified in this study were clustered in exon 7 which encodes portion of a transcription activation domain of this transcription factor (20). After a mutation was identified, significant effort was made to characterize family members, albeit with limited success.
The MEF2A mutation N263S was identified in two independent patients, GB04146 and GB05072 (Fig. 1; Tables 2 and 3). GB04146 is a 49-year-old male patient who was diagnosed with CAD at the age of 41 years, and had coronary artery bypass grafting. His risk factors include history of occasional tobacco use. GB05072 is a 47-year-old male who was diagnosed with CAD at the age of 45 years. A stent was placed in the right coronary artery (RCA) at that time. In 2003, the patient had a coronary procedure with a cutting balloon, brachytherapy and second stents placed to the mid-RCA. Risk factors include hyperlipidemia (Table 3), hypertension with age of onset at 30 years and history of smoking one pack per day for 17 years (Table 2). Family history includes a father who died of a massive MI at age 38.
Mutation P279L was identified in patient GB00305 (Fig. 2). The patient is a 39-year-old male with history of CAD (diagnosed at 36), and a stent was placed to the proximal LAD and balloon angioplasty was performed in the first diagonal artery in 2001. In 2002, the patient had a coronary procedure with unsuccessful attempt to cross a proximal total occlusion in the RCA. Risk factors include dyslipidemia (triglycerides level of 252 mg/dl), history of diabetes mellitus and history of one pack per day smoking with cessation in 2001 (Tables 2 and 3). Further analysis of family members revealed that the 66-year-old father of the patient also carried the mutation and had a clear diagnosis of CAD. The father was diagnosed with CAD in 2001 and had coronary artery bypass grafting. Other risk factors include hyperlipidemia, hypertension, obesity, diet controlled diabetes and a remote history of smoking.
Mutation G283D was identified in patient GB04583 (Fig. 3). This patient is a 50-year-old female with history of CAD first diagnosed in 2002 after a catheterization revealed left main trunk disease with 70–80% stenosis. The patient had coronary artery bypass grafting. Risk factors include hypertension, hyperlipidemia, and one pack per day smoking for 32 years (Tables 2 and 3). The patient additionally was treated for CAD with right internal carotid endarterectomy in 2000.
Functional defects of MEF2A missense mutations in transcriptional activation
As all three MEF2A mutations identified in this study (N263S, P279L and G283D) are located close to or within the transcription activation domain (amino acids 274–373) (20), we assessed the functional effect of these mutations on transcriptional activation using a reporter gene (ANFp700/Luc, Fig. 4A). The reporter gene has the ANF promoter (region from −700 bp to +1 bp) fused to the luciferase gene (16). The ANFp700/Luc reporter gene was co-transfected with wild-type or various mutant MEF2A expression constructs into HeLa cells. Western blot analysis showed that wild-type and mutant MEF2A proteins were successfully expressed in transfected cells (Fig. 4B). Transcriptional activity was examined and expressed as relative luciferase units. As shown in Figure 4B, expression of wild-type MEF2A activated transcription of the ANF promoter. Mutations N263S, P279L and G283D significantly reduced transcription activation by MEF2A. Transcription activation activity in cells co-transfected with wild-type MEF2A plus each mutant MEF2A was indistinguishable from those transfected with wild-type MEF2A alone (data not shown). These data indicate that mutant N263S, P279L and G283D MEF2A proteins did not form functional transcription factor, and did not cause dominant-negative suppression of wild-type MEF2A function. Thus, patients with the missense mutations are expected to express half of the normal amount of MEF2A.
We previously showed that MEF2A alone can activate transcription of its target genes, but in the presence of transcription factor GATA-1, synergy in transcriptional activation by these two proteins was observed (16). Accordingly, we then investigated whether the missense mutations in MEF2A could disrupt the synergistic transactivation between MEF2A and GATA-1. As shown in Figure 4C, either wild-type MEF2A or GATA-1 alone activated expression of the ANF promoter, but co-transfection of MEF2A and GATA-1 into HeLa cells showed synergistic activation of the ANF promoter. However, mutations N263S, P279L and G283D significantly reduced synergistic transcription activation by MEF2A and GATA-1.
DISCUSSION
Previously, we studied a large family with CAD and MI, and mapped a genetic locus for the autosomal dominant form of CAD to chromosome 15q26 (adCAD1) (16). Further studies identified a seven-amio acid deletion (Δ7aa) in the myocyte enhancer factor 2A (MEF2A) that is responsible for the disease in the family (16). In this study, we identified three new mutations in MEF2A. These mutations include N263S in two independent CAD patients, P279L in a family and G283D in a single patient. These three or other MEF2A mutations were not identified through SSCP analysis in 191 control individuals with normal angiograms (lack of any significant coronary arterial narrowings). Functional studies indicate that all three mutations significantly reduce the transcription activation activity, suggesting that they are associated with CAD.
The pathophysiology of chromosome 15-linked CAD is related to the deregulation of transcription programs by MEF2A in coronary arteries. We have detected high levels of MEF2A protein expression in human coronary arteries by western blot analysis (S. Arckacki and Q. Wang, data not shown). Our immunostaining experiment also demonstrated the co-localization of MEF2A protein signal with endothelial cell marker CD31 signal in coronary arteries (16). These results suggest that MEF2A protein is expressed at significant levels in the endothelium, a single layer of endothelial cells that act as a barrier between the blood and coronary vessels. The detailed molecular mechanism of the development of CAD and MI associated with chromosome 15-linked CAD is uncertain. If CAD-associated mutations in MEF2A cause dysfunctional or abnormally developed endothelium, the coronary vessels could be prone to inflammation and thrombosis, ultimately leading to development of CAD or MI.
Chromosome 15-linked CAD/MI results from multiple molecular mechanisms that reduce the transcriptional activity of MEF2A. Coexpression of mutant and wild-type MEF2A led to loss or dominant suppression of MEF2A transcription function. The Δ7aa mutation, reported previously (16), acts by a dominant-negative mechanism, whereas the three mutations identified in this study (N263S, P279L and G283D) act by a loss-of-function mechanism. Similar findings of the spectrum of functional effects by disease-causing mutations have been reported in other cardiovascular diseases, including Holt–Oram syndrome (21,22) and long QT syndrome (LQTS) (23,24). Sanguinetti et al. (25) found that LQTS-causing mutations Δbp1261 and ΔI500–F508 of HERG act by a loss-of-function mechanism, but mutations G628S and A561V act by a dominant-negative mechanism. Some LQTS-causing mutations in the LQT1 gene KCNQ1, A341V, R190Q or G189R did not affect the function of wild-type KCNQ1 (loss-of-function) (26). The other mutations A177P, V254M, L272F, T311I, G306R, A341E and G345E resulted in dominant-negative suppression of KCNQ1 function (26,27). No clear correlation was identified between the degree of HERG or KCNQ1 dysfunction and the clinical outcome. Interestingly, a difference in pattern of the clinical features was observed between MEF2A gene carriers with a dominant-negative mutation and those with a loss-of-function mutation. The carriers with the Δ7aa mutation appear to have a more severe form of CAD than those with the three missense mutations. Nine of the 13 carriers with the Δ7aa mutation developed MI; however, none of the five carriers with missense mutations developed MI (Table 2).
The frequency of MEF2A mutations in the CAD population is unknown. This study offers a preliminary estimate. Identification of four mutation carriers among 207 independent CAD patients suggest that as many as 1.93% of CAD population may carry a MEF2A mutation. These numbers need to be viewed as preliminary since the sample size is small and we limited our studies to affected male patients of <55 years and females of <60 years of age, characteristics of premature CAD. More studies with large sample sizes are now needed.
A limitation of the present study is the lack of more comprehensive family studies due to difficulties in expanding the families with MEF2A mutations. A high rate of sudden death due to MI prevents enrollment of patients in the earlier generations. The late-onset characteristic of CAD results in the lack of clinical diagnosis of CAD for people in the younger generations. A potential confounding issue of phenocopy may occur in a complex disease of multifactorial basis, like CAD. Future studies with knock-in mice with the mutations identified in this study will further validate our conclusion. Another limitation of the study is that the sensitivity of the mutation detection method used in the study, SSCP analysis, is not 100%, which may fail to identify some mutations. Furthermore, mutations in the promoter, 5′- and 3′-untranslated regions and some intronic regions will not be identified as the present study focused on the exons and intron–exon boundaries of MEF2A only. A comprehensive, larger scale sequencing study may be needed to accurately estimate the true prevalence frequency of MEF2A in the CAD/MI as well as the general populations.
In summary, the present study and our earlier report (16) together establish that MEF2A mutations cause one form of CAD/MI. The diagnosis of CAD is based primarily on significant findings of coronary stenosis by an angiogram or by development of MI. Since the first sign of CAD can be a fatal MI, early diagnosis and identification of high-risk individuals is important. In the future, genetic testing with incorporation of MEF2A mutations may prove to be useful in identifying patients with particular increased risk of CAD/MI. If such patients can be identified, aggressive lifestyle improvement and pharmacologic strategies may be implemented early to offset the risk.
MATERIALS AND METHODS
Study subjects and isolation of genomic DNA
The study participants were identified and enrolled at the Cleveland Clinic Foundation through the GeneBank program at the Cleveland Clinic Heart Center, which is a registry of data in conjunction with a repository of DNA/serum/plasma for the individuals undergoing coronary catheterization. Family data and blood samples were also obtained as much as possible from family members if a positive mutation was identified in the proband. This study was approved by the Cleveland Clinic Foundation Institutional Review Board on Human Subjects.
The diagnostic criteria for CAD include: (1) any preceding or existing indication of MI which is based on the existence of at least two of the following: prolonged chest pain, ECG patterns consistent with acute MI or significant elevation of cardiac enzymes; (2) percutaneous coronary angioplasty (PTCA); (3) coronary artery bypass surgery (CABG); or (4) coronary angiography with >70% stenosis (16). The controls were selected as those individuals with coronary angiography showing no luminal stenosis, and were ≥55 years of age.
Genomic DNA was isolated with the DNA Isolation Kit for Mammalian Blood (Roche Diagnostic Co., Indianapolis, IN, USA).
Mutational analysis
Mutation screening was carried out using SSCP analysis as described (23,28–31). The PCR primers for SSCP were as described previously (16). Abnormal SSCP bands were isolated from gels, dehydrated, re-amplified using the original primers and sequenced by the BigDye Terminator cycle sequencing reaction and an ABI PRISM 3100 Genetic Analyzer.
Plasmid constructs and mutagenesis
The MEF2A expression construct was created by cloning the full-length MEF2A cDNA into plasmid vector pcDNA3, and was kindly provided by Dr Eric N. Olson at University of Texas Southwestern Medical Center. Each new missense mutation of MEF2A was introduced into the wild-type construct by PCR-based site-directed mutagenesis, and verified by DNA sequence analysis.
The full-length GATA-1 cDNA was isolated by RT–PCR, and cloned into pcDNA3 (16), resulting in the GATA-1 expression construct. The reporter gene for transcriptional activation assays ANFp-700/Luc has −700 bp to +1 bp upstream region from the transcription start site of the human atrial natriuretic factor (ANF) gene fused to the luciferase gene (16).
Transcriptional activation (luciferase) assays
The effect of missense mutations of MEF2A on transcriptional activation was analyzed using the ANFp-700/Luc reporter gene. The reporter gene was co-transfected into HeLa cells with either the wild-type or mutant MEF2A expression construct alone or in combination with the GATA-1 expression construct. Transcriptional activation activity was assayed with luciferase activity.
HeLa cells were grown to 95% confluence in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and 50 ng of DNA for the expression construct, 1 µg of DNA for the reporter gene and 50 ng of internal control plasmid pSV-galactosidase. Luciferase assays were performed using a Dual-Luciferase assay kit according to the manufacturer's instructions (Promega, Madison, WI, USA). The β-galactosidase activity expressed from pSV-galactosidase was used to normalize the transfection efficiency. The experiments were repeated two times in triplicate. Data are expressed as mean ±SE.
Western blot analysis
The efficiency of transfection was examined by western blot analysis. Cells were transfected as described earlier, harvested and lysed 24 h after transfection. Forty micrograms of total cellular lysates were separated by 12% SDS–PAGE and electro-transferred to a polyvinylidene fluoride membrane. The membrane was probed with goat polyclonal anti-MEF2A antiserum (Santa Cruz Biotechnology, Santa Cruz, CA, USA) as the primary antibody and the rabbit anti-goat IgG horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology). ECL western blotting detection reagents (Amersham Pharmacia Biotech) were used to visualize the protein signal.
ACKNOWLEDGEMENTS
We thank the study participants for their enthusiasm and support to this study, M. Goormastic (the manager of C5 Clinical Data Management) for help with Table 1 and A. Timur, R. Kadaba, S. Chen, S. Rao and other laboratory members for help and discussions. E.J.T. is the co-principal investigator on this study. This work was supported by the National Heart, Lung, and Blood Institute grants R01 HL73817, R01 HL65630, R01 HL66251 (Q.W.), an American Heart Association Established Investigator award (Q.W.) and in part by Public Health Service National Center for Research Resources grant no. M01-RR18390-01 at the Cleveland Clinic Foundation.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
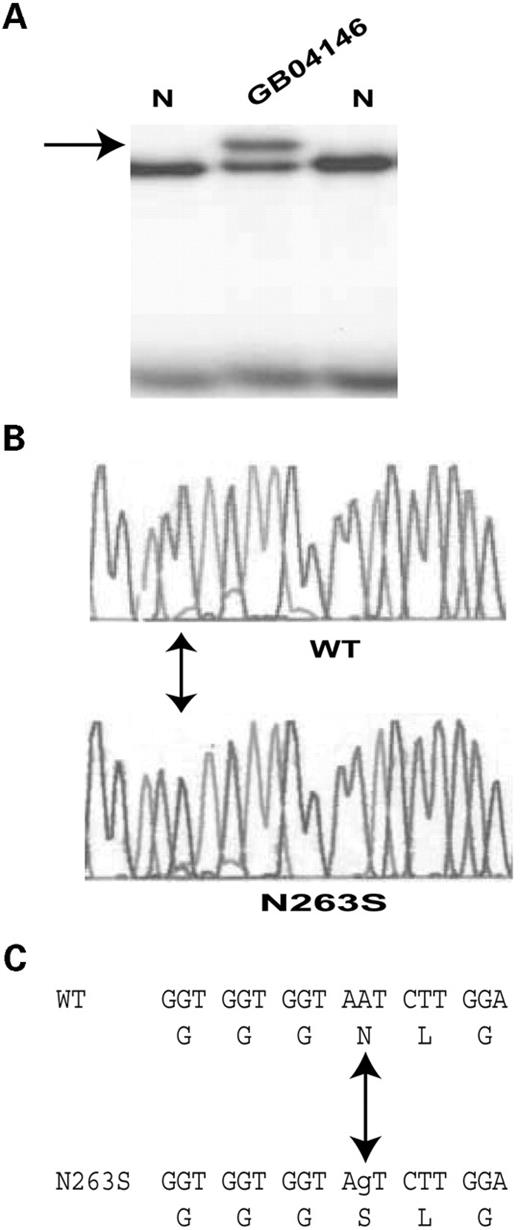
Figure 1. MEF2A missense mutation G263S in CAD patient GB04146. (A) Results of SSCP analysis showing an aberrant SSCP conformer (indicated by an arrow). N, normal control. The abnormal SSCP band was also detected in CAD patient GB05072, but was not seen in 200 controls. (B) Sequence analysis of the normal (WT) and aberrant (G263S) SSCP conformers revealed an A to G substitution at codon 263 in exon 7 of MEF2A. This mutation causes substitution of amino acid residue asparagines by serine (C).
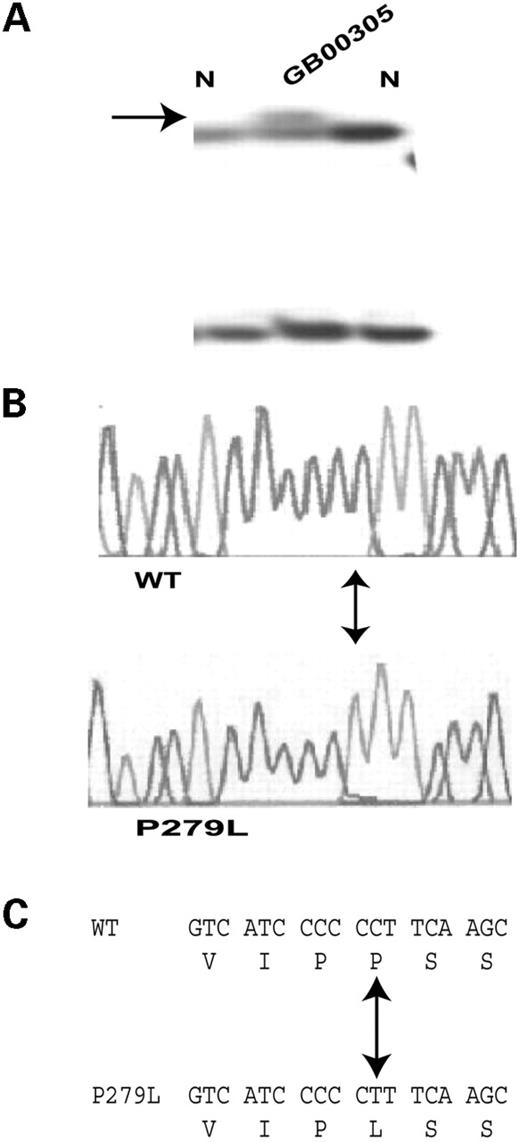
Figure 2. Identification of MEF2A mutation P279L in CAD patient GB00305. Figure caption is as described in the legend to Figure 1.
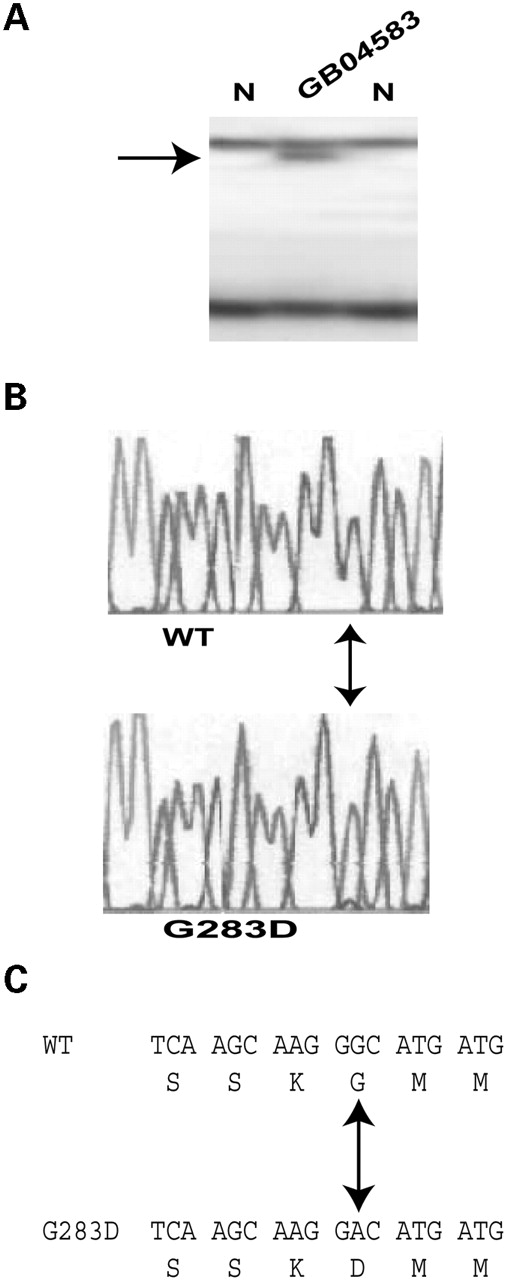
Figure 3. Identification of MEF2A mutation G283D in CAD patient GB04583. Figure caption is as described in the legend to Figure 1.
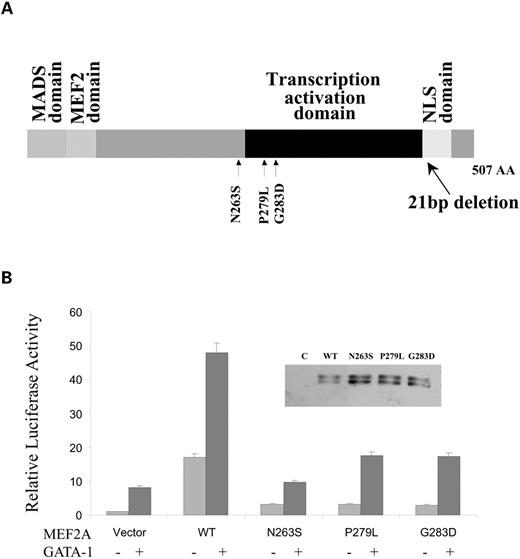
Figure 4. (A) Structure of MEF2A protein with CAD/MI-associated mutations indicated. The MEF2A gene consists of 11 exons and encodes a 507 amino acid protein. The MADS domain and MEF2 domain at the N-terminal region are responsible for DNA binding, dimerization and interaction with other transcription factor. The transcription activation domain is locate in the middle portion and the C-terminal region is responsible for nuclear localization site (NLS). (B) The effect of missense mutations of MEF2A on transcription activation activity in the presence (+) or absence (−) of GATA-1. Transcriptional activity is shown as relative luciferase activity on the y-axis. The transcriptional activity for the vector alone was set arbitrarily to 1. WT, wild-type. Inset: western blot analysis to determine whether mutant MEF2A were successfully expressed in transfected HeLa cells. C, empty vector control; WT, lane with lysate from HeLa cells containing expressed wild-type MEF2A; N263S, P279L, G283D, lanes with lysates from HeLa cells containing expressed MEF2A proteins with corresponding mutation.
Characteristics of the study population
| Characteristic | CAD cases | Normal controls | P-value |
|---|---|---|---|
| Sample size | 207 | 191 | – |
| Mean current age (SD) (years) | 44.8 (4.8) | 43.9 (7.8) | 0.18a |
| Mean body mass index (SD) (kg/m2) | 31.0 (6.8) | 30.6 (8.4) | 0.60a |
| Race | |||
| Caucasian | 77.8% | 74.4% | 0.63b |
| African-American | 17.9% | 19.4% | |
| Native American | 2.9% | 5.2% | |
| Asian | 1.5% | 1.1% | |
| % Male | 77.3 | 49.2 | <0.001b |
| % Hypertension | 69.6 | 38.2 | <0.001b |
| % Diabetes | 27.5 | 8.4 | <0.001b |
| % Current smokers | 31.9 | 19.4 | 0.004b |
| Qualifying events | |||
| Angiogram>70% stenosis | 77.8 | 0 | <0.001b |
| Coronary artery bypass graft (CABG) | 40.1 | 0 | <0.001b |
| Myocardial infarction | 17.9 | 0 | <0.001b |
| Angioplasty (PTCA) | 40.5 | 0 | <0.001b |
| Characteristic | CAD cases | Normal controls | P-value |
|---|---|---|---|
| Sample size | 207 | 191 | – |
| Mean current age (SD) (years) | 44.8 (4.8) | 43.9 (7.8) | 0.18a |
| Mean body mass index (SD) (kg/m2) | 31.0 (6.8) | 30.6 (8.4) | 0.60a |
| Race | |||
| Caucasian | 77.8% | 74.4% | 0.63b |
| African-American | 17.9% | 19.4% | |
| Native American | 2.9% | 5.2% | |
| Asian | 1.5% | 1.1% | |
| % Male | 77.3 | 49.2 | <0.001b |
| % Hypertension | 69.6 | 38.2 | <0.001b |
| % Diabetes | 27.5 | 8.4 | <0.001b |
| % Current smokers | 31.9 | 19.4 | 0.004b |
| Qualifying events | |||
| Angiogram>70% stenosis | 77.8 | 0 | <0.001b |
| Coronary artery bypass graft (CABG) | 40.1 | 0 | <0.001b |
| Myocardial infarction | 17.9 | 0 | <0.001b |
| Angioplasty (PTCA) | 40.5 | 0 | <0.001b |
aAn unpaired t-test was used for the continuous factors (age and body mass index).
bA chi-square test was used for the categorical factors.
Characteristics of the study population
| Characteristic | CAD cases | Normal controls | P-value |
|---|---|---|---|
| Sample size | 207 | 191 | – |
| Mean current age (SD) (years) | 44.8 (4.8) | 43.9 (7.8) | 0.18a |
| Mean body mass index (SD) (kg/m2) | 31.0 (6.8) | 30.6 (8.4) | 0.60a |
| Race | |||
| Caucasian | 77.8% | 74.4% | 0.63b |
| African-American | 17.9% | 19.4% | |
| Native American | 2.9% | 5.2% | |
| Asian | 1.5% | 1.1% | |
| % Male | 77.3 | 49.2 | <0.001b |
| % Hypertension | 69.6 | 38.2 | <0.001b |
| % Diabetes | 27.5 | 8.4 | <0.001b |
| % Current smokers | 31.9 | 19.4 | 0.004b |
| Qualifying events | |||
| Angiogram>70% stenosis | 77.8 | 0 | <0.001b |
| Coronary artery bypass graft (CABG) | 40.1 | 0 | <0.001b |
| Myocardial infarction | 17.9 | 0 | <0.001b |
| Angioplasty (PTCA) | 40.5 | 0 | <0.001b |
| Characteristic | CAD cases | Normal controls | P-value |
|---|---|---|---|
| Sample size | 207 | 191 | – |
| Mean current age (SD) (years) | 44.8 (4.8) | 43.9 (7.8) | 0.18a |
| Mean body mass index (SD) (kg/m2) | 31.0 (6.8) | 30.6 (8.4) | 0.60a |
| Race | |||
| Caucasian | 77.8% | 74.4% | 0.63b |
| African-American | 17.9% | 19.4% | |
| Native American | 2.9% | 5.2% | |
| Asian | 1.5% | 1.1% | |
| % Male | 77.3 | 49.2 | <0.001b |
| % Hypertension | 69.6 | 38.2 | <0.001b |
| % Diabetes | 27.5 | 8.4 | <0.001b |
| % Current smokers | 31.9 | 19.4 | 0.004b |
| Qualifying events | |||
| Angiogram>70% stenosis | 77.8 | 0 | <0.001b |
| Coronary artery bypass graft (CABG) | 40.1 | 0 | <0.001b |
| Myocardial infarction | 17.9 | 0 | <0.001b |
| Angioplasty (PTCA) | 40.5 | 0 | <0.001b |
aAn unpaired t-test was used for the continuous factors (age and body mass index).
bA chi-square test was used for the categorical factors.
Demographics of the patients
| Individual ID | MEF2A mutation | Current age | Age at diagnosis | Clinical diagnosis for CAD | Risk factors |
|---|---|---|---|---|---|
| GB04146 | N263S | 49 | 43 | CABG | SM |
| Angina class IV | |||||
| GB05072 | N263S | 47 | 43 | PTCA/stent | DL, HTN (onset at 30), SM, FH (father, MI at 38) |
| GB00305 | P279L | 39 | 36 | PTCA/stent | DL, DB (onset at 35), SM |
| CAD669 | P279L | 66 | 63 | CABG | DL, HTN, OB, DB, SM |
| GB04583 | G283D | 50 | 48 | CABG | DL, HTN (onset at 43), SM |
| Angina class IV 70–80% stenosis |
| Individual ID | MEF2A mutation | Current age | Age at diagnosis | Clinical diagnosis for CAD | Risk factors |
|---|---|---|---|---|---|
| GB04146 | N263S | 49 | 43 | CABG | SM |
| Angina class IV | |||||
| GB05072 | N263S | 47 | 43 | PTCA/stent | DL, HTN (onset at 30), SM, FH (father, MI at 38) |
| GB00305 | P279L | 39 | 36 | PTCA/stent | DL, DB (onset at 35), SM |
| CAD669 | P279L | 66 | 63 | CABG | DL, HTN, OB, DB, SM |
| GB04583 | G283D | 50 | 48 | CABG | DL, HTN (onset at 43), SM |
| Angina class IV 70–80% stenosis |
PTCA, percutaneous coronary angioplasty; CABG, coronary artery bypass surgery; DL, dyslipidemia; HTN, hypertension; DB, diabetes; SM, smoking; FH, family history; OB, obesity.
Demographics of the patients
| Individual ID | MEF2A mutation | Current age | Age at diagnosis | Clinical diagnosis for CAD | Risk factors |
|---|---|---|---|---|---|
| GB04146 | N263S | 49 | 43 | CABG | SM |
| Angina class IV | |||||
| GB05072 | N263S | 47 | 43 | PTCA/stent | DL, HTN (onset at 30), SM, FH (father, MI at 38) |
| GB00305 | P279L | 39 | 36 | PTCA/stent | DL, DB (onset at 35), SM |
| CAD669 | P279L | 66 | 63 | CABG | DL, HTN, OB, DB, SM |
| GB04583 | G283D | 50 | 48 | CABG | DL, HTN (onset at 43), SM |
| Angina class IV 70–80% stenosis |
| Individual ID | MEF2A mutation | Current age | Age at diagnosis | Clinical diagnosis for CAD | Risk factors |
|---|---|---|---|---|---|
| GB04146 | N263S | 49 | 43 | CABG | SM |
| Angina class IV | |||||
| GB05072 | N263S | 47 | 43 | PTCA/stent | DL, HTN (onset at 30), SM, FH (father, MI at 38) |
| GB00305 | P279L | 39 | 36 | PTCA/stent | DL, DB (onset at 35), SM |
| CAD669 | P279L | 66 | 63 | CABG | DL, HTN, OB, DB, SM |
| GB04583 | G283D | 50 | 48 | CABG | DL, HTN (onset at 43), SM |
| Angina class IV 70–80% stenosis |
PTCA, percutaneous coronary angioplasty; CABG, coronary artery bypass surgery; DL, dyslipidemia; HTN, hypertension; DB, diabetes; SM, smoking; FH, family history; OB, obesity.
Lipid profile of MEF2A mutation carriers
| Individual ID | MEF2A mutation | Lipid level before medication | Treatment (medication) | Lipid level after medication |
|---|---|---|---|---|
| GB04146 | N263S | Cho, 177 | Fluvastatin (Welchol) | Cho, 160 |
| Tri, 54 | Tri, 116 | |||
| HDL, 40 | HDL, 33 | |||
| LDL, 126 | LDL, 110 | |||
| GB05072 | N263S | Cho, 280 | Atorvastatin (Lipitor) | Cho, 148 |
| Tri, 515 | Tri, 340 | |||
| HDL, 39 | HDL, 31 | |||
| LDL, 164 | LDL, 49 | |||
| GB00305 | P279L | Cho, 182 | Simvastatin (Zocor) | Cho, 167 |
| Tri, 252 | Tri, 134 | |||
| HDL, 27 | HDL, 30 | |||
| LDL, 130 | LDL, 110 | |||
| CAD669 | P279L | N/A | N/A | N/A |
| GB04583 | G283D | Cho, 274 | Atorvastatin (Lipitor) | Cho, 165 |
| Tri, 107 | Tri, 93 | |||
| HDL, 71 | HDL, 61 | |||
| LDL, 182 | LDL, 85 |
| Individual ID | MEF2A mutation | Lipid level before medication | Treatment (medication) | Lipid level after medication |
|---|---|---|---|---|
| GB04146 | N263S | Cho, 177 | Fluvastatin (Welchol) | Cho, 160 |
| Tri, 54 | Tri, 116 | |||
| HDL, 40 | HDL, 33 | |||
| LDL, 126 | LDL, 110 | |||
| GB05072 | N263S | Cho, 280 | Atorvastatin (Lipitor) | Cho, 148 |
| Tri, 515 | Tri, 340 | |||
| HDL, 39 | HDL, 31 | |||
| LDL, 164 | LDL, 49 | |||
| GB00305 | P279L | Cho, 182 | Simvastatin (Zocor) | Cho, 167 |
| Tri, 252 | Tri, 134 | |||
| HDL, 27 | HDL, 30 | |||
| LDL, 130 | LDL, 110 | |||
| CAD669 | P279L | N/A | N/A | N/A |
| GB04583 | G283D | Cho, 274 | Atorvastatin (Lipitor) | Cho, 165 |
| Tri, 107 | Tri, 93 | |||
| HDL, 71 | HDL, 61 | |||
| LDL, 182 | LDL, 85 |
Lipid levels in mg/dl; Cho, cholesterol; Tri, triglycerides; HDL, high-density lipoprotein; LDL, low-density lipoprotein. Normal Cho, Tri, HDL and LDL levels are <200, <150,>45 and <130, respectively. N/A, no data.
Lipid profile of MEF2A mutation carriers
| Individual ID | MEF2A mutation | Lipid level before medication | Treatment (medication) | Lipid level after medication |
|---|---|---|---|---|
| GB04146 | N263S | Cho, 177 | Fluvastatin (Welchol) | Cho, 160 |
| Tri, 54 | Tri, 116 | |||
| HDL, 40 | HDL, 33 | |||
| LDL, 126 | LDL, 110 | |||
| GB05072 | N263S | Cho, 280 | Atorvastatin (Lipitor) | Cho, 148 |
| Tri, 515 | Tri, 340 | |||
| HDL, 39 | HDL, 31 | |||
| LDL, 164 | LDL, 49 | |||
| GB00305 | P279L | Cho, 182 | Simvastatin (Zocor) | Cho, 167 |
| Tri, 252 | Tri, 134 | |||
| HDL, 27 | HDL, 30 | |||
| LDL, 130 | LDL, 110 | |||
| CAD669 | P279L | N/A | N/A | N/A |
| GB04583 | G283D | Cho, 274 | Atorvastatin (Lipitor) | Cho, 165 |
| Tri, 107 | Tri, 93 | |||
| HDL, 71 | HDL, 61 | |||
| LDL, 182 | LDL, 85 |
| Individual ID | MEF2A mutation | Lipid level before medication | Treatment (medication) | Lipid level after medication |
|---|---|---|---|---|
| GB04146 | N263S | Cho, 177 | Fluvastatin (Welchol) | Cho, 160 |
| Tri, 54 | Tri, 116 | |||
| HDL, 40 | HDL, 33 | |||
| LDL, 126 | LDL, 110 | |||
| GB05072 | N263S | Cho, 280 | Atorvastatin (Lipitor) | Cho, 148 |
| Tri, 515 | Tri, 340 | |||
| HDL, 39 | HDL, 31 | |||
| LDL, 164 | LDL, 49 | |||
| GB00305 | P279L | Cho, 182 | Simvastatin (Zocor) | Cho, 167 |
| Tri, 252 | Tri, 134 | |||
| HDL, 27 | HDL, 30 | |||
| LDL, 130 | LDL, 110 | |||
| CAD669 | P279L | N/A | N/A | N/A |
| GB04583 | G283D | Cho, 274 | Atorvastatin (Lipitor) | Cho, 165 |
| Tri, 107 | Tri, 93 | |||
| HDL, 71 | HDL, 61 | |||
| LDL, 182 | LDL, 85 |
Lipid levels in mg/dl; Cho, cholesterol; Tri, triglycerides; HDL, high-density lipoprotein; LDL, low-density lipoprotein. Normal Cho, Tri, HDL and LDL levels are <200, <150,>45 and <130, respectively. N/A, no data.
References
Wang, Q. and Chen, Q. (
Wang, Q. and Chen. Q. (
Lewis, D., Wang, Q. and Topol, E.J. (
American Heart Association (
American Heart Association (
Nora, J.J., Lortscher, R.H., Spangler, R.D., Nora, A.H. and Kimberling, W.J. (
Schildkraut, J.M., Myers, R.H., Cupples, L.A., Kiely, D.K. and Kannel, W.B. (
Wang, Q. and Pyeritz, R.E. (
Wang, Q., Rao, S., Shen, G.Q., Li, L., Moliterno, D.J., Newby, L.K., Rogers, W.J., Cannata, R., Zirzow, E., Elston, R.C. and Topol, E.J. (
Broeckel, U., Hengstenberg, C., Mayer, B., Holmer, S., Martin, L.J., Comuzzie, A.G., Blangero, J., Nurnberg, P., Reis, A., Riegger, G.A. et al. (
Pajukanta, P., Cargill, M., Viitanen, L., Nuotio, I., Kareinen, A., Perola, M., Terwilliger, J.D., Kempas, E., Daly, M., Lilja, H. et al. (
Harrap, S.B., Zammit, K.S., Wong, Z.Y., Williams, F.M., Bahlo, M., Tonkin, A.M. and Anderson, S.T. (
Hauser, E.R., Crossman, D.C., Granger, C.B., Haines, J.L., Jones, C.J., Mooser, V., McAdam, B., Winkelmann, B.R., Wiseman, A.H., Muhlestein, J.B. et al. (
Francke, S., Manraj, M., Lacquemant, C., Lecoeur, C., Lepretre, F., Passa, P., Hebe, A., Corset, L., Yan, S.L., Lahmidi, S. et al. (
Wang, L., Fan, C., Topol, S.E., Topol, E.J. and Wang, Q. (
Black, B.L., Molkentin, J.D. and Olson, E.N. (
McKinsey, T.A., Zhang, C.L. and Olson, E.N. (
Naya, F.J., Black, B.L., Wu, H., Bassel-Duby, R., Richardson, J.A., Hill, J.A. and Olson, E.N. (
Yu, Y.T. (
Fan, C., Duhagon, M.A., Oberti, C., Chen, S., Hiroi, Y., Komuro, I., Duhagon, P.I., Canessa, R. and Wang, Q. (
Fan, C., Liu, M. and Wang, Q. (
Chen, S., Zhang, L., Bryant, R.M., Vincent, G.M., Flippin, M., Lee, J.C., Brown, E., Zimmerman, F., Rozich, R., Szafranski, P. et al. (
Yong, S., Tian, X. and Wang, Q. (
Sanguinetti, M.C., Curran, M.E., Spector, P.S. and Keating, M.T. (
Wang, Z., Tristani-Firouzi, M., Xu, Q., Lin, M., Keating, M.T. and Sanguinetti, M.C. (
Shalaby, F.Y., Levesque, P.C., Yang, W.P., Little, W.A., Conder, M.L., Jenkins-West, T. and Blanar, M.A. (
Chen, Q., Kirsch, G.E., Zhang, D., Brugada, R., Brugada, J., Brugada, P., Potenza, D., Moya, A., Borggrefe, M., Breithardt, G. et al. (
Wang, Q., Shen, J., Li, Z., Timothy, K., Vincent, G.M., Priori, S.G., Schwartz, P.J. and Keating, M.T. (
Wang, Q., Shen, J., Splawski, I., Atkinson, D., Li, Z., Robinson, J.L., Moss, A.J., Towbin, J.A. and Keating, M.T. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}