




Abstract
Laminopathies comprise a group of inherited diseases with variable clinical phenotypes, caused by mutations in the lamin A/C gene (LMNA). A prominent feature in several of these diseases is muscle wasting, as seen in Emery–Dreifuss muscle dystrophy, dilated cardiomyopathy and limb-girdle muscular dystrophy. Although the mechanisms underlying this phenotype remain largely obscure, two major working hypotheses are currently being investigated, namely, defects in gene regulation and/or abnormalities in nuclear architecture causing cellular fragility. In this study, using a newly developed cell compression device we have tested the latter hypothesis. The device allows controlled application of mechanical load onto single living cells, with simultaneous visualization of cellular deformation and quantitation of resistance. With the device, we have compared wild-type (MEF+/+) and LMNA knockout (MEF−/−) mouse embryonic fibroblasts (MEFs), and found that MEF−/− cells show a significantly decreased mechanical stiffness and a significantly lower bursting force. Partial rescue of the phenotype by transfection with either lamin A or lamin C prevented gross nuclear disruption, as seen in MEF−/− cells, but was unable to fully restore mechanical stiffness in these cells. Our studies show a direct correlation between absence of LMNA proteins and nuclear fragility in living cells. Simultaneous recordings by confocal microscopy revealed that the nuclei in MEF−/− cells, in contrast to MEF+/+ cells, exhibited an isotropic deformation upon indentation, despite an anisotropic deformation of the cell as a whole. This nuclear behaviour is indicative for a loss of interaction of the disturbed nucleus with the surrounding cytoskeleton. In addition, careful investigation of the three-dimensional organization of actin-, vimentin- and tubulin-based filaments showed a disturbed interaction of these structures in MEF−/− cells. Therefore, we suggest that in addition to the loss of nuclear stiffness, the loss of a physical interaction between nuclear structures (i.e. lamins) and the cytoskeleton is causing more general cellular weakness and emphasizes a potential key function for lamins in maintaining cellular tensegrity.
INTRODUCTION
Nuclear lamins are karyoskeletal proteins located at the inner nuclear membrane and belonging to the family of intermediate filament proteins. Two main classes can be distinguished, i.e. A- and B-type lamins. B-Type lamins are either products of the LMNB1 gene, encoding lamin B1 (1), or the LMNB2 (LMN2) gene (2), encoding lamins B2 and B3 (3). Until now at least four different proteins are known to arise from the A-type lamin gene (LMNA) (4), i.e. lamin A, lamin AΔ10, lamin C (5) and lamin C2 in (mouse) spermatocytes (6). Unlike B-type lamins, which are expressed in most eukaryotic cells (7,8), A-type lamins are differentially expressed, i.e. A-type lamins are reduced or absent in early embryonic cells, in cells of a low degree of differentiation, in cells of the cellular immune and hematopoietic system, and in proliferating cells (7–9).
Although some functional aspects of the nuclear lamins have been established, several other proposed functions remain to be confirmed. For B-type lamins, it has become clear that the absence of these proteins is not compatible with life. Silencing of lamin B1 or lamin B2 by siRNA produces apoptotic features both in mammalian cells and in cells from Caenorhabditis elegans (10,11). Apparently, the presence of B-type lamins is critical for vital cellular processes, such as the elongation phase during DNA synthesis (12). As mutations in either LMNB1 or LMNB2 can cause cell death, this can explain why no mutations in these genes have been found in human heritable diseases.
In contrast to B-type lamins, the loss of A-type lamins leads to specific loss of function. This can be seen in human heritable diseases resulting from LMNA mutations and in LMNA knockout mice, where the loss of A-type lamins leads eventually to death (13,14). However, the exact impact of the impairment of LMNA expression on the functioning of cells is not yet well understood.
A variety of inherited diseases are associated with mutations in the LMNA gene. These diseases are collectively called laminopathies, and include Emery–Dreifuss muscular dystrophy (EDMD) (15), dilated cardiomyopathy type 1A (16), limb-girdle muscular dystrophy type 1B (17), familial partial lipodystrophy (18,19), Charcot–Marie–Tooth disease type 2 (20) mandibuloacral dysplasia (21) and rare childhood syndromes of premature ageing, the Hutchinson–Gilford progeria (22–24) and atypical Werner's progeria (25). To explain this relatively heterogeneous spectrum of diseases, two main working hypotheses are currently exploited. First, it is suggested that patients with laminopathies experience mechanical weakness in cells, caused by a disturbed nuclear architecture. This mechanical stress hypothesis could explain the loss of muscle tissues seen in the group of muscle failure-related diseases. The second hypothesis trying to explain development of symptoms deals with the loss of gene regulatory functions of the A-type lamins. Studies have suggested that A-type lamins can play a role in gene regulation by interaction with the retinoblastoma protein (26,27), possibly mediated by the Lamina Associated Protein 2 alpha (LAP2alpha) (28) or by the prevention of transcriptional complex formation by binding to transcription factors such as SREBP1 (29) and MOK2 (30).
In this article, we have examined the role of A-type lamins in providing structural support to the nucleus and to cells in general, by performing compression experiments on mouse embryonic fibroblasts (MEFs) with normal expression of the LMNA gene (MEF+/+ cells), and on cells lacking expression of the LMNA gene (MEF−/− cells). As the nucleus seems to account for most of the mechanical stiffness of a cell (31,32), a disturbance in the nuclear architecture should be reflected by a decreased mechanical stiffness of these cells.
To test this hypothesis, unconfined compression experiments were performed on single cells, using a recently developed loading device (Fig. 1). The device enables quantification of the mechanical resistance to compression, and during the experiments cell deformation and cell damage can be observed using confocal microscopy (33). Our results show that, using this device, we can indeed measure a significantly reduced mechanical stiffness in cells lacking expression of the LMNA gene. In contrast to nuclei of normal cells, the integrity of MEF−/− nuclei was lost after a compression experiment. Moreover, the preferred deformation direction as seen in the nuclei of MEF+/+ cells was not observed in MEF−/− nuclei. This behaviour is explained by the absence of a link between the nucleus and the cytoskeleton scaffolds in MEF−/− cells. Rescue of MEF−/− cells with lamin A or lamin C restored some but not all mechanical characteristics of the MEF−/− cells.
RESULTS
Decreased resistance to compression in LMNA null cells
Cell cultures from MEF+/+ and MEF−/− cells were subjected to compression at different passages, at variable densities, and at different time points after seeding.
The low variation between measurements of the individual cell types under these different conditions allowed us to group them. Cellular stiffness of 36 MEF+/+ cells and 31 MEF−/− cells was evaluated. Figure 2 shows confocal recordings of two representative cells from a MEF+/+ and an MEF−/− cell culture during compression. Full recordings of the behaviour of these cells during indentation are available as Supplementary Material. During compression, the cell flattens and enlarges dramatically in lateral directions (Fig. 2A and B). Immediately after cellular disruption (seen in all cells during compression) propidium iodide (PI) enters the cytoplasm followed by an almost immediate concentration in the nucleus, probably via the nuclear pores complexes. Binding of PI to the chromatin shows a morphologically intact nucleus after compression in the MEF+/+ cell, and large strands of DNA protruding into the cytoplasm in the case of the MEF−/− cell (compare Fig. 2A and B). Comparison of the mean force needed to induce indentation of a cell in z-direction (axial deformation) shows that, as expected, the force needed to cause an axial deformation increased with increasing indentation of the cells (Fig. 2C). Furthermore, these mean force versus axial strain curves show that the forces needed to compress a cell were larger for MEF++, than for MEF− cells. These differences became statistically significant from the point where the indentor caused more than 27.5% axial deformation (Student's t-test, P<0.05).
Beyond 75% of axial deformation, the resistance of both cell types increased rapidly, probably owing to the fact that the tip of the probe reached the surface of the slide and/or that the presence of cellular debris (∼20% of the initial cellular height) prevented further compression.
In 50% of the cells, a typical decrease in the force could be observed during compression, caused by rupture of the cellular membrane. The maximum force at cell membrane bursting is referred to as the bursting force. Two typical curves of individual MEF+/+ and MEF−/− cells exhibiting this characteristic decrease in force are shown in Figure 2D (arrows). The mean bursting force for MEF+/+ cells (8.7±3.0 µN) was significantly higher than that for MEF−/− cells (2.9±1.9 µN, Student's t-test, P<0.0001). The accompanying axial deformation at cellular rupture is significantly different for both cell types, i.e. 68.8±7.5% for MEF+/+ cells and 62.0±5.8% for MEF−/− (Student's t-test, P<0.05). For rescued cells, no clear cellular membrane rupture point could be calculated from the force versus deformation plots. Possibly, transfection influences the cellular membrane properties.
Lamin A and/or lamin C rescue does not restore mechanical resistance to deformation
In order to investigate whether lamin A or lamin C tagged with the green fluorescent protein (GFP) are capable of rescuing the MEF−/− phenotype, we transfected lamin A or lamin C into MEF−/− cells. In order to examine whether a correct nuclear structure was restored, we studied the (re)distribution of other nuclear markers next to lamins. The localization of emerin, which is known to be largely dependent on the presence of lamin A and/or lamin C (13), shows the expected staining pattern at the nuclear membrane in unaffected MEF+/+ cells (Fig. 3A), whereas in MEF−/− cells, emerin is distributed throughout the endoplasmic reticulum with only a minor portion of emerin present in the nuclear periphery (Fig. 3B). Another marker for verification of a correct nuclear membrane organization is the nuclear pore complex protein nucleoporin p62. In normal cells, nuclear pores are evenly distributed over the nuclear membrane surface. In lamin null cells, nucleoporin can be absent from large areas of the nuclear membrane, as seen in Figure 3C (arrow). Transient restoration of lamin A or lamin C expression or both could not fully restore the normal architecture of affected nuclei. First, the typical honeycomb appearance of lamins at the distal poles of the nuclei, seen in many cases of mutated lamins, was still apparent after cotransfection with (GFP-tagged) lamin A and lamin C (Fig. 3D), with a relatively heavy decoration of lamin A– and lamin C–GFP in these areas. These findings indicate that distorted MEF−/− nuclei did not revert to their normal phenotype after rescue. Indeed, the percentage of abnormal nuclei on the basis of nuclear DNA staining remained unaltered (∼5% of the cells). In addition, in the transfected cells, emerin was not concentrated at the nuclear membrane but was still mainly dispersed throughout the endoplasmic reticulum (Fig. 3E), similar to cells lacking A-type lamins (compare Fig. 3C and E).
Further, co-localization studies of cells transfected with lamin A and stained for p62 expression showed that nuclear pore complex distribution remained depleted in some areas of the nuclear envelope (Fig. 3F, arrow). Similarly, lamin B remained absent in the same regions (data not shown).
Measuring resistance to indentation of MEF−/− cells transfected with A-type lamins did not show significant differences when compared with untransfected MEF−/− cells (Fig. 3G). Strikingly, no significant differences in cellular stiffness were observed, when comparing rescued cells with morphologically normal nuclei to cells with aberrantly localized A-type lamins. Apparently, restoring A-type lamin expression and incorporation into the nuclear lamina is not sufficient to restore cellular stiffness.
Differences in nuclear damage resulting from compression
On the basis of the abnormalities in nuclear morphology, previously seen in MEF−/− cells (13), we anticipated that lamina and nuclear membrane aberrations would cause premature disruption of nuclei in affected cells during compression. Indeed, most nuclei of MEF−/− cells could not endure the complete compression regimen without bursting or showing otherwise irreversible nuclear damage. Figure 4 shows the nuclear morphology of MEF+/+ and MEF−/− cells after compression, and compares the number of affected nuclei in the two types of cell culture. Although the majority of MEF+/+ nuclei remained largely intact throughout the complete compression regimen (Fig. 4A and B), most MEF−/− nuclei became damaged, resulting in chromatin protruding into the cytoplasm as seen by PI staining. Figure 4C shows a massive eruption of DNA outside of the nucleus, whereas in Figure 4D more subtle DNA protrusions into the cytoplasm are observed (arrows in Fig. 4D).
A-Type lamin rescue reduces nuclear damage upon compression
Rescue with either lamin A(–GFP) or lamin C(–GFP) had a positive impact on maintaining the nuclear shape during compression. After transfection of A-type lamin–GFP into MEF−/− cells, a morphologically intact nuclear surface was observed in many cells, highlighted by the lamin GFP signal lining the nuclear lamina (Fig. 4H). These nuclei in general remained intact after compression, as deduced from the PI staining. In cells with an irregular lamina after rescue, nuclear areas with a relatively strong A-type lamin–GFP expression would not burst, whereas simultaneously the nuclear areas with less A-type lamin expression did burst upon indentation, revealed by PI labelling protruding into the cytoplasm after compression (Fig. 4E and F). Strikingly, a cell with a predictable weak spot in the nucleus before compression (Fig. 4G, inset, arrow), did burst in this area before any noticeable damage was seen in the remaining parts of the nucleus (Fig. 4G; Supplementary Material). Quantitative comparisons of visual nuclear damage among MEF−/−, MEF+/+ and transfected MEF−/− cells (Fig. 4I) show that, indeed, nuclear coherence is increased after rescue, resulting in fewer cells with prominent nuclear damage after compression.
MEF+/+ nuclei deform anisotropically, MEF−/− nuclei isotropically
To explain the apparent discrepancy between the restoration of nuclear coherence and simultaneous lack of restoration of cellular stiffness, we more closely examined the deformation behaviour of cells and nuclei during compression. In MEF+/+ cells, nuclear deformation was mainly anisotropic, showing nuclear enlargement perpendicular to the long axis in cells with a prevalent growth orientation (Fig. 5A). However, careful inspection of nuclear deformation direction of MEF−/− cells did not reveal this correlation in most cells (Fig. 5B). The nuclei deformed equally into all directions (isotropic), with no correlation with the initial cellular orientation. Comparing the width to length ratio in MEF+/+ cells before indentation and at the time of maximum indentation, an average relative increase of the width to length ratio of 40% was observed (Fig. 5C). The initial shape of the nucleus did not predict the anisotropic behaviour, as both round and oval nuclei deformed with similar changes in width to length ratios. MEF−/− cells or A-type lamin-rescued cells showed only an average increase in the length to width ratio of 8–11%, indicating an almost isotropic deformation in most of these cells.
The anisotropic deformation of nuclei is not caused by the shape or structure of the nuclei in itself. When isolated nuclei, obtained by treatment of MEF+/+ or MEF−/− cells with acetic acid, were subjected to compression, both round and oval nuclei of both cell lines deformed strictly isotropically (data not shown).
Cytoskeletal disorganization in MEF−/− cells
The direction of nuclear deformation is most likely determined by extranuclear factors involved in maintaining cellular shape and orientation. As cytoskeletal structures are the likely candidates in this respect, we examined their organization in MEF−/− and MEF+/+ cells. Figure 6 shows representative cells from these populations. At the peripheral regions of these cells, no obvious differences in the organization of stress fibres were observed. However, in areas close to the nucleus, the actin stress fibres seemed to be disrupted in MEF−/− cells and not in MEF+/+ cells (compare Fig. 6A and B). In these areas, numerous dots were observed consisting of actin not correctly organized into fibres (Fig. 6B). We observed actin abnormalities in 5–10% of MEF−/− cells, with considerable regional variations within the same sample and even larger variations between samples. Careful inspection of the microtubule network (Fig. 6C and D) in perinuclear areas showed that the characteristic concentration of microtubules around the nucleus starting at the centrosomes, as seen in MEF+/+ cells (Fig. 6C), is disturbed in MEF−/− cells (Fig. 6D). From the centrosomes, microtubules were diverted into all directions, showing little or no orientation at the nuclear periphery. Although in most normal cells the nuclear membrane seemed to be completely covered by microtubules, large nuclear membrane areas in MEF−/− cells were devoid of microtubule attachments. The number of cells with abnormal tubulin organization was similar to the number of cells with actin disturbances. However, as actin and tubulin proteins abnormalities only became fully apparent after confocal microscope recording at high magnification of individual cells, the number of abnormal cells was most likely underestimated. An even more disturbed vimentin network was seen (Fig. 6E–H). MEF+/+ cells show a characteristic perinuclear filament organization (Fig. 6E and F), whereas in MEF−/− cells several disturbances were noted (Fig. 6G and H). A striking loss of vimentin organization in the cytoplasm was seen as regions with very bright patches of vimentin, whereas other areas were almost completely devoid of vimentin fibres. The typical wrapping of vimentin around the nucleus was almost absent in these cells. This vimentin disorganization was much more obvious, and could be readily observed even with standard fluorescence microscopy. The percentage of MEF−/− cells with these vimentin abnormalities was between 20 and 30%.
DISCUSSION
The phenotype of several of the diseases associated with A-type lamin mutations, points to a correlation between cellular and/or nuclear weakness, and the loss in mechano-sensitive tissues, such as heart and skeletal muscle. EDMD, limb-girdle dystrophy and dilated cardiomyopathy are all characterized by the loss of muscle tissue (reviewed in 34). Next to stretching forces, muscle cell nuclei also encounter heavy local compression forces during muscle activity. As muscle strength increases with age, nuclear strain will increase with age as well, possibly explaining the gradual increase of symptoms in most patients (34). B-Type lamins are crucial for cellular survival, and cells depleted of B-type lamins will die via apoptosis (10). Also, in cell lines with A-type lamin mutations, increased apoptosis can be observed, especially after triggering of differentiation (35,36). Organisms lacking A-type lamin expression, including LMNA null mice (13) or humans expressing no functional lamins (14), die shortly after birth. The occurrence of apoptotic cells in cardiac muscle cells of LMNA deficient mice is probably a late effect, which would imply that these cells die as a result from ‘wear and tear’ effects of repeated contractions (37).
A possible explanation would be the loss of cellular stiffness due to A-type lamin mutations. On the basis of the localization of the nuclear lamina, it has already been postulated long ago that lamins play a prominent role in maintaining nuclear integrity (38). Indeed, cellular extraction studies in cell lines harbouring A-type lamin mutations indicate that cells with mutations display increased nuclear fragility upon extraction (39).
More direct evidence for nuclear weakness as a result of A-type lamins abnormalities was recently supplied by Lammerding et al. (40), who showed that, indeed, defective nuclear mechanics are observed in LMNA null fibroblasts after stretching of cells. These authors concluded that the absence of lamins causes loss of nuclear stability, resulting in cellular weakness. In order to examine directly the effects of strain onto the nucleus, we chose an alternative method to mechanically challenge cells, i.e. by using cellular compression rather than mechanical stretch. Our set-up enables the direct visualization of the response of the nucleus during compression, with simultaneous recording of the force needed to exert this indentation. We measured the mechanical stiffness in normal fibroblasts and fibroblasts completely lacking LMNA gene expression. We chose fibroblasts, as these cells are easy to obtain from (future) patient samples and these cells should reflect general mechanical weakness in cells, if caused by structural abnormalities. Moreover, loss of fibroblasts themselves could induce weakening of connective tissues at several sites in the body. Using a newly developed cell loading device (33), we found a significantly higher resistance against indentation force in MEF+/+ cells than in MEF−/− cells. Moreover, the force needed to cause cellular bursting was significantly higher in MEF+/+ cells.
Simultaneous confocal microscopic imaging allowed us to more closely examine the type and direction of deformation of individual nuclei and cells. Although most wild-type nuclei exhibited an intact morphology after compression, 90% of the LMNA null nuclei burst completely after compression, leaving a disrupted nuclear mass spreading into the cytoplasm. This percentage is considerably higher than the rupture observed in 3–5% of the cells after cell strain using cellular stretching (40), and implies that the mechanical weakness of these cells could be an important factor in the development of laminopathies.
This nuclear damage due to compression could be largely overcome by transfection of A-type lamins into these cells. However, in these rescue experiments, mechanical stiffness of the affected cells was not restored. In order to find an explanation for this phenomenon, we investigated the protein organization of these rescued cells more closely. It became clear that the presence of lamin A– and/or lamin C–GFP in the lamina did not result in the localization of emerin to the nuclear membrane. Moreover, part of the cells transfected with A-type lamins still showed an abnormal nuclear morphology characteristic for LMNA−/− cells (13). A possible explanation for not restoring the normal phenotype in these cells was the inability to form stable transfectants. Although we did get an efficiency of transfection of up to 10%, these numbers are significantly lower than the efficiency obtained by electroporation (up to 58%) (41). As a result, the expression levels of A-type lamins in transfected cells might be too low for obtaining restoration of nuclear architecture (41). Alternatively, the manifestation of changes in the nuclear structure phenotype might require one or more cycles of nuclear disassembly and reformation as occurs during mitosis (42). In our transfected cell, we never saw fluorescent cells forming colonies, i.e. going through mitosis. Some cells would remain single fluorescent cells for up to 1 month, before disappearing from the culture.
This partial rescue indicated that the diminished cellular strength in the LMNA−/− cells results from factors other than the absence of A-type lamins in the nucleus. Although nuclear strength is a major component of total cellular stiffness (31,32), the interaction of cytoskeletal and nucleoskeletal elements is important for maintenance of the full cellular stiffness. According to a previously postulated tensegrity model (43,44) for cellular and tissue strength, this can only be achieved if all cyto- and nucleoskeletal components are correctly assembled. A strong indication that, indeed, the partially rescued cells still lacked tensegrity came from our observations on the direction of deformation of compressed nuclei. As observed in other cell lines (data not shown), normal nuclei exhibit anisotropic deformation during compression; if cells have a prevalent direction of orientation (the length axis), the nuclei will deform in a direction perpendicular to this axis. Indeed, most MEF+/+ showed this type of anisotropic deformation during compression. However, MEF−/− cells as well as A-type lamin-rescued cells show an isotropic deformation. Apparently, the interconnection between nuclear components such as lamins and the cytoplasm was lacking in these cells. Anisotropic deformation is most likely caused by the attachment of the cytoskeletal components to the nucleus, including microfilaments, intermediate filaments and possibly microtubules. Indeed, when cytoskeletal connections are disrupted by acetic acid and detergent extraction, isolated intact nuclei deform isotropically during compression. These findings are in line with the viscoelastic behaviour of isolated nuclei (31). The observation of decreased cytoskeletal stiffness in MEF−/− cells is in this respect very intriguing (40).
There is growing evidence that, indeed, the cytoskeleton is closely connected to the nucleus. Microfilaments are indirectly connected to the nucleus, most likely via nesprins. The recently discovered giant proteins spanning the complete cytoplasm of the cell (45). Of these, nesprin1α is localized in the intranuclear compartment of the cell, and interacts with A-type lamins as well as emerin at the nuclear envelope (46). Binding of nesprin1α to these molecules occurs at the most C-terminal part of nesprin to A-type lamins and at a their N-terminal part to emerin. A more recent study indicates that nesprin 1α is the C-terminal part of a much larger protein, nesprin 1, which is not present in the nucleus but spans large areas of the cytoplasm (45). Interestingly, the N-terminal part of this large protein binds prominently to actin (45). A second member of the nesprin family, called NUANCE (47) or nesprin-2 (45,48), shows a similar topology, i.e. binding to actin in the N-terminal region of the molecule and anchoring to the nuclear membrane via the common KLS motif (45). In cells lacking A-type lamin expression, the proper binding of nesprin 1α is disturbed (14). Re-localization of nesprin 1α from the nucleus to the cytoplasm occurs in human fibroblasts lacking expression of the LMNA gene. The correct localization of this protein is restored if these cells are rescued with either lamin A or lamin C (14). In these experiments, in contrast to our study, the correct localization of other nuclear proteins such as emerin is restored as well.
Although it is not yet shown that nesprin contributes to mechanical stiffness, it is well known that actin is a major component in maintaining cellular stiffness (49,50). Moreover, disruption of the actin cytoskeleton by cytochalasin-D causes nuclei to respond differently to compression (51), suggesting a connection of actin to the nucleus. Careful examination of actin architecture in LMNA−/− cells did reveal abnormalities in actin filament formation not seen in control cells. These abnormalities in actin molecules, especially present in perinuclear areas of affected cells, could have been easily overlooked in previous studies, since only confocal microscopy allows the detailed inspection of the cellular interior covered with a dense layer of brightly fluorescent actin fibres. Next to an indirect binding of actin to the nucleus via nesprins, other studies showed direct interaction between cytoplasmic actin and emerin (52), thought to be localized exclusively at the inner nuclear membrane after transport from the endoplasmic reticulum to the nucleus. However, the presence of emerin (or similar transmembrane proteins containing a LEM domain) at the outer nuclear membrane could be the missing link in connecting the inner and the outer nuclear membranes. Such a link is necessary if direct mechanical strain is to be transduced from the cellular membrane to the nucleus. This hypothesis is supported by the finding that similar muscle dystrophic diseases are caused by mutations in the actin gene itself (myopathies; reviewed in 53), or by inappropriate attachment of actin to the periphery of the cell. For instance, limb-girdle muscular dystrophy type 1A is associated with the actin-binding protein myotilin (54); sporadic dilated cardiomyopathies can be associated with dystrophin gene mutations (55) and actinin mutations can be associated with congenital muscular dystrophy (56).
The importance of the other two main cytoskeletal components, microtubules and vimentin intermediate filaments, for cellular stiffness has also to be considered. Both components are important for resisting mechanical load (43), and are clearly disorganized in MEF−/− cells. Direct binding of microtubules to the nuclear surface has recently been suggested (57). In addition, Patterson et al. (58) showed in Drosophila the close interaction between lamins, Klarsicht and the microtubule organization centre. The proper localization of Klarsicht appeared to be dependent on the presence of lamins. Similarly, the correct targeting of nesprins 1α, containing a Klarsicht-like signal, to the nuclear membrane is dependent on the presence of A-type lamins in the nuclear lamina (14). Disruption of the desmin intermediate filament in muscle cells of LMNA null mice, seen as the loss of attachment sites for desmin at the nuclear envelope (37) suggests a close correlation between A-type lamin expression and a proper desmin organization. Although for intermediate filaments, similar to the other cytoskeletal components, no direct connection with the nuclear membrane is shown, these EM studies as well as other cellular extraction studies (59) do indicate the existence of such a connection. The disorganization of vimentin intermediate filaments, as seen in our study, confirms this correlation. Moreover, mutations in intermediate filaments themselves, in this case in the neurofilament light subunit, can cause Charcot–Marie–Tooth neuropathy type 2 (60), similar to the disease caused by a LMNA mutation (20).
We observed the most dramatic cytoskeletal changes in the vimentin network. Clearly, more research should solve the exact (loss of) interaction between these cellular components.
In summary, we have demonstrated that fibroblasts lacking A-type lamin expression have a significantly reduced mechanical stiffness and reduced bursting force, when compared with normal fibroblasts. Next to a loss in nuclear stiffness, we also observed aberrant deformation behaviour during compression. This points towards a loss of cytoskeletal–nucleoskeletal interactions (via emerin and nesprins), which could explain the general cellular weakness in laminopathies.
MATERIALS AND METHODS
Cell culture
Wild-type mouse embryonic fibroblasts (MEF+/+) as well as LMNA knockout mouse embryonic fibroblasts (MEF−/−) were obtained as described previously (13). Cells were grown at 37°C in a humidified incubator containing 5% CO2 in DMEM (ICN Biomedicals BV, Zoetermeer, the Netherlands) containing 10% fetal bovine serum. Cells were passaged by splitting at 1 : 3 to 1 : 5 ratios using a 0.125% trypsin/0.02 m EDTA/0.02% glucose solution in PBS.
Transfection
In order to restore LMNA protein expression in LMNA null cells, MEF−/− cells were transfected using Genejammer (Invitrogen) according to the manufacturer's instructions. Typically, 10–20% of the MEF−/− cells were transfected. Cells were transfected either with lamin A–EGFP or with lamin C–GFP for mechanical stiffness studies. The GFP-tag was used to identify transfected cells in living cell cultures.
Both lamin A–EGFP and lamin C–EGFP were obtained by cloning the lamin A or lamin C cDNA fragment from the PS65T-C1 vector (61) into the EGFP-vector (Clontech Laboratories Inc., Palo Alto, CA, USA). In order to study morphological nuclear changes by immunofluorescence, cells were transfected with lamin A–EGFP, lamin C–EGFP alone, or were co-transfected with lamin A–EGFP and lamin C cDNA cloned into pcDNA3 (Invitrogen Life Technologies, Breda, The Netherlands). No stable transfectants could be generated from this cell line.
Immunofluorescence studies
After 2–5 days of transfection, cells were plated onto glass slides, grown overnight and fixed with 4% formaldehyde in PBS for 15 min, followed by permeabilization in 0.1% Triton X-100 for 10 min at room temperature (RT). Alternatively, cells were fixed in methanol (−20°C) for 5 min. Primary antibodies were applied onto the cells for 1 h.
The following primary antibodies were used:
Mouse monoclonal antibody (MoAb) 133A2 (IgG3, 3 µg/ml purified immunoglobulin) (62,63) was a kind gift from Dr Y. Raymond (Montréal, Canada). This antibody recognizes lamin A and is reacting with the epitope consisting of amino acids 598–611.
Affinity-purified rabbit polyclonal antiserum RaLC (1 : 50 dilution), kindly provided by Dr C. J. Hutchison (Durham, UK), directed against the C-terminal sequence VSGSRR (position 567–572) of human lamin C, and exclusively reactive with lamin C and not lamin A (64).
Affinity-purified rabbit polyclonal antiserum to lamin B1, kindly provided by Dr J.C. Courvalin (INSERM, Paris, France).
Mouse MoAb to nucleoporin p62 (IgG2b, dilution 1 : 300; Transduction Laboratories, Lexington, KY, USA) (65).
Mouse MoAb NCL–emerin to emerin (IgG1, dilution 1 : 60; Novocastra, Newcastle upon Tyne, UK).
Mouse MoAb RV202 to vimentin (IgG1, undiluted; Mubio Products B.V., Maastricht, The Netherlands).
Mouse MoAb BV1118 to vimentin (IgG1, diluted 1 : 5; kindly supplied by Dr C. Viebahn, Bonn, Germany).
Mouse MoAb E7 against tubulin (IgG1, diluted 1 : 10; Developmental Studies Hybridoma Bank, Iowa City, IA, USA).
After washing in PBS, secondary antibodies were applied for 1 h at RT. Secondary antibodies used are FITC-conjugated rabbit anti-mouse Ig (1 : 100, DAKO, Glostrup, DK), FITC-conjugated goat anti-rabbit Ig (1 : 50, SBA/ITK, Birmingham, AL, USA), Texas Red conjugated rabbit anti-mouse Ig (SBA/ITK) and Texas Red conjugated goat anti-rabbit Ig (1 : 50, SBA/ITK). After final washings in PBS slides were mounted in 90% glycerol, 0.02 m Tris–HCl pH 8.0, 0.8% NaN3 and 2%1,4-di-azobicyclo-(2,2,2)-octane (DABCO; Merck, Darmstadt, Germany) containing 0.5 µg/ml diamidino-2-phenylindole (DAPI, Sigma) or 1 µg/ml PI and 0.1 mg/ml RNase for DNA staining.
Microfilament detection
Cells were cultured on cover slips, fixed and permeabilized as described earlier, and incubated with Texas Red-conjugated Phalloidin (dilution 1 : 50; Molecular Probes, Leiden, The Netherlands). Nuclei were counterstained for 15 min with Syto-13 (1 µm; Molecular Probes), recognizing nucleic acids.
Confocal laser scanning microscopy
Fluorescent samples were imaged using a Bio-Rad MRC600 confocal microscope (Bio-Rad Laboratories Ltd, Hemel Hempstead, UK) equipped with an air-cooled Argon–Krypton mixed gas laser and mounted onto an Axiophote microscope (Zeiss), using oil-immersion objectives (40×, NA=3D1.3 or 63×, NA=3D1.4). The laser-scanning microscope was used in the dual parameter set-up, according to the manufacturer's specifications, using dual wavelength excitation at 488 and 568 nm. Emission spectra were separated by the standard sets of dichroic mirrors and barrier filters. Optical sections were recorded in the Kalman filtering mode using 4–8 scans for each picture. Z-Series were generated by collecting a stack consisting of optical sections using a step size of 0.18 or 0.36 µm in the z-direction.
The Huygens System image restoration software (Scientific Volume Imaging B.V., Hilversum, The Netherlands) was used to improve the effective resolution of some of the confocal images and to reduce background noise. Because of the photon limited character of the data a maximum likelihood estimation algorithm (66) was used.
Mechanical compression studies using a single cell loading device
After 24 or 48 h of transfection, cells were seeded onto glass cover slips and allowed to adhere to the glass surface for 18 h. Cells were seeded at a density low enough to allow compression of single cells. Cellular viability was examined by the addition of 5 µg/ml PI to the culture medium. In order to visualize cellular deformation during the experiments, living cells were labelled with 7.5 µm Cell Tracker Green (CTG, Molecular Probes). This green fluorescent dye (excitation 488 nm, emission 520 nm) shows a diffuse staining in all cellular compartments, with often an increased intensity in the nuclei of cells. In cells, transfected with EGFP-tagged lamins, contours of the complete cells were visualized by addition of 10 µm Cell Tracker Orange (Molecular Probes) to the culture medium.
Next to cultured cells, isolated nuclei were subjected to compression. For this purpose nuclei were extracted from cell cultures using an extraction solution containing 1% acetic acid and 0.01% NP40 (Fluka) in water. After extraction for 5 min at RT, nuclei were washed and transferred to the culture medium.
The set-up used to study the response of cells to indentation forces has been described previously (33). Briefly, this set-up consists of a cell-loading device, a cell culture chamber in which cells can be grown at optimal cell culturing conditions (37°C, 5% CO2 in an humidified atmosphere), an inverted confocal microscope (Zeiss LSM510; Zeiss, Jena, Germany) and computerized electronic control of microscope and compression device, using a 40× 295 NA air objective. The complete set-up is shown in Figure 1. Full characteristics and technical details about the newly developed cell loading device have been published elsewhere (33). Summarizing these data, we can state that the device is capable of controlled compression of a single cell while simultaneously recording the force needed to induce cellular deformation. A combination of three micromanipulators and a piezoelectric system allows a controlled displacement of the glass indentor (diameter 60 µm) in three dimensions with a displacement range of 5 nm–15 mm and a frequency range of 0–25 Hz. The resolution of the force transducer is 10 nN. The device enables very rapid compression and relaxation, as well as slow or repetitive compression of cells.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
The authors wish to thank Dr Brian B. Burke (Department of Anatomy and Cell Biology, University of Florida, Gainesville, FL, USA) and Dr Colin L. Stewart (Laboratory of Cancer and Developmental Biology, NCI-FCRDC, Frederick, MD, USA) for providing the embryonic fibroblast cells from LMNA+/+ and LMNA−/− mice. The authors thank Dr Jean-Claude Courvalin (INSERM, Institut Jacques Monod, Paris) and Dr Christoph Viebahn (Department of Anatomy, University of Bonn) for kindly providing antibodies to lamin B1 and vimentin, respectively. We thank Dr Christopher J. Hutchison (School of Biological and Biomedical Sciences, University of Durham, UK) for helpful suggestions and critical reviewing of the manuscript. The vital imaging studies were financially supported by a grant from the Netherlands Organization for Scientific Research (NWO Grant 901-28-134).
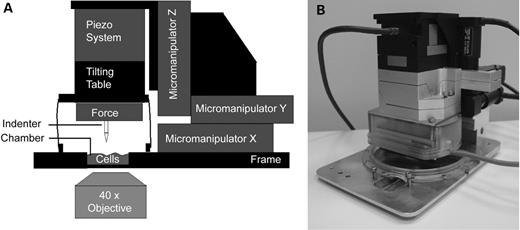
Figure 1. Schematic representation (A) and picture (B) of the cellular compression device. Cells grown on cover slips are placed in the incubation chamber, and individual cells are tracked by confocal imaging using a 40× air objective. Next, the indentor is placed on top of a single cell, and the cell is compressed.
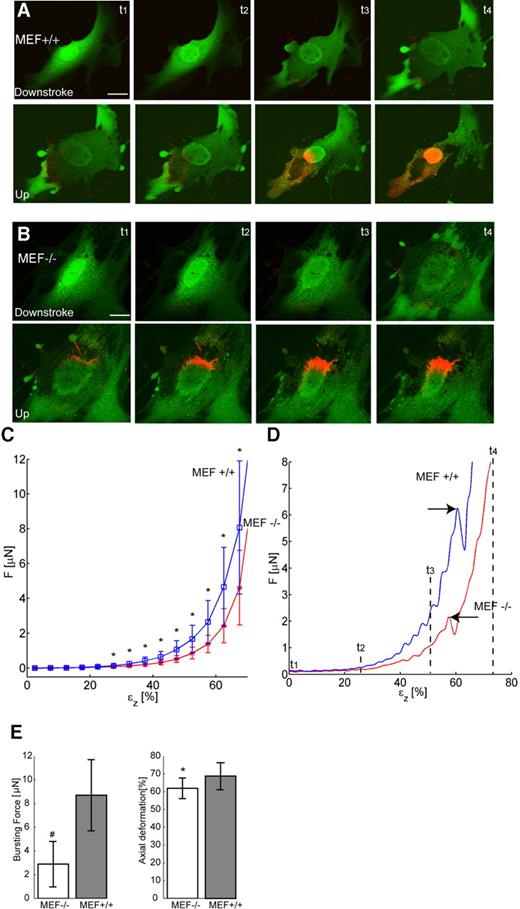
Figure 2. Confocal image and force recordings of cellular deformation during the compression routine. (A) Confocal images of a characteristic MEF+/+ cell, stained with Cell Tracker green and propidium iodide (PI) during compression and subsequent relaxation. The influx of PI into the cell starts immediately after cellular rupture and rapidly concentrates at the nuclear DNA. (B) Idem for a MEF−/− cell. Note the presence of large strands of DNA protruding from the damaged nucleus into the cytoplasm after compression. (C) Plot of compression force versus axial deformation of MEF+/+ and MEF−/− cells. Asterisks indicate the statistically significant differences between both cell types. Average of n=36 (MEF+/+) and n=31 (MEF−/−). (D) Force plots of MEF+/+ cell from (A) and of MEF−/− cell from (B), showing the difference in bursting force for both cell types (arrows). t1–t4 correspond to time points t1–t4 in (A) and (B). (E) Bursting force plots showing average force needed to induce cellular bursting and corresponding axial deformation at which cellular bursting took place. Statistically significant differences were noted between MEF+/+ and MEF−/− cells, both for force needed (# denotes P<0.001, Student's t-test) and axial deformation (* denotes P<0.05, Student's t-test). Bar represents 20 µm in all figures.
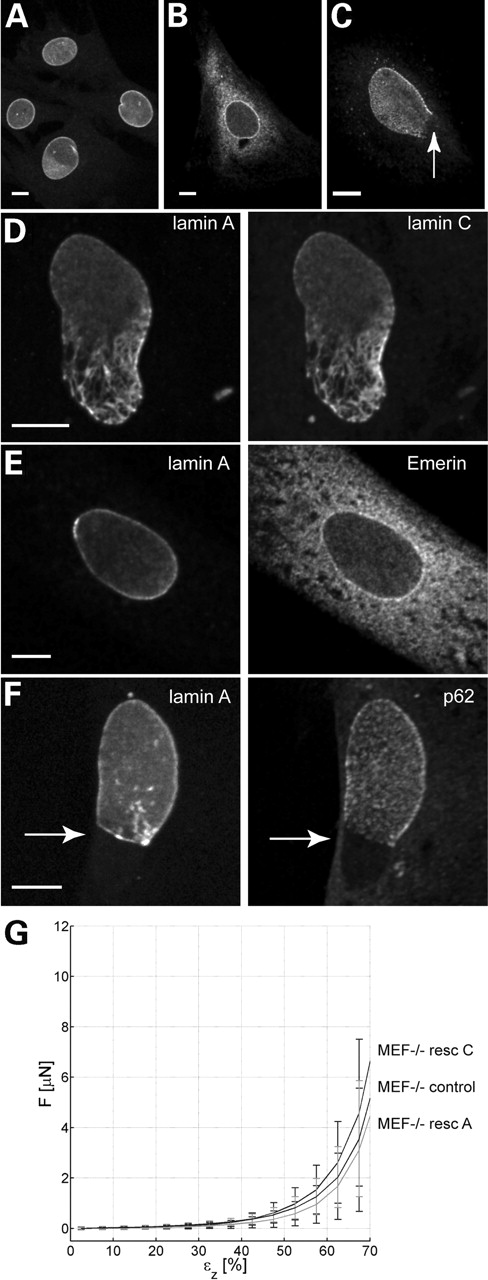
Figure 3. Rescue experiment of MEF−/− cells using lamin A–GFP and lamin C cDNA in pcDNA3. (A and B) Immunofluorescence patterns of untransfected MEF+/+ (A) and MEF−/− cells (B) showing the pronounced difference in emerin localization. Note that a MEF−/− cell shows mainly endoplasmic reticulum staining, next to some fluorescent signal at the nuclear membrane (B), whereas in the MEF+/+ cell, emerin is exclusively localized at the nuclear membrane (A). (C) p62 is unevenly distributed in a small percentage (3–5%) of untransfected MEF−/− cells, being absent at the lower part of the nucleus (arrow). (D) Fluorescence of a MEF−/− cell co-transfected with lamin A–GFP (left) and lamin C cDNA (right). Although both lamins are present in the nucleus, the nuclear shape of this cell still shows abnormalities characteristic of MEF−/− cells, even 6 days after transfection. (E) lamin A–GFP fluorescence (left) and emerin immunostaining (right) showing that in the lamin A rescued cell, emerin is not relocalized to the nuclear membrane but is still distributed throughout the endoplasmic reticulum. (F) lamin A–GFP fluorescence (left) and p62 immunostaining (right). In this cell, transfected with lamin A–GFP (left), nuclear pore complexes visualized by the p62 antibody (right) are still confined to the lamin-containing part of the nuclear membrane, and absent from areas devoid of lamin A (arrow). (G) Force versus axial deformation curves of MEF−/− cells and cells rescued with either lamin A–GFP or lamin C-GFP. No significant differences between different MEF−/− cells and rescued cells are seen. Bar represents 10 µm in all figures.
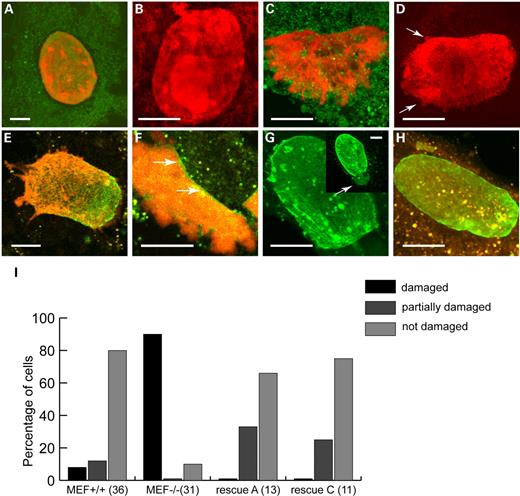
Figure 4. Nuclear damage during compression, visualized with Cell Tracker Green combined with PI (A–D) or with Cell Tracker Orange combined with PI (E–H). In all samples, the prominent fluorescent signal in the central region of the cell results from PI fluorescence. Note that especially Cell Tracker Orange forms cytoplasmic aggregates in these cells. (A and B) Absence of visual nuclear damage in two MEF+/+ cells after compression. The nuclear shape of these nuclei remains intact, with chromatin still located in a sharply delineated nuclear compartment. (C and D) Damaged nuclei of MEF−/− cells that did burst during compression, with chromatin protruding into the cytoplasm as large (C) or small irregular lobules (arrows in D). (E–H) Depending on the amount of transfected lamins at the nuclear membrane, rescued nuclei transfected with lamin A(–EGFP) showed no disruption, or partial disruption at areas with so-called honeycomb lamin patterns (E), and less disruption in areas with high levels of lamin A (F, arrow). Similar observations were made for lamin C(–EGFP) rescued cells with preservation of nuclear shape in cells, with high levels of lamin C (H) and disruption of areas lacking lamin C (G, arrow). Inset in (G) shows lamin C fluorescence before compression and the predicted weak spot in the nucleus, which indeed did burst during compression (Supplementary Material, Movie S3). (I) Quantitative comparison of visual nuclear damage between MEF+/+ and MEF−/− cells and A-type lamin-rescued cells. The number of analysed cells for each category is shown in parentheses. Undamaged nuclei: all DNA remains in nucleus after compression. Partial damage: one or more areas of the nucleus show protrusions of DNA into the cytoplasm, whereas the remaining nucleus remains intact. Damage: DNA protrudes into the cytoplasm into all directions. Note that nuclear damage as seen in MEF−/− is largely prevented after rescue with either lamin A or lamin C. Bar represents 10 µm in all figures.
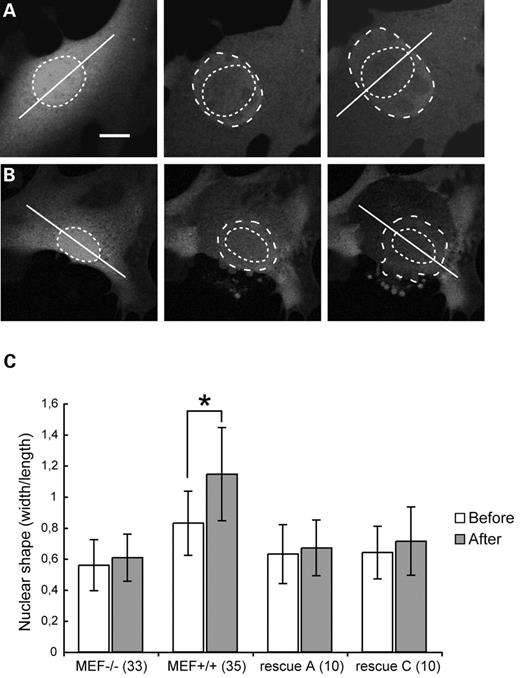
Figure 5. Determination of direction of deformation of nuclei during compression. (A) MEF+/+ cell showing anisotropic deformation (hatched ovals) compared to the long axis of the cell (straight line). (B) MEF−/− cell showing isotropic deformation during compression, i.e. an even expansion in lateral directions. (C) Comparison of the type of nuclear deformation in MEF+/+ cells, MEF−/− cells and A-type lamin-rescued cells. The number of analysed cells is shown in parentheses. The degree of (an-)isotropic nuclear deformation during compression is expressed in width to length ratio in cells with a well-defined growth direction. On the basis of this length axis, the width to length ratio before compression and at the moment of maximal expansion are compared. This resulted in an average increase of 40% of the width compared with the length in MEF+/+ cells, and 8–11% in other cell types. Asterisk indicates significant differences in ratio before and after full indentation (P<0.05, Student's t-test). Before, before compression; After, after full indentation, i.e. at maximum compression. Bar represents 10 µm in (A) and (B).
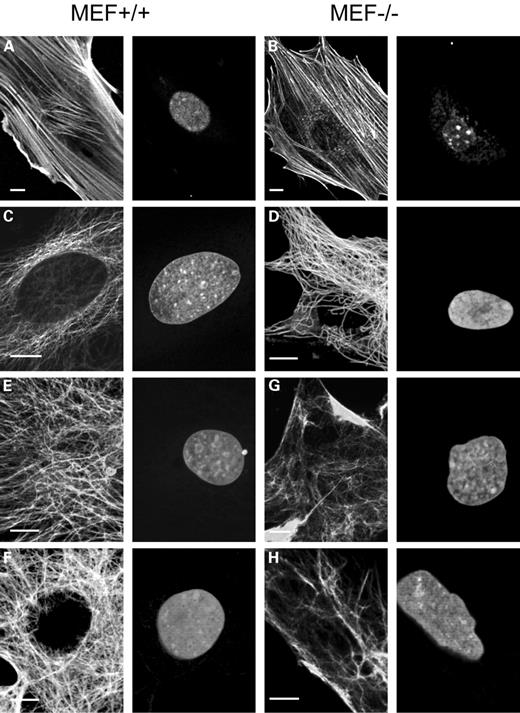
Figure 6. (A and B) Actin staining in MEF+/+ (A) and MEF−/− (B) cells with Phalloidin-Texas Red (left) and counterstained with Syto-13 (right). In both the cases, a single slice from a confocal z-series is shown. Note the apparently truncated actin filaments and granular actin staining patterns in the perinuclear region of a MEF−/− cell, whereas intact actin fibres are seen in the MEF+/+ cell. (C and D) Microtubular organization in a MEF+/+ (C) and a MEF−/− cell (D). In both the cases, a single slice from a confocal z-series is shown. Microtubule staining with the E7 antibody shows a close association of microtubule endings with the nucleus in the MEF+/+ cell (C, left panel), whereas the perinuclear area of the MEF−/− cell lacks microtubules in part of the perinuclear area (D, left panel). Nuclei are counterstained with PI (C and D, right panels). (E–H) Vimentin organization of MEF+/+ (E and F) and MEF−/− cells (G and H). Vimentin immunofluorescence with antibody BV118 shows disturbed vimentin organization in MEF−/− cells (G and H, left panels), when compared with vimentin labelling in MEF+/+ cells (E and F, left panels). Note the absence of vimentin in large areas at the nuclear periphery and the patched staining in other areas. In (E) and (G) projections of confocal z-series are shown, stressing the dramatic differences in vimentin distribution between MEF+/+ and MEF−/− cells, whereas in (F) and (H) a single slice from a confocal z-series is shown. Bar represents 10 µm in all figures.
References
Lin, F. and Worman, H.J. (
Biamonti, G., Giacca, M., Perini, G., Contreas, G., Zentilin, L., Weighardt, F., Guerra, M., Della Valle, G., Saccone, S., Riva, S. et al. (
Furukawa, K. and Hotta, Y. (
Lin, F. and Worman, H.J. (
Machiels, B.M., Zorenc, A.H.G., Endert, J.M., Kuijpers, H.J.H., van Eys, G.J.J.M., Ramaekers, F.C.S. and Broers, J.L.V. (
Furukawa, K., Inagaki, H. and Hotta, Y. (
Röber, R.-A., Sauter, H., Weber, K. and Osborn, M. (
Röber, R.A., Weber, K. and Osborn, M. (
Broers, J.L.V., Machiels, B.M., Kuijpers, H.J.H., Smedts, F., van den Kieboom, R., Raymond, Y. and Ramaekers, F.C.S. (
Harborth, J., Elbashir, S.M., Bechert, K., Tuschl, T. and Weber, K. (
Cohen, M., Tzur, Y.B., Neufeld, E., Feinstein, N., Delannoy, M.R., Wilson, K.L. and Gruenbaum, Y. (
Moir, R.D., Spann, T.P., Herrmann, H. and Goldman, R.D. (
Sullivan, T., Escalante-Alcalde, D., Bhatt, H., Anver, M., Bhat, N., Nagashima, K., Stewart, C.L. and Burke, B. (
Muchir, A., van Engelen, B.G., Lammens, M., Mislow, J.M., McNally, E., Schwartz, K. and Bonne, G. (
Bonne, G., Di Barletta, M.R., Varnous, S., Becane, H.M., Hammouda, E.H., Merlini, L., Muntoni, F., Greenberg, C.R., Gary, F., Urtizberea, J.A. et al. (
Fatkin, D., MacRae, C., Sasaki, T., Wolff, M.R., Porcu, M., Frenneaux, M., Atherton, J., Vidaillet, H.J., Jr, Spudich, S., De Girolami, U. et al. (
Muchir, A., Bonne, G., van der Kool, A.J., van Meegen, M., Baas, F., Bolhuis, P.A., de Visser, M. and Schwartz, K. (
Shackleton, S., Lloyd, D.J., Jackson, S.N., Evans, R., Niermeijer, M.F., Singh, B.M., Schmidt, H., Brabant, G., Kumar, S., Durrington, P.N. et al. (
Cao, H. and Hegele, R.A. (
De Sandre-Giovannoli, A., Chaouch, M., Kozlov, S., Vallat, J.M., Tazir, M., Kassouri, N., Szepetowski, P., Hammadouche, T., Vandenberghe, A., Stewart, C.L. et al. (
Novelli, G., Muchir, A., Sangiuolo, F., Helbling-Leclerc, A., D'Apice, M.R., Massart, C., Capon, F., Sbraccia, P., Federici, M., Lauro, R. et al. (
Cao, H. and Hegele, R.A. (
De Sandre-Giovannoli, A., Bernard, R., Cau, P., Navarro, C., Amiel, J., Boccaccio, I., Lyonnet, S., Stewart, C.L., Munnich, A., Le Merrer, M. et al. (
Eriksson, M., Brown, W.T., Gordon, L.B., Glynn, M.W., Singer, J., Scott, L., Erdos, M.R., Robbins, C.M., Moses, T.Y., Berglund, P. et al. (
Chen, L., Lee, L., Kudlow, B.A., Dos Santos, H.G., Sletvold, O., Shafeghati, Y., Botha, E.G., Garg, A., Hanson, N.B., Martin, G.M. et al. (
Mancini, M.A., Shan, B., Nickerson, J.A., Penman, S. and Lee, W.-H. (
Ozaki, T., Saijo, M., Murakami, K., Enomoto, H., Taya, Y. and Sakiyama, S. (
Markiewicz, E., Dechat, T., Foisner, R., Quinlan, R.A. and Hutchison, C.J. (
Lloyd, D.J., Trembath, R.C. and Shackleton, S. (
Dreuillet, C., Tillit, J., Kress, M. and Ernoult-Lange, M. (
Guilak, F., Tedrow, J.R. and Burgkart, R. (
Caille, N., Thoumine, O., Tardy, Y. and Meister, J.J. (
Peeters, E.A., Bouten, C.V., Oomens, C.W. and Baaijens, F.P. (
Bridger, J.M. and Kill, I.R. (
Favreau, C., Higuet, D., Courvalin, J.C. and Buendia, B. (
Nikolova, V., Leimena, C., McMahon, A.C., Tan, J.C., Chandar, S., Jogia, D., Kesteven, S.H., Michalicek, J., Otway, R., Verheyen, F. et al. (
Lehner, C.F., Kurer, V., Eppenberger, H.M. and Nigg, E.A. (
Vigouroux, C., Auclair, M., Dubosclard, E., Pouchelet, M., Capeau, J., Courvalin, J.C. and Buendia, B. (
Lammerding, J., Schulze, P.C., Takahashi, T., Kozlov, S., Sullivan, T., Kamm, R.D., Stewart, C.L. and Lee, R.T. (
Holt, I., Ostlund, C., Stewart, C.L., Man, N., Worman, H.J. and Morris, G.E. (
Raharjo, W.H., Enarson, P., Sullivan, T., Stewart, C.L. and Burke, B. (
Ingber, D.E. (
Ingber, D.E. (
Zhang, Q., Ragnauth, C., Greener, M.J., Shanahan, C.M. and Roberts, R.G. (
Mislow, J.M., Holaska, J.M., Kim, M.S., Lee, K.K., Segura-Totten, M., Wilson, K.L. and McNally, E.M. (
Zhen, Y.Y., Libotte, T., Munck, M., Noegel, A.A. and Korenbaum, E. (
Zhang, Q., Skepper, J.N., Yang, F., Davies, J.D., Hegyi, L., Roberts, R.G., Weissberg, P.L., Ellis, J.A. and Shanahan, C.M. (
Janmey, P.A. (
Sato, M., Theret, D.P., Wheeler, L.T., Ohshima, N. and Nerem, R.M. (
Guilak, F. (
Lattanzi, G., Cenni, V., Marmiroli, S., Capanni, C., Mattioli, E., Merlini, L., Squarzoni, S. and Maraldi, N.M. (
Schroder, R., Reimann, J., Salmikangas, P., Clemen, C.S., Hayashi, Y.K., Nonaka, I., Arahata, K. and Carpen, O. (
Feng, J., Yan, J., Buzin, C.H., Towbin, J.A. and Sommer, S.S. (
North, K.N. and Beggs, A.H. (
Beaudouin, J., Gerlich, D., Daigle, N., Eils, R. and Ellenberg, J. (
Patterson, K., Molofsky, A.B., Robinson, C., Acosta, S., Cater, C. and Fischer, J.A. (
Tolstonog, G.V., Sabasch, M. and Traub, P. (
Perez-Olle, R., Leung, C.L. and Liem, R.K. (
Broers, J.L.V., Machiels, B.M., van Eys, G.J.J., Kuijpers, H.J.H., Manders, E.M.M., van Driel, R. and Ramaekers, F.C.S. (
Hozak, P., Sasseville, A.M., Raymond, Y. and Cook, P.R. (
Machiels, B.M., Broers, J.L.V., Raymond, Y., de Ley, L., Kuijpers, H.J.H., Caberg, N.E. and Ramaekers, F.C.S. (
Venables, R.S., McLean, S., Luny, D., Moteleb, E., Morley, S., Quinlan, R.A., Lane, E.B. and Hutchison, C.J. (
Carmo-Fonseca, M., Kern, H. and Hurt, E.C. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}