




Abstract
Farnesoid X receptor (FXR) is a transcription factor that controls bile acid homeostasis. The phenotype of Fxr null mice is characterized by hypercholanaemia, impaired secretion of bile acids and failure to thrive. Human disorders with these characteristics include FIC1 disease (caused by mutations in ATP8B1, which encodes a putative aminophospholipid translocase, FIC1, whose function in bile handling is unknown) and bile salt export pump (BSEP) disease (caused by mutation in ABCB11, which encodes BSEP, the primary canalicular bile salt export pump). We investigated the possibility of hepatic down-regulation of FXR in FIC1 disease and BSEP disease. Three siblings with this phenotype, born to consanguine parents, were initially studied. The children were demonstrated to be compound heterozygotes for missense and nonsense mutations in ATP8B1. Expression of specific genes in liver was analysed, comparing one of these siblings with a child homozygous for missense mutation in ABCB11, as well as with a child having idiopathic cholestatic liver disease, a child with extrahepatic biliary atresia and a normal organ donor. The expression of two main FXR isoforms was specifically decreased in the liver of the FIC1 disease patient. A consistent and concomitant reduction in messenger RNA levels of FXR targets, such as BSEP and small heterodimer partner, was also found. Gene-profiling experiments identified 163 transcripts whose expression changed significantly in FIC1-disease liver. Of note was that several genes involved in synthesis, conjugation and transport of bile acids were down-regulated. A cluster of genes involved in lipid metabolism was also differentially expressed. Our findings suggest that hepatic down-regulation of FXR contributes to the severe cholestasis of FIC1 disease.
INTRODUCTION
Farnesoid X receptor (FXR, NR1H4) is a nuclear receptor that functions as a physiological sensor of bile acids (1,2). Upon binding to bile acids or their conjugates, it coordinately regulates the expression of critical target genes involved in the synthesis and transport of bile acids. Thus, in the liver, FXR induces the expression of the canalicular adenosine triphosphate-binding cassette (ABC) transporters ABCB11, multidrug resistance transport protein 3 (MDR3, ABCB4) and multidrug resistance-related protein 2 (MRP2, cMOAT; ABCC2), whilst repressing that of basolateral sodium taurocholate cotransporting polypeptide (NTCP; SLC10A1) (3–7). In the intestine, FXR up-regulates expression of ileal bile acid binding protein (I-BABP) (8) and down-regulates that of apical sodium-dependent bile acid transporter (ASBT; SLC10A2) (9,10). In addition, FXR regulates the expression of genes encoding enzymes involved in bile acid synthesis and conjugation (11–13), as well as key target genes controlling cholesterol homeostasis (14,15). In vivo evidence of the central role of Fxr in bile acid and lipid homeostasis has come from Nr1h4 knockout mice, which have increased levels of bile salts, cholesterol and triglycerides in serum (14).
FXR expression is decreased in intestine of patients with FIC1 disease (10), a form of inherited cholestasis caused by mutation in ATP8B1 (16,17). FIC1 disease is clinically similar in some respects to bile salt export pump (BSEP) disease, another form of inherited cholestasis. Both FIC1 disease and BSEP disease, when severe, are manifest in infancy. They are characterized by low serum concentrations of gamma-glutamyl transpeptidase (GGT) activity and cholesterol despite conjugated hyperbilirubinaemia and by decreased concentrations of bile salts in bile (18,19). BSEP disease is caused by mutation in ABCB11, which is expressed only in the hepatocyte and encodes BSEP, the primary canalicular bile salt export pump (20). Hepatocellular retention of bile salts owing to absence or dysfunction of BSEP can be invoked to explain liver injury and cholestasis in BSEP disease. FIC1 is a P-type ATPase expressed at many body sites, with putative activity as an aminophospholipid translocase (16,21,22). Its precise function and how its dysfunction leads to cholestasis are not understood. To understand better the association of FIC1 disease with cholestasis, we investigated expression of FXR and FXR target genes in liver of a patient with genetically documented FIC1 disease and of a patient with genetically documented BSEP disease, and compared our findings with those in other forms of liver disease and in normal liver. Our results support a significant role for FXR in the pathogenesis of FIC1 disease.
RESULTS
Three of five gypsy children (patients 1–3, Table 1), born to first cousins, had cholestatic liver disease with hypercholanaemia, low serum GGT and low serum cholesterol. They were treated by liver transplantation. All three suffered from chronic diarrhoea after transplantation, with failure of ‘catch-up’ growth. These post-transplant complications are associated with FIC1 disease (23,24). Genomic screening suggested linkage of the disorder to the ATP8B1 locus, albeit without homozygosity. Single-strand conformation polymorphism (SSCP) and direct sequencing revealed that each affected child was a compound heterozygote for the same missense and nonsense mutations in ATP8B1. The paternal allele shows a C→T transition at nucleotide 1367 that results in a substitution of a threonine for methionine at codon 456 (Fig. 1). This change was not found in 94 chromosomes from a control population. A C→T transition of nucleotide 1804 is transmitted from the mother and results in a substitution of an arginine for a stop codon, leading to a truncated protein at amino acid 602.
We examined mRNA expression of ATP8B1, as well as ABCB11 and ABCB4 [encoding a canalicular transporter, MDR3, deficient in a form of high-GGT cholestatic liver disease (25)], in liver from patient 1 and from four other age-matched children. One (patient 5, Table 1) had cholestatic liver disease with hypercholanaemia, low serum GGT and low serum cholesterol. This patient was homozygous for a missense mutation (3692G>A, R1231Q) in ABCB11 and lacked immunohistochemically demonstrable BSEP at canaliculi; MRP2 marked normally (Fig. 2). Another (patient 4) had high-GGT cholestatic liver disease of undetermined aetiology; in particular, no mutation was found in the entire coding sequence of ABCB4. A third had extrahepatic biliary atresia (EHBA). A fourth was normal; donor liver excess to need at transplantation was used. As shown in Figure 3A, no consistent differences in ATP8B1 or ABCB4 mRNA expression, as determined by real-time quantitative RT–PCR, were observed but, unexpectedly, a 4-fold reduction in the expression of ABCB11 was detected only in the patient with FIC1 disease. In this line, in liver sections of this patient, immunohistochemical staining for BSEP was only evident at an antibody dilution of 1 : 20 (Fig. 2), whereas in simultaneous assays with sections from patient 4 and normal liver, protein could be also detected at a 1 : 50 antibody dilution (data not shown). As ABCB11 expression strictly depends on FXR (3,4), we next examined mRNA levels of this transcription factor as well as the liver-X receptor (LXR), a structurally and functionally closely related nuclear receptor (26). A marked decrease in FXR mRNA levels was observed specifically in patient 1 (Fig. 3B). The expression of small heterodimer partner (SHP, NR0B2), a primary FXR target gene (11,12) was also evaluated. Consistent with the above results, SHP mRNA levels were found to be decreased only in FIC1 disease patient (Fig. 3C). The expression of NTCP, which is inhibited by SHP (7), did not change in patient 1 when compared with samples from normal liver or from patients 4 and 5 (Fig. 2). However, a 2.5-fold decrease in its mRNA levels was observed in biliary atresia, which is consistent with a previous finding indicating a down-regulation in the expression of this gene in patients with this disorder (27). Altogether, results from patient 1 are in agreement with those reported for Fxr null mice, which exhibit a marked decrease in ABCB11 and SHP expression, but show no differences in NTCP and ABCB4 (14). Moreover, these findings suggest that, unlike ABCB11 and SHP, FXR transcriptional activity is not required for constitutive expression of NTCP and ABCB4.
The occurrence of two main FXR isoforms (FXRα and FXRβ) has been reported in mice and human (28,29). They are spliced variants that differ at the N-terminus and are expressed in a developmental and tissue-specific pattern. Because both isoforms may differentially regulate target genes in various tissues (28,29), we decided to determine the precise contribution of each one to the overall decreased FXR expression detected in patient 1. Initially, to unequivocally discriminate the two variants, RT–PCR assays were carried out by using different primer sets. Two of them corresponded to sequences at the divergent N-termini and 5′ untranslated regions (5′-UTR) (Fig. 4A), and have been previously used for the specific detection of each of the two human FXR isoforms (28). In the remaining ones, which were predicted to lead to amplified products markedly different in length, the same sequence corresponding to the common coding region was used as a reverse primer (Fig. 4A). RT–PCR assays resulted in amplified fragments of the expected sizes for each FXR variant and indicated that both isoforms decreased to a similar extent specifically in patient 1 (Fig. 4B). The relative expression of each one was then assessed by real-time PCR (Fig. 4C). In all the samples, FXRβ expression was higher than FXRα, showing a ratio of 1.9, and an ∼4-fold decrease in the expression of both isoforms was detected in patient 1.
To this end, we investigated whether liver disease was also linked to FXR in the three affected children in the gypsy sibship. This possibility was excluded by both genotyping of six members of this family (patients 1–3, their parents and a healthy brother) with five polymorphic markers flanking FXR and direct sequencing of the coding region of FXR in patient 1 (data not shown).
Having established the specific decrease in FXR expression in patient 1, we hypothesized that phenotype of FIC1 disease might in part result from reduced transcriptional activity of this nuclear receptor. To assess this, gene-profiling experiments were carried out with the five liver samples. Expression data from patient 4, patient 5, the child with EHBA and the normal child were compared with those from patient 1, resulting in four pair-wise comparisons. A total of 163 genes, out of approximately 8300, whose expression changed at least 2-fold specifically in patient 1 were identified. These genes were then subjected to hierarchical clustering (30) for visualization. This analysis divided the samples into distinct groups and, as expected, clustered data from patient 1 in a separate branch of the dendrogram (Fig. 5, left panel). Various sets of genes involved in bile acid and lipid metabolism, as well as transporters and detoxifying enzymes were, among others, found to be differentially expressed in patient 1 (Fig. 5, right panel). Notably, genes encoding enzymes involved in bile acid synthesis, as well as hepatic basolateral and canalicular transporters were found to be down-regulated in this patient. They included CYP27A1, which catalyses the initial step of an alternative biosynthetic pathway (31), bile acid-CoA–amino acid N-acyltransferase (BAAT), which is involved in conjugation of bile acids to taurine and glycine (31), the canalicular transporters ABCC2 (MRP2), which mediates export of organic anions including conjugated bilirubin and ABCG2, an alternative canalicular efflux pump for sulphated conjugates, as well as OATP-C, the major basolateral Na+-independent bile salt uptake system (32). In addition, the expression of biliverdin reductase A, which catalyses the recycling of biliverdin to bilirubin (33), was also decreased. A cluster of genes involved in various aspects of lipid metabolism was also found to be down-regulated in patient 1. Among them are key modulators and enzymes of cholesterol synthesis, such as 3-hydroxy-3-methylglutaryl-Coenzyme A reductase (HMG-CoA) reductase, intracellular trafficking, such as sterol carrier protein 2, and removal of its cellular excess, such as vigilin (34). Noteworthy, most of these genes, as well as hepatic lipase, microsomal triglyceride transfer protein, stearoyl-CoA desaturase, phenylalanine hydroxylase and delta 4-3-ketosteroid 5 beta reductase, among others, have been reported to be down-regulated in Fxr null mice (14,15,35), and/or up-regulated in culture systems upon treatment with FXR agonists (13,36–38).
DISCUSSION
Mutations in ATP8B1 have been identified in individuals with low-GGT cholestatic liver disease (17,39). Here we described two mutations in ATP8B1 present in compound heterozygous form in three children from a gypsy family. One mutation is predicted to lead to truncation of synthesis at amino acid 602 (of 1251), with loss of six (of 10) transmembrane domains in FIC1, the P-type ATPase encoded by ATP8B1. The other mutation, T456M, lies in the phosphorylation domain, a DKTG motif highly conserved among all P-type ATPases (40). Both mutations are likely to abolish protein function.
In liver from patient 1, a child with low-GGT cholestatic liver disease who carried both these mutations, predicted absence of FIC1 activity was correlated with down-regulation of FXR. This nuclear receptor is highly expressed in the enterohepatic system, where it acts as a bile acid sensor that maintains a regular bile salt pool size and protects the hepatocyte from excessive intracellular bile acid concentration. This is largely achieved through the regulation of the expression of specific transporters that drive enterohepatic circulation and key enzymes involved in bile acid biosynthesis (41,42). Specific down-regulation of various genes in this group was observed, concomitant with decreased FXR expression, in liver with FIC1 disease. This may be associated with abnormalities that characterize the disease; in particular, impaired canalicular secretion of bile salts, organic anions and other bile constituents could result from reduced ABCB11, ABCC2 and ABCG2 expression. An imbalance in biosynthesis of bile salts species might follow from down-regulation of CYP27A1, as this enzyme catalyses the first step in a pathway producing mainly chenodeoxycholic acid (31). However, the physiological significance of this finding is unclear, because a marked reduction of the secretion of chenodeoxycholic relative to cholic acid conjugates has been found not only in children with FIC1 disease (43,44), but also in BSEP disease patients (45,46). Regarding the basolateral influx of bile salts, the apparent normal expression of NTCP, the main transport system for the uptake of conjugated bile salts, does not seem to correlate with the elevated serum levels of bile acids. This holds for patients 4 and 5 as well. However, because NTCP is also subjected to posttranscriptional regulation (32), a reduced functional activity of this transporter cannot be excluded. On the other hand, in patient 1, diminished uptake of unconjugated species of bile acids, as well as unconjugated bilirubin and other compounds, could be predicted from the detected down-regulation of the basolateral transporter OATP-C.
A number of genes differentially expressed in liver from patient 1, specially those involved in lipid metabolism, are similarly altered in Nr1h4 knockout mice (14,15). These animals exhibit a phenotype of cholestatic liver disease, with hypercholanaemia, impaired canalicular bile salt secretion and failure to thrive (14). The partial overlap of phenotypes between Fxr knockout mice and children with severe FIC1 disease further supports involvement of FXR down-regulation in the pathogenesis of FIC1 disease. Decreased expression of FXR has been reported in the small intestine of children with FIC1 disease, and the link between FIC1 deficiency and FXR down-regulation has been demonstrated in Caco-2 cells using specific antisense RNA oligonucleotides (10). Intriguingly, no changes in Fxr gene expression have been detected, either in liver or in intestine, in Atp8b1 mutant mice expressing a non-functional protein (47). Moreover, unlike patients with FIC1 disease, these mice neither have impaired bile secretion nor develop cholestatic disease (47). This contrast in phenotypes appears related to the ability of mice to rehydroxylate secondary bile acids (47). Hydrophobic secondary bile acids, such as lithocholic or deoxycholic acids, are formed in the intestine by bacterial dehydroxylation of primary bile acids. An abnormal accumulation of lithocholic acid was documented in an Amish patient meeting clinical, histopathologic and ultrastructural criteria for FIC1 disease (48). Bearing in mind the putative function of FIC1 as an aminophospholipid flippase, a role for this protein accordingly has been proposed in the elimination of secondary bile acids or other hydrophobic substances from enterohepatic circulation (49). As lithocholic acid is a potent FXR antagonist (50), it is tempting to speculate that in humans the accumulation of secondary bile acids due to FIC1 dysfunction would lead to reduced FXR activity. This in turn would alter expression of FXR targets and of FXR itself (10,28). The ability of mice to rehydroxylate secondary bile acids would prevent Fxr from inactivation, despite the absence of functional Fic1. An alternative explanation for differences between FXR and Nr1h4 expression in humans with FIC1 disease and Atp8b1 mutant mice might invoke direct molecular links, present in humans but not in mice, between FIC1 dysfunction and FXR down-regulation, with FIC1 influencing signal transduction pathways involved in the regulation of expression of FXR (10).
Our findings, like those of Chen et al. (10), suggest that in FIC1 disease FXR down-regulation leads to defects in bile salt transport at both hepatic and intestinal levels. This likely accounts for complications that persist or arise in children with FIC1 disease after successful liver transplantation (24,51). Exacerbation of diarrhoea and no catch-up in growth were observed in the three siblings whom we studied. These complications can be ascribed to persistent alterations in ileal bile acid transport owing to FXR down-regulation, with enhanced presentation of bile salts to the ileum after liver transplant leading to exacerbation of diarrhoea. That FXR and ATP8B1 are co-expressed in many other tissues (16,22,28,52) raises the possibility that FXR down-regulation is involved in systemic manifestations of ATP8B1 disease.
The implications of these findings for possible therapy deserve further consideration. Specific FXR agonists have been developed, and have been assayed in both culture systems and animal models (12,36,53,54). Treatment with a selective non-steroidal FXR agonist has been reported to be hepatoprotective in two rat models of cholestatic liver disease (38). Perhaps, the residual pool of functional FXR in children with ATP8B1 disease can be further activated by these compounds. This might offer an additional pharmacological approach to management of this disease.
MATERIALS AND METHODS
Patients
Clinical and clinical-biochemistry findings are summarized in Table 1. All patients received ursodeoxycholic acid treatment, without resolution of cholestasis. All allografts placed were cadaveric whole livers.
Patient 1, a male, is the fifth of five children born to first-cousin gypsy (ROMÁ) parents. Two brothers (patients 2 and 3, Table 1) had chronic cholestasic liver disease; a brother and a sister are healthy. Liver disease in all three boys was manifest as neonatal jaundice with pruritus in early infancy. All were growth-impaired with hepatic enlargement but without signs of portal hypertension. Serum aminotransferase, cholesterol and GGT values were near-normal; serum bile acids and bilirubin values were elevated. Liver transplantation was undertaken at ages 3, 5 and 8 years, respectively, to treat severe cholestasis and pruritus. Severe cholestasis and fibrosis, without steatosis, were found in each explant. Catch-up growth has not ensued; although quality of life has improved, all are affected by persistent idiopathic diarrhoea. Serum bilirubin values are normal; aminotransferase activity is slightly increased, with fluctuating hypocholesterolaemia and depressed prothrombin activity in two of the three.
Patient 4 was jaundiced as a neonate, with elevated serum GGT values. His parents denied consanguinity. Severe fibrosis and proliferated bile ducts were seen in a needle biopsy specimen. No aetiology was identified. Signs of severe liver disease developed, with gastrointestinal bleeding ascribed to portal hypertension and with growth delay. Transient pruritus was observed at the age of 2 years. Liver transplantation was undertaken at the age of 33 months. The explant was cirrhotic, with mildly proliferated ducts; the extrahepatic biliary tract appeared normal. Catch-up growth occurred. Patient 5 came to medical attention at the age of 5 months, with mild jaundice, elevated serum aminotransferase and bile acid values and normal GGT values. Small bile ducts, cholestasis and mild fibrosis were seen in a needle biopsy specimen. Pruritus developed at 11 months. Gallbladder stones were seen at 19 months. Hepatic synthetic function declined from 2 years onward. Liver transplantation was undertaken at 35 months. Cirrhotic transformation was seen in the explant; the stones were of mixed type, containing cholesterol and pigment. Catch-up growth occurred.
Liver samples obtained from patients 1, 4 and 5 at transplantation were immediately submerged in RNAlater solution (Ambion Inc., Austin, TX, USA), as was liver obtained at transplantation from a 3-year-old child with EHBA and, at donor-organ harvesting, from another 3-year-old child without liver disease. Informed consent and ethics committee approval were obtained before tissue collection.
Genetic analysis
Genomic DNA was isolated from peripheral blood leukocytes using the PureGene® DNA Isolation kit (Gentra Systems, Minneapolis, MN, USA). Linkage analysis was performed in patients 1, 2 and 3, their parents and one unaffected brother, with polymorphic markers surrounding ATP8B1 gene (D18S1152, D18S1117, D18S1144, D18S1129, D18S1401, D18S977 and D18S381) and FXR gene (D12S332, D12S1588, D12S306, D12S318 and D12S78). The 27 exons of the ATP8B1 gene and intron boundaries regions were amplified in all family members with primers specifically designed (primer sequences and PCR conditions available upon request). Fragments were electrophoresed through the SSCP technique. Abnormal conformers were sequenced on both strands using the Big Dye® Terminator RRMix (Applied Biosystems Inc., Foster City, CA, USA) and an ABI PRISM® 377 or 3100 sequencer (Applied Biosystems Inc.). A control group of 47 unrelated subjects (94 alleles) was used to exclude DNA polymorphisms. DNA sequences were analysed by both direct observation of the electrophoretographs and comparison using the sequence alignment of the DNA ClustalW® program with the available sequence of the gene (http://www.ncbi.nlm.nih.gov). For sequencing the entire coding regions of ATP8B1, ABCB11 and ABCB4, cDNAs obtained from liver samples of patients 4 and 5, as well as from normal tissue, were subjected to PCR, using Pfu DNA polymerase (Biotools, Madrid, Spain) and several sets of internal primers corresponding to the following nucleotides (numbered according to the published cDNA sequences). ATP8B1 (16): 1–22, 1091–1112; 681–701, 1681–1702; 1549–1569, 2710–2731; 2584–2607, 3733–3756. ABCB11 (20): 43–62, 1047–1066; 761–780, 1800–1819; 1572–1591, 2659–2678; 2401–2420, 3431–3450; 3202–3221, 4111–4131. ABCB4 (55): 3–22, 664–683; 542–561, 1641–1660; 1445–1464, 2513–2532; 2371–2390, 3399–3418; 3255–3274, 3849–3868. The coding sequences for FXRα and FXRβ isoforms were amplified from cDNA from hepatic tissue of patient 1, by using specific forward primers detailed in what follows and a sequence complementary to nucleotides 1997–2016 as a reverse primer (56).
Immunohistochemistry
Tissue specimens from normal and explanted livers were fixed in 10% formalin and paraffin embedded. Standard immunohistochemical procedures were used to detect BSEP and MRP2. Briefly, 5 µm thick sections were treated with 0.3% hydrogen peroxide for 30 min, to reduce endogenous peroxidase activity, and then subjected to antigen retrieval by autoclaving in 0.1 M citrate buffer (pH 6.0) at 121°C for 3 min. Sections were incubated overnight at 37°C with the primary antibodies, a mouse monoclonal anti-MRP2 (clone M2III-6; Chemicon International, Temecula, CA, USA) and a goat polyclonal anti-BSEP (Santa Cruz Biotechnology, Santa Cruz, CA, USA), at 1 : 20 and 1 : 50 dilutions. After 1 h incubation with a secondary biotinylated universal antibody (Vector, Burlingame, CA, USA), sections were incubated with a standard streptavidin–peroxidase complex (Vector). The enzymatic reaction was developed with 3,3′-diaminobenzidine tetrahydrochloride (Zymed Laboratories, San Francisco, CA, USA). Counterstaining was carried out with haematoxylin. For negative controls, Tris-buffered saline (TBS) replaced the primary antibodies.
Real-time PCR
Total RNA was extracted from liver samples with TRI REAGENT® (Molecular Research Center, Inc., Cincinnati, OH, USA) and poly(A)+ RNA was isolated using oligotex resin (Qiagen Inc., Valencia, CA, USA). Complementary DNAs (cDNAs) were prepared from poly(A)+ RNA using AMV (Roche Applied Science, Indianapolis, IN, USA) and random hexamers. Real-time quantitative PCR was performed using LightCycler FastStart DNA Master SYBR Green I (Roche Applied Science, Indianapolis, IN, USA) and LightCycler detector. Assays were conducted in duplicate using three different cDNA preparations from each mRNA sample. Quantitative expression values were extrapolated from separate standard curves, and normalized to β2-microglobulin. Specific oligonucleotide primers used were as follows: ATP8B1, 5′-AACAGCAGAACTGGACGGAG-3′ (forward primer, F), 5′-AGAGGAATTGCCCACCTGTG-3′ (reverse primer, R); ABCB11, 5′-CATCAGTTGACCCAATGCAA-3′ (F), 5′-ACTGGCTCCTGCACAGAGAA-3′ (R); ABCB4, 5′-ATCGAGACGTTACCCCACAA-3′ (F), 5′-CATTCTGGATGGTGGACAGG-3′ (R); FXR, 5′-AGGAGCCACTTCTTGATGTG-3′ (F), 5′-ATTAGGCAAACAGGGCTTGC-3′ (R); NTCP, 5′-CTCTGGGAAATGGCACCTACA-3′ (F), 5′-GTTGGGGATAGGTGAGGCTTC-3′ (R); SHP, 5′-AACACAGAGCCAGAGAGCTG-3′ (F), 5′-CCACCTCAAAGGTCACAGCA-3′ (R); LXR, 5′-GGAGAGGCTGCAGCACACAT-3′ (F), 5′-TAATGCCACGGGAGGATCTC-3′ (R); FXRα, 5′-GCTGGGATCTGGAGAGGAAGA-3′ (F), 5′-TGGGTCAGAGATGGACTTTC-3′ (R); FXRβ, 5′-GCTGTACGCCGTCAGGATTT-3′ (F), 5′-GGACCTGCCACTTGTTCTGT-3′ (R); FXR (primer C), 5′-TGTTGTCGAGGTCACTTGTC-3′; β2-microglobulin, 5′-CCAGCAGAGAATGGAAAGTC-3′ (F), 5′-GATGCTGCTTACATGTCTCG-3′ (R).
Analysis of differential gene expression by the use of oligonucleotide microarrays
‘AtlasTM Plastic Human 8 k Microarrays’ (BD Biosciences Clontech, Palo Alto, CA, USA) from the same lot were used throughout the study. These include approximately 8300 unique genes and corresponding oligonucleotides are gridded in a double-spotted pattern. Poly (A)+ RNA samples were reverse-transcribed with Moloney murine leukaemia virus reverse transcriptase in the presence of [α-33P]dATP. Purified radiolabelled probes were hybridized to microarrays according to the manufacturer's instructions. Hybridization signals were detected by phosphorimager and analysed by Phoretix array software (Nonlinear Dynamics Inc., NUT, UK). Expression data from patient 1 were compared with those from patients 4, 5, the child with biliary atresia and the normal control, resulting in four pair-wise comparisons. Differentially expressed genes in patient 1 were identified by using Student's t‐test (P<0.005). The hierarchical clustering method from Eisen et al. (30) was used to cluster the gene expression changes. Median centering and normalization of the data were performed before clustering. TreeView program was used for visualization.
ACKNOWLEDGEMENTS
We would like to acknowledge the patients and their relatives who participated in this study. Dolores Arjona and María T. Vallejo are acknowledged for technical assistance. We thank the reviewers of this manuscript for thorough revision and valuable suggestions. This work has been supported by grants from FIS (01/0281) and CAM (08.6/0015.1/2003).
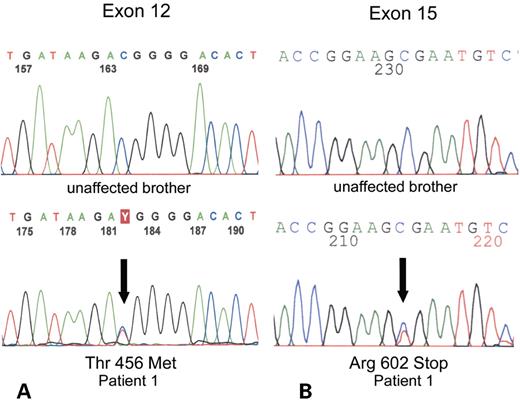
Figure 1. Partial sequences of exon 12 (A) and exon 15 (B) of ATP8B1 gene of patient 1 and his unaffected brother. Note heterozigosity (both T/C; arrows) in exons 12 and 15 of patient 1, leading to Thr456Met and Arg602Stop changes, respectively. Both mutations were also present in the two affected siblings (patients 2 and 3).
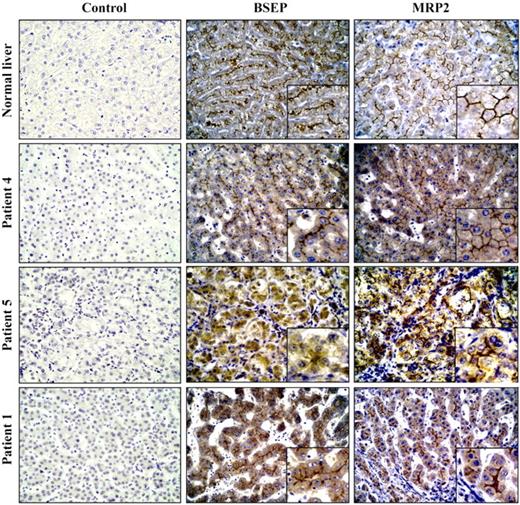
Figure 2. Immunohistochemical detection of BSEP and MRP2 in livers of children with cholestatic disease and in normal tissue. Paraffin embedded sections were stained with primary antibodies against BSEP and MRP2 (dilution 1 : 20), as indicated. For negative controls, primary antibodies were omitted (control). Note the absence of canalicular staining with BSEP antibody in tissue section from patient 5. Magnifications are 400×.
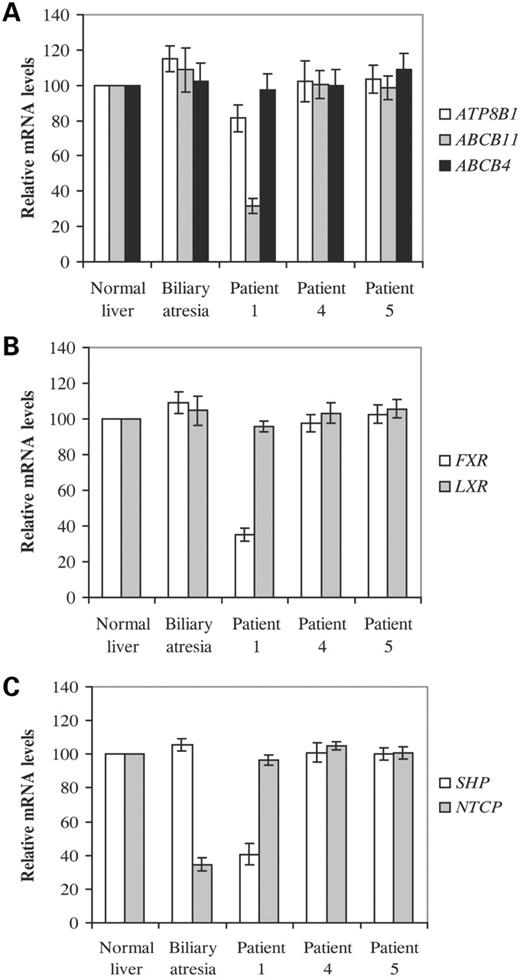
Figure 3. Relative expression of specific genes in liver from patients with cholestasis. Poly(A)+ RNA was isolated from the indicated liver samples and subjected to quantitative real-time PCR. Assays were performed in duplicate and normalized to β2-microglobulin. Expression of ABC transporters is shown in (A) nuclear receptors in (B) and SHP and NTCP in (C). Data represent the mean ±SEM of three experiments performed with three separate cDNA preparations from each sample. Values in normal liver were arbitrarily set to 100.
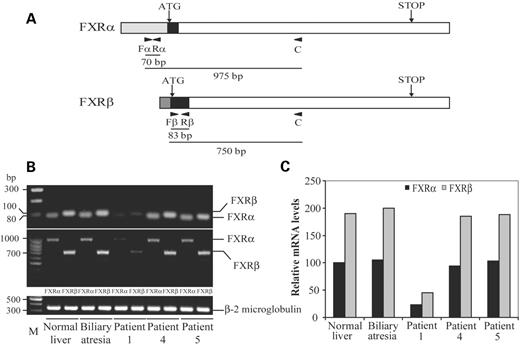
Figure 4. Decreased expression of FXR isoforms in patient 1. (A) schematic representation of FXRα and FXRβ mRNAs (adapted from 28,29). Shadowed and black areas denote the divergent 5′-UTR and N-terminus, respectively. Specific primers for each FXR isoform (Fα, Rα, Fβ and Rβ) and primer C are indicated by arrowheads. Below it is shown the predicted lengths of amplified products. (B) Electrophoresis bands after RT–PCR performed on equal amounts of poly(A)+ RNA from each sample and using specific primer pairs for FXR variants (upper panel), forward primers (Fα and Fβ) and primer C (middle panel) and β2-microglobulin primers (lower panel). (C) Quantitative real-time PCR was carried out with the sets of primers specific for each isoform. Values are represented relative to that for FXRα in normal liver, which was set to 100.
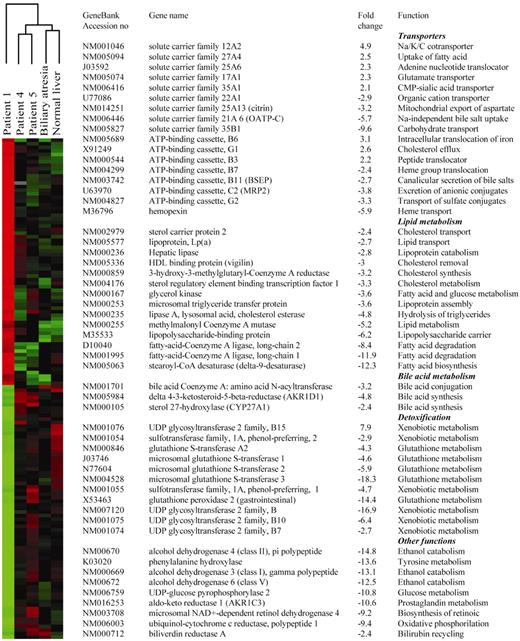
Figure 5. Gene array studies. The pattern of expression of 8327 genes were compared in the indicated samples. (Left panel) Hierarchical clustering of 163 human transcripts with differential expression in patient 1. The expression data from each patient are represented by a colour gradient from red (relatively high expression) to green (relatively low expression). Black denotes no change. (Right panel) List of representative genes differentially expressed in patient 1. Fold changes represent the average change over the other four samples.
Clinical and laboratory data of the patients
| Patient no. | Year of birth | Age at OLT (years) | ALTa (U/l) | Total bilirubina (mg/dl) | Serum bile salts (µmol/l) | GGTa (U/l) | Chola (mg/dl) | PreOLT height (in SDS) | Explanted liver | Outcome after OLT | Post OLT height (in SDS) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1997 | 3 | 18/121 | 12.6/17.1 | 112 | 21/35 | 166/132 | −2.6 | Severe fibrosis | Steatosis, diarrhoea | −4.2 |
| 2 | 1990 | 8.25 | 30/32 | 8.8/7.7 | 216 | 31/61 | 170/120 | −3.5 | Severe fibrosis | Diarrhoea | −4.1 |
| 3 | 1994 | 5.1 | 28/45 | 10.2/4.2 | 209 | 39/49 | 179/118 | −2.7 | Severe fibrosis | Steatosis, diarrhoea | −3.6 |
| 4 | 1999 | 2.75 | 81/219 | 7.4/2.9 | 23 | 802/241 | 177/234 | −1.1 | Cirrhosis | Normal | −0.8 |
| 5 | 2000 | 2.9 | 170/694 | 2.4/11.2 | 328.9 | 20/40 | 214/265 | −2.1 | Severe fibrosis, lithiasis | Normal | −1.3 |
| Patient no. | Year of birth | Age at OLT (years) | ALTa (U/l) | Total bilirubina (mg/dl) | Serum bile salts (µmol/l) | GGTa (U/l) | Chola (mg/dl) | PreOLT height (in SDS) | Explanted liver | Outcome after OLT | Post OLT height (in SDS) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1997 | 3 | 18/121 | 12.6/17.1 | 112 | 21/35 | 166/132 | −2.6 | Severe fibrosis | Steatosis, diarrhoea | −4.2 |
| 2 | 1990 | 8.25 | 30/32 | 8.8/7.7 | 216 | 31/61 | 170/120 | −3.5 | Severe fibrosis | Diarrhoea | −4.1 |
| 3 | 1994 | 5.1 | 28/45 | 10.2/4.2 | 209 | 39/49 | 179/118 | −2.7 | Severe fibrosis | Steatosis, diarrhoea | −3.6 |
| 4 | 1999 | 2.75 | 81/219 | 7.4/2.9 | 23 | 802/241 | 177/234 | −1.1 | Cirrhosis | Normal | −0.8 |
| 5 | 2000 | 2.9 | 170/694 | 2.4/11.2 | 328.9 | 20/40 | 214/265 | −2.1 | Severe fibrosis, lithiasis | Normal | −1.3 |
OLT, orthotopic liver transplantation; Chol, cholesterol; SDS, standard deviation score.
aAnalytical values at first assessment (1–5 months of age) and last follow-up visit before transplantation (all of them having received ursodeoxycholic acid therapy). Normal reference values: ALT, 5–45 U/l; total bilirubin, 0.1–1 mg/dl; serum bile salts, <14 µmol/l; GGT, 5–30 U/l; cholesterol, 80–180 mg/dl.
Clinical and laboratory data of the patients
| Patient no. | Year of birth | Age at OLT (years) | ALTa (U/l) | Total bilirubina (mg/dl) | Serum bile salts (µmol/l) | GGTa (U/l) | Chola (mg/dl) | PreOLT height (in SDS) | Explanted liver | Outcome after OLT | Post OLT height (in SDS) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1997 | 3 | 18/121 | 12.6/17.1 | 112 | 21/35 | 166/132 | −2.6 | Severe fibrosis | Steatosis, diarrhoea | −4.2 |
| 2 | 1990 | 8.25 | 30/32 | 8.8/7.7 | 216 | 31/61 | 170/120 | −3.5 | Severe fibrosis | Diarrhoea | −4.1 |
| 3 | 1994 | 5.1 | 28/45 | 10.2/4.2 | 209 | 39/49 | 179/118 | −2.7 | Severe fibrosis | Steatosis, diarrhoea | −3.6 |
| 4 | 1999 | 2.75 | 81/219 | 7.4/2.9 | 23 | 802/241 | 177/234 | −1.1 | Cirrhosis | Normal | −0.8 |
| 5 | 2000 | 2.9 | 170/694 | 2.4/11.2 | 328.9 | 20/40 | 214/265 | −2.1 | Severe fibrosis, lithiasis | Normal | −1.3 |
| Patient no. | Year of birth | Age at OLT (years) | ALTa (U/l) | Total bilirubina (mg/dl) | Serum bile salts (µmol/l) | GGTa (U/l) | Chola (mg/dl) | PreOLT height (in SDS) | Explanted liver | Outcome after OLT | Post OLT height (in SDS) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1997 | 3 | 18/121 | 12.6/17.1 | 112 | 21/35 | 166/132 | −2.6 | Severe fibrosis | Steatosis, diarrhoea | −4.2 |
| 2 | 1990 | 8.25 | 30/32 | 8.8/7.7 | 216 | 31/61 | 170/120 | −3.5 | Severe fibrosis | Diarrhoea | −4.1 |
| 3 | 1994 | 5.1 | 28/45 | 10.2/4.2 | 209 | 39/49 | 179/118 | −2.7 | Severe fibrosis | Steatosis, diarrhoea | −3.6 |
| 4 | 1999 | 2.75 | 81/219 | 7.4/2.9 | 23 | 802/241 | 177/234 | −1.1 | Cirrhosis | Normal | −0.8 |
| 5 | 2000 | 2.9 | 170/694 | 2.4/11.2 | 328.9 | 20/40 | 214/265 | −2.1 | Severe fibrosis, lithiasis | Normal | −1.3 |
OLT, orthotopic liver transplantation; Chol, cholesterol; SDS, standard deviation score.
aAnalytical values at first assessment (1–5 months of age) and last follow-up visit before transplantation (all of them having received ursodeoxycholic acid therapy). Normal reference values: ALT, 5–45 U/l; total bilirubin, 0.1–1 mg/dl; serum bile salts, <14 µmol/l; GGT, 5–30 U/l; cholesterol, 80–180 mg/dl.
References
Makishima, M., Okamoto, A.Y., Repa, J.J., Tu, H., Learned, R.M., Luk, A., Hull, M.V., Lustig, K.D., Mangelsdorf, D.J. and Shan, B. (
Parks, D.J., Blanchard, S.G., Bledsoe, R.K., Chandra, G., Consler, T.G., Kliewer, S.A., Stimmel, J.B., Willson, T.M., Zavacki, A.M., Moore, D.D. et al. (
Ananthanarayanan, M., Balasubramanian, N., Makishima, M., Mangelsdorf, D.J. and Suchy F.J. (
Plass, J.R., Mol, O., Heegsma, J., Geuken, M., Faber, K.N., Jansen, P.L. and Muller, M. (
Huang, L., Zhao, A., Lew, J.L., Zhang, T., Hrywna, Y., Thompson, J.R., De Pedro, N., Royo, I., Blevins, R.A., Pelaez, F. et al. (
Kast, H.R., Goodwin, B., Tarr, P.T., Jones, S.A., Anisfeld, A.M., Stoltz, C.M., Tontonoz, P., Kliewer, S., Willson, T.M. and Edwards, P.A. (
Denson, L.A., Sturm, E., Echevarria, W., Zimmerman, T.L., Makishima, M., Mangelsdorf, D.J. and Karpen, S.J. (
Grober, J., Zaghini, I., Fujii, H., Jones, S.A., Kliewer, S.A., Willson, T.M., Ono, T. and Besnard, P. (
Chen, F., Ma, L., Dawson, P.A., Sinal, C.J., Sehayek, E., Gonzalez, F.J., Breslow, J., Ananthanarayanan, M. and Shneider, B.L. (
Chen, F., Ananthanarayanan, M., Emre, S., Neimark, E., Bull, L., Knisely, A.S., Strautnieks, S., Thompson, R., Magid, M., Gordon, R. et al. (
Lu, T.T., Makishima, M., Repa, J.J., Schoonjans, K., Kerr, T.A., Auwerx, J. and Mangelsdorf, D.J. (
Goodwin, B., Jones, S.A., Price, R.R., Watson, M.A., McKee, D.D., Moore, L.B., Galardi, C., Wilson, J.G., Lewis, M.C., Roth, M.E. et al. (
Pircher, P.C., Kitto, J.L., Petrowski, M.L., Tangirala, R.K., Bischoff, E.D., Schulman, I.G. and Westin, S.K. (
Sinal, C.J., Tohkin, M.M., Miyata, M., Ward, J.M., Lambert, G. and Gonzalez, F.J. (
Lambert, G., Amar, M.J., Guo, G., Brewer, H.B., Jr, Gonzalez, F.J. and Sinal, C.J. (
Bull, L.N., van Eijk, M.J., Pawlikowska, L., DeYoung, J.A., Juijn, J.A., Liao, M., Klomp, L.W., Lomri, N., Berger, R., Scharschmidt, B.F. et al. (
Klomp, L.W.J., Vargas, J.C., van Mil, S.W.C., Pawlikowska, L., Strautnieks, S.S., van Eijk, M.J.T., Juijn, J.A., Pabón-Peña, C., Smith, L.B., DeYoung, J.A. et al. (
Bezerra, J.A. and Balistreri, W.F. (
Thompson, R. and Jansen, P. (
Strautnieks, S.S., Bull, L.N., Knisely, A.S., Kocoshis, S.A., Dahl, N., Arnell, H., Sokal, E., Dahan, K., Childs, S., Ling, V. et al. (
Ujhazy, P., Ortiz, D., Misra, S., Li, S., Moseley, J., Jones, H. and Arias, I.M. (
Harris, M.J. and Arias, I.M. (
van Mil, S.W., Klomp, L.W., Bull, L.N. and Houwen, R.H. (
Lykavieris, P., van Mil, S., Cresteil, D., Fabre, M., Hadchouel, M., Klomp, L., Bernard, O. and Jacquemin, E. (
de Vree, J.M., Jacquemin, E., Sturm, E., Cresteil, D., Bosma, P.J., Aten, J., Deleuze, J.F., Desrochers, M., Burdelski, M., Bernard, O. et al. (
Francis, G.A., Fayard, E., Picard, F. and Auwerx, J. (
Shneider, B.L., Fox, V.L., Schwarz, K.B., Watson, C.L., Ananthanarayanan, M., Thevananther, S., Christie, D.M., Hardikar, W., Setchell, K.D., Mieli-Vergani, G. et al. (
Huber, R.M., Murphy, K., Miao, B., Link, J.R., Cunningham, M.R., Rupar, M.J., Gunyuzlu, P.L., Haws, T.F., Kassam, A., Powell, F. et al. (
Zhang, Y., Kast-Woelbern, H.R. and Edwards, P.A. (
Eisen, M.B., Spellman, P.T., Brown, P.O. and Botstein, D. (
Russell, D.W. (
Kullak-Ublick, G.A., Stieger, B. and Meier, P.J. (
Horton, J.D., Goldstein, J.L. and Brown, M.S. (
Kok, T., Hulzebos, C.V., Wolters, H., Havinga, R., Agellon, L.B., Stellaard, F., Shan, B., Schwarz, M. and Kuipers, F. (
Downes, M., Verdecia, M.A., Roecker, A.J., Hughes, R., Hogenesch, J.B., Kast-Woelbern, H.R., Bowman, M.E., Ferrer, J.L., Anisfeld, A.M., Edwards, P.A. et al. (
Claudel, T., Inoue, Y., Barbier, O., Duran Sandoval, D., Kosykh, V., Fruchart, J., Fruchart, J.C., Gonzalez, F.J. and Staels, B. (
Liu, Y., Binz, J., Numerick, M.J., Dennis, S., Luo, G., Desai, B., MacKenzie, K.I., Mansfield, T.A., Kliewer, S.A., Goodwin, B. et al. (
Chen, H.L., Chang, P.S., Hsu, H.C., Ni, Y.H., Hsu, H.Y., Lee, J.H., Jeng, Y.M., Shau, W.Y. and Chang, M.H. (
Chiang, J.Y. (
Tazawa, Y., Yamada, M., Nakagawa, M., Konno, T. and Tasa, K. (
Bull, L.N., Juijn, J.A., Liao, M., van Eijk, M.J.T., Sinke, R.J., Stricker, N.L., DeYoung, J.A., Carlton, V.E.H., Baharloo, S., Klomp, L.W.J. et al. (
Bull, L.N., Carlton, V.E., Stricker, N.L., Baharloo, S., De Young, J.A., Freimer, N.B., Magid, M.S., Kahn, E., Markowitz, J., DiCarlo, F.J. et al. (
Jansen, P.L.M., Strautnieks, S.S., Jacquemin, E., Hadchouel, M., Sokal, E.M., Hooiveld, G.J.E.J., Koning, J.H., De Jager-Krikken, A., Kuipers, F., Stellaard, F. et al. (
Pawlikowska, L., Groen, A., Eppens, E.F., Kunne, C., Ottenhoff, R., Looije, N., Knisely, A.S., Killeen, N.P., Bull, L.N., Oude Elferink, R.P. et al. (
Linarelli, L.G., Williams, C.N. and Phillips, M.J. (
Stieger, B. (
Yu, J., Lo, J.L., Huang, L., Zhao, A., Metzger, E., Adams, A., Meinke, P.T., Wright, S.D. and Cui, J. (
Egawa, H., Yorifuji, T., Sumazaki, R., Kimura, A., Hasegawa, M. and Tanaka, K. (
Bishop-Bailey, D., Walsh, D.T. and Warner, T.D. (
Pellicciari, R., Fiorucci, S., Camaioni, E., Clerici, C., Costantino, G., Maloney, P.R., Morelli, A., Parks, D.J. and Willson, T.M. (
Dussault, I., Beard, R., Lin, M., Hollister, K., Chen, J., Xiao, J.H., Chandraratna, R. and Forman, B.M. (
van der Bliek, A.M., Kooiman, P.M., Schneider, C. and Borst, P. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}