





Abstract
Sepsis-induced lung injury is associated with significant morbidity and mortality. Previously, we showed that heterozygous deletion of polymerase δ-interacting protein 2 (Poldip2) was protective against sepsis-induced lung injury. Since endothelial barrier disruption is thought to be the main mechanism of sepsis-induced lung injury, we sought to determine if the observed protection was specifically due to the effect of reduced endothelial Poldip2.
Endothelial-specific Poldip2 knock-out mice (EC−/−) and their wild-type littermates (EC+/+) were injected with saline or lipopolysaccharide (18 mg/kg) to model sepsis-induced lung injury. At 18 h post-injection mice, were euthanized and bronchoalveolar lavage (BAL) fluid and lung tissue were collected to assess leucocyte infiltration. Poldip2 EC−/− mice showed reduced lung leucocyte infiltration in BAL (0.21 ± 0.9×106 vs. 1.29 ± 1.8×106 cells/mL) and lung tissue (12.7 ± 1.8 vs. 23 ± 3.7% neutrophils of total number of cells) compared to Poldip2 EC+/+ mice. qPCR analysis of the lung tissue revealed a significantly dampened induction of inflammatory gene expression (TNFα 2.23 ± 0.39 vs. 4.15 ± 0.5-fold, IκBα 4.32 ± 1.53 vs. 8.97 ± 1.59-fold), neutrophil chemoattractant gene expression (CXCL1 68.8 ± 29.6 vs. 147 ± 25.7-fold, CXCL2 65 ± 25.6 vs. 215 ± 27.3-fold) and a marker of endothelial activation (VCAM1 1.25 ± 0.25 vs. 3.8 ± 0.38-fold) in Poldip2 EC−/− compared to Poldip2 EC+/+ lungs. An in vitro model using human pulmonary microvascular endothelial cells was used to assess the effect of Poldip2 knock-down on endothelial activation and permeability. TNFα-induced endothelial permeability and VE-cadherin disruption were significantly reduced with siRNA-mediated knock-down of Poldip2 (5 ± 0.5 vs. 17.5 ± 3-fold for permeability, 1.5 ± 0.4 vs. 10.9 ± 1.3-fold for proportion of disrupted VE-cadherin). Poldip2 knock-down altered expression of Rho-GTPase-related genes, which correlated with reduced RhoA activation by TNFα (0.94 ± 0.05 vs. 1.29 ± 0.01 of relative RhoA activity) accompanied by redistribution of active-RhoA staining to the centre of the cell.
Poldip2 is a potent regulator of endothelial dysfunction during sepsis-induced lung injury, and its endothelium-specific inhibition may provide clinical benefit.
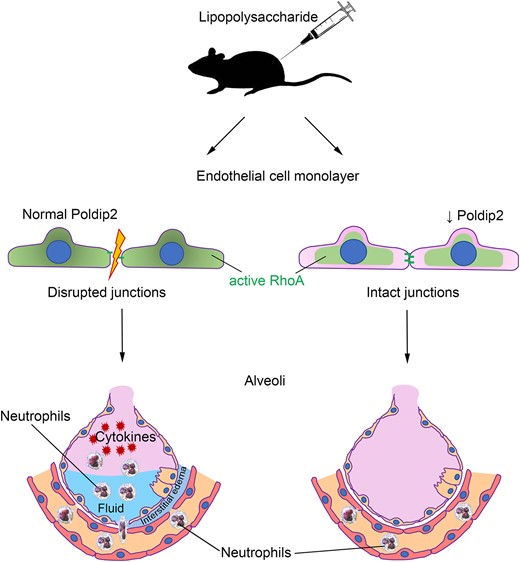
1. Introduction
Sepsis consists of life-threatening organ dysfunction caused by a dysregulated host response to infection1 and is associated with significant morbidity and mortality. Approximately 50% of patients with sepsis will develop acute lung injury (ALI).2 While multiple conditions can result in ALI, patients with sepsis-induced ALI have higher mortality than those with ALI from other causes.3 This suggests that there may be sepsis-specific molecular pathways leading to ALI that are distinct from the pathways activated by other conditions. Unlike the direct ALI arising from pneumonia, the mechanisms of indirect ALI seen in sepsis originating from other sources are not as well understood. Current perception can be summarized as follows: the highly vascular nature of the lung predisposes it to injury by circulating toxins leading to disruption of the endothelial barrier, microthrombi formation, extravasation of immune cells, and secretion of inflammatory mediators, such as tumour necrosis factor-α (TNFα) that facilitate further loss of alveolar-capillary barrier.4 TNFα is not only one of the main cytokines involved in sepsis5 but is also considered to be one of the effectors of lipopolysaccharide (LPS)5 and causes endothelial barrier disruption.6 However, the exact molecular mechanisms of sepsis-induced ALI remain unclear and only a handful of potential interventions have been investigated.7–9
Polymerase δ-interacting protein 2 (Poldip2) is a multifunctional protein that initially was implicated in DNA repair10 and was suggested to play a pathological role in cancer development.11,12 The importance of Poldip2 in DNA repair was suggested based on its interaction with POLD2 (p50), a subunit of both Polδ and Polζ.13,14 Its exact role in DNA repair is, however, not completely understood, with recent data suggesting that Poldip2 might control the relative usage of error-prone translesion DNA synthesis and error-free template switching by homologous DNA recombination.15 Our previous work focusing on the role of Poldip2 in sepsis demonstrated that mice with heterozygous whole-body deletion of Poldip2 were protected against sepsis-induced ALI. We also showed that Poldip2 knock-down decreased the expression of adhesion molecules and leucocyte adhesion to lung endothelial cells in response to LPS stimulation.16 These results suggested an important role for Poldip2 in endothelial barrier function during sepsis and a beneficial effect of reduced Poldip2, presumably in endothelium. We, therefore, hypothesized that endothelial-specific knock-out of Poldip2 would be also protective against sepsis-induced ALI. We also wished to probe the potential mechanisms whereby endothelial Poldip2 can mediate the response to sepsis. Endothelial cells are a compelling target for therapeutic intervention given their direct contact with the bloodstream and ability to absorb viral particles, siRNA, and circulating compounds. If endothelial-specific reduction of Poldip2 is sufficient to protect against sepsis-induced ALI, an endothelium-specific therapy could be developed, thus avoiding potential off-target effects.
2. Methods
2.1 Animals
Adult male and female (20–30 g) mice on the C57BL/6J background were used in this study. Endothelial-specific knock-out mice (Poldip2 EC−/−) were generated as detailed elsewhere.17 Briefly, floxed Poldip2 mice were crossed with cdh5-Cre mice on the C57BL/6 background to generate Poldip2 EC−/− mice and their littermate controls (Poldip2-fl/fl). Mice were genotyped using a conventional PCR method. All animal experiments were conducted with the approval of the Institutional Animal Care and Use Committee at Emory University. Ethical approval was received before conducting the study. All reported procedures conform to the NIH Guide for the Care and Use of Laboratory Animals. For reported data, researchers performing analyses were blinded to genotype and experimental groupings of mice. RNA was extracted from endothelial, smooth muscle (media + adventitia) fractions (see below) as well as isolated monocytes. Poldip2 expression was assessed by qPCR to confirm the endothelial specificity of the knock-out (Supplementary material online, Figure S1A).
2.2 Ex-vivo carotid artery endothelial-enriched and smooth muscle cell-enriched RNA collection
Animals were sacrificed and their carotid arteries were explanted after retrograde perfusion with ice-cold saline solution and transferred to a 35 mm dish containing ice-cold HBSS (Hank’s balanced salt solution). Using an insulin syringe fitted with a 29-gauge needle, 150 µL of Trizol (Cat# 15596026, Thermo Fisher Scientific) solution was quickly flushed to extract the intimal (endothelial-enriched) RNA.18 The leftover carotid tissue (comprised of the medial smooth muscle layer and adventitia) were separately shredded in Trizol using stainless steel beads. RNA extracts from both fractions were purified separately using the miRNeasy Kit (Cat# 217004, Qiagen, USA). RNA integrity was assessed with the RNA Nano 6000 Assay Kit of the Agilent Bio analyzer 2100 system (Life technologies, CA, USA). Purity of the fractions was assessed by comparison of the expression of endothelial and smooth muscle-specific genes in each fraction (Supplementary material online, Figure S1C and D).
2.3 LPS-induced ALI
Poldip2-EC+/+ and Poldip2-EC−/− mice were randomly divided into phosphate buffered saline (PBS) (n = 5) and LPS (n = 5–6) groups. Animals received an intraperitoneal (i.p.) injection of PBS or LPS (18 mg/kg) from Escherichia coli 0111: B4 (Cat# tlrl-eblps, InvivoGen) diluted in PBS. The majority of the in vivo data was collected at 18 h post-injection. A separate experiment (n = 5 per group) was performed for survival analysis. Mortality was monitored every 8–12 h after the injection. Our previous studies showed that ∼40% of wild-type mice treated with LPS did not survive past 24 h; therefore, all the measurements were conducted upon euthanasia with carbon dioxide inhalation at 18 h after the injection. Euthanasia was induced using CO2 supplied as a metered gas to a ventilated chamber. Air was displaced with 100% CO2 at a constant flow rate of 10–30% of the chamber volume per minute. Mice were kept in the chamber at least 1 min after they stopped breathing. Mice were checked by palpation to verify that the heart stopped beating. At 18 h post-injection, just prior to euthanasia, core temperature was measured using a rectal thermoprobe under light anaesthesia (1–1.5% isoflurane).
2.4 BAL collection
After euthanasia, mouse tracheas were exposed through a small skin incision on the anterior neck and cannulated using a 21-gauge lavage needle. For determination of total cell counts in BAL, 1.5 mL of PBS was instilled in the tracheal lavage needle and retrieved. BAL cell counts were obtained with a TC20 automated cell counter (Bio-Rad) using a 7–15 µm window. In some experiments, BAL was centrifuged and the protein content of the supernatant was assessed using Bio-Rad Protein Assay Dye (Cat#5000006).
2.5 ELISA
Interleukin (IL)-1β, IL-6, chemokine (C-X-C motif) ligand 1 (CXCL-1), and chemokine (C-X-C motif) ligand 2 (CXCL-2) levels in BAL fluids and TNF-α in the lung tissue were measured using specific ELISA kits according to the manufacturer’s instructions (R&D Systems). TNF-α tissue levels were normalized by the protein concentration measured by Bio-Rad Protein Assay Dye (Cat#5000006).
2.6 Flow cytometry and data analysis
Cells from lavage fluid were resuspended in FACS Buffer and then blocked with anti-mouse CD16/32 (Fc Block BD Pharmigen, 1:100) for 5 min. Cells were then incubated with antibodies against CD45-APC-FITC, Ly6G-APC, Ly6C-APC-eFluor-780, and CD14-PerCP-Cy5.5 or CD45-APC-e780, CD11c-FITC, CD68-PE, and F4/80-APC in FACS buffer for 30 min on ice. Cells were washed with FACS buffer, measured by flow cytometry on an Aurora cytometer (Cytek Biosciences) and analysed with FlowJo_V10 software. Single cells were gated based on forward scatter area (size) and forward scatter height. CD45+ leucocytes were selected and gated vs. Ly6G (PMN), Ly6C+ (myeloid), CD14 (monocyte), F4/80+ (macrophage), and CD68 (macrophage). Cells positive for CD45 were also selected and gated by Ly6G vs. CD14, Ly6G vs. Ly6C, CD11C vs. F4/80, and CD11c vs. CD68.
2.7 Lung permeability measurement in vivo
To quantify albumin extravasation, we used Evans blue dye as previously described.19 Evans blue dye (2% solution in PBS, 4 mL/kg; MP Biomedicals; Cat No. 151108) was injected intravenously 18 h after LPS injection and allowed to circulate for 30 min. Animals were then perfused transcardially with PBS until fluid from the left atrium became colourless. Lungs were isolated, and the dye was extracted with formamide overnight at 50°C. Subsequently, lungs were allowed to dry for 1 h at room temperature before being weighed. Evans blue concentration was quantified spectrophotometrically at 611 nm and normalized to the dry weight of both lungs.
2.8 RNA extraction and RT–qPCR
Total RNA was purified with QIAzol (Cat# 79306, Qiagen) and the RNeasy Plus kit (Cat# 74104, Qiagen). Reverse transcription was performed using Protoscript reverse transcriptase (New England Biolabs) with random primers. The resulting cDNA was amplified with previously validated primers against Iκbα, Tnfα, Cxcl1, Cxcl2, Vcam1, Hprt and Ppia. Hprt, and Ppia (Supplementary material online, Table S1) were used as housekeeping genes as their expression was not affected by LPS treatment. Amplification was performed in 96-well plates using Forget-Me-Not EvaGreen qPCR Master Mix with low ROX (Cat # 31045, Biotium) in a QuantStudio 7 instrument (Invitrogen). Data analysis was performed using the mak3i module of the qpcR software library (version 1.4-0)20,21 in the R-environment.22 Final quantification results were divided by the average of PBS-treated Poldip2 EC+/+ values and expressed in arbitrary units.
2.9 Cell culture
Human lung microvascular endothelial cells (HLMECs) were purchased from Lonza (Cat# CC-2527) and were cultured in EC growth medium (EBM-2, Cat# CC-3156, Lonza) supplemented with Microvascular Endothelial Cell Growth Medium SingleQuots supplements (Cat# CC-4147, Lonza). Rat brain microvascular endothelial cells (RBMECs) were purchased from CellBiologics (Cat# RA-6023) and cultured in supplemented Endothelial Cell medium (Cat# M1266, Cell Biologics). Media was changed every 2 days until cells reached confluence. All experiments were conducted between Passages 4 and 7. In each experiment, cultures exposed to species-specific TNFα [recombinant rat TNFα (Cat# 510-RT-050, R&D Systems) or recombinant human TNFα (Cat# 300-01A, PeproTech, Inc)] (10 ng/mL) were compared with PBS control conditions.
2.10 Small interfering RNA
For Poldip2 silencing studies, confluent HLMECs were transfected with human small interfering RNA against Poldip2 (siPoldip2; sense: 5′- CGUGAGGUUUGAUCAGUAAdTdT-3′, antisense: 5′- UUACUGAUCAAACCUCACGdTdG-3′; Qiagen) or Mission siRNA Universal Negative Control #1 (Sigma). Cells were washed with HBSS, and incubated with siRNA (100 nmol/L) + RNAiMAX (Invitrogen) complexes in OPTIMEM containing 2% serum for 5 h. After transfection, OPTIMEM was changed to complete EBM-2 media for an additional 48 h, at which point experiments were conducted.
Silencing experiments in RBMECs were conducted using small interfering RNA against rat Poldip2 (siPoldip2; sense: 5′-GUCUAUUGGUGGCGAUACUdTdT-3′, antisense: 5′- AGUAUCGCCACCAAUAGACdTdT-3′; Sigma) or the Mission siRNA Universal Negative Control #1 (Sigma). Cells were washed with HBSS and incubated with siRNA (100 nmol/L) + RNAiMAX (Invitrogen) complexes in OPTIMEM containing 2% serum for 5 h. After transfection, OPTIMEM was changed to serum free endothelial cell media without supplements for an additional 40 h.
2.11 RNASeq and bioinformatics analysis
RNA from carotid endothelial and smooth muscle-enriched layers was collected as described above. RNA sequencing was performed using a low-input RNASeq pipeline from Novogene. The pooled libraries were read using an Illumina NextSeq 550 sequencer. Read mapping and quantification of gene expression level were also performed by Novogene. The reference mouse genome (mm9) was used to count the read numbers mapped to each gene. Quantification was achieved by assembling Digital Gene Expression reads into transcripts, and their abundance was estimated in fragments per kilobase of exon model per million reads mapped.23 Bowtie (v2.2.3) was used to build an index of the reference genome, and TopHat (v2.0.12) was used to align paired-end clean reads. The differential expression of the two conditions/groups (two biological replicates per group) was obtained using the DESeq R package (1.18.0). DESeq provided a statistical routine for determining differential expression in DGE data using a model based on the negative binomial distribution. The resulting P-value was adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate.24 The P-value was adjusted using corrected P-values (q value), q < 0.005 and a log2 (fold change) >1 was set as the threshold for significant differential expression. Gene Ontology enrichment of differentially expressed genes was executed by the GOSeq R Package based on the Wallenius hyper distribution, which can adjust for gene length bias in DGEs.25 FDR < 0.05 indicates a significant enrichment of differentially expressed genes in that pathway. We used the REVIGO tool to visualize the gene ontology terms associated with the differentially expressed genes (Supplementary material online, Figure S1B).26 Data are deposited at GSE165939.
RNA from HLMECs from three independent experiments treated as described in detail above (siControl or siPoldip2 followed with stimulation with TNFα or PBS for 6 h) was purified with the QIAzol and RNeasy Plus kit (Qiagen). RNASeq library preparation and analysis were performed by Novogene, Inc. similarly to describe above. GRCh38/hg38 was used as a reference genome. Reactome analysis was performed by Novogene, Inc. using the Reactome database27 and ClusterProfiler software.28 GeneRatio was calculated as a ratio of differentially expressed genes to all genes, which are concerned with a certain pathway. Padj < 0.05 indicates a significant enrichment of differentially expressed genes in that pathway. Data are deposited at GSE165410. TNFAIP, TNFRSF1A, and TNFRSF1B expression was confirmed by qPCR (for primer sequences see Supplementary material online, Table S1).
2.12 Permeability assay
HLMECs were grown to confluence in 12-well plates precoated with biotinylated collagen I (prepared as described in Reference29) siRNA transfection was performed as described above. At 48 h post-transfection, cells were treated with TNFα (10 ng/mL) or PBS for 3 h. FITC-avidin (Cat# A821, Molecular Probes, concentration 25 μg/mL) was then added and incubated for 210 s. Cultures were rinsed, fixed with 4% paraformaldehyde in HBSS and imaged (6 images per condition) using an Olympus epifluorescence inverted microscope with a 10× objective by a blinded investigator. Gaps between cells were measured by imaging of FITC-avidin binding to the substrate.
2.13 Transendothelial electrical resistance measurement
HLMECs were seeded (2.4× 104 cells/well) in the upper chamber of a transwell permeable support (0.4 μm pore size, Greiner Bio-one, Cat No. 662641). At 24 h later, monolayers were transfected with siRNA as described above. After another 48 h, cells were exposed to TNF-α (10 ng/mL) added to both the bottom and top chambers for 3 h at 37°C. Resistance was measured using STX2 chopstick electrodes connected to an EVOM2 voltohmmeter (World Precision Instruments, Berlin, Germany). The resistance value of a blank culture insert was subtracted from the total resistance measured, and the resulting value was multiplied by the membrane area to obtain the transendothelial electrical resistance (TEER) measurement in Ω × cm2. Each individual experiment consisted of two replicates (derived from a single preparation of HPMECs). Data are reported as fold change of control (siControl PBS-treated cells).
2.14 RhoA activity measurement
RBMECs were grown in 6-well plates and siRNA transfection was performed as described above. Cells were then serum-starved for 40 h and treated with rat TNFα (10 ng/mL) for 3 h. G-LISA for RhoA activity was performed using the G-LISA kit (Cat# BK124, Cytoskeleton, Inc.) as per the manufacturer’s instructions. Activity values were normalized to protein levels measured using the Precision Red reagent (Cat# ADV02, Cytoskeleton, Inc.). Total RhoA levels were evaluated via western-blotting using anti-RhoA antibody (Cat# 2117S, Cell Signalling).
2.15 Immunohistochemistry and immunofluorescence
After euthanasia, lungs were instilled with 10% formalin through the trachea, dissected out and placed in 10% formalin for 24 h, and then transferred to 70% ethanol, paraffin embedded and sectioned. Sections were stained with haematoxylin eosin for morphology. Neutrophil staining was carried out on 5 μm sections of paraffin-embedded tissues. Antigen retrieval was performed using Proteinase K (10 µg/mL) before incubating with anti-LY6G and anti-LY6C antibody (1:200 in 3% BSA Cat#2557; Abcam) followed by incubation with biotinylated anti-rat secondary antibody (Cat# BA-9400-1.5; Vector Laboratories Inc.) and then with Streptavadin QDot 655 (1:200 in 2% BSA Cat# Q10123MP; Invitrogen). Sections were then mounted using Vectashield antifade medium with DAPI (Cat# H-1200; Vector Laboratories Inc.). Images were acquired by a separate investigator blinded to the experimental groups and the hypothesis with a Zeiss LSM 800 Airyscan laser scanning confocal microscope using a Plan-Apochromat 20×/0.80 NA objective. Quantification was performed using ImageJ software by a blinded investigator (five consecutive sections per animal were analysed). The ratio of Ly6G+Ly6C staining area to DAPI staining area was used to quantify neutrophil infiltration.
For in vitro studies, HLMECs were treated with TNFα or PBS for 6 h, rinsed with HBSS++ (Cat# 14025092, Gibco), fixed with 4% PFA in HBSS++ for 15 min. Cells were then permeabilized with 0.2% triton-X100 for 5 min, and blocked with 0.5% BSA for 30 min. Coverslips were incubated with antibodies against active-RhoA (Cat# 26904; NewEast Biosciences) and VE-cadherin (Cat# ab33168; Abcam) diluted in blocking solution overnight. Alexa Fluor 568 and 488 (Cat# A10042 and A32766; Invitrogen) secondary antibodies were used for detection. Alexa Fluor 350 Phalloidin (Cat# A22281, Invitrogen) staining for actin cytoskeleton was performed during the secondary antibody incubation. Coverslips were mounted on slides with Immu-Mount (Cat# 9990402, Thermo Fisher Scientific) and imaged with a Zeiss LSM 800 Airyscan laser scanning confocal microscope using a Plan-Apochromat 63×/1.40 objective by an investigator blinded to the treatment conditions. Quantification of VE-cadherin integrity was performed from maximum intensity projections of the Z-stacks using ImageJ software by a blinded investigator (six cells per condition were analysed). The length of the cell border with disrupted VE-cadherin staining was measured and normalized to the cell perimeter. Changes in localization of active-RhoA staining were assessed by measuring the area stained with active-RhoA relative to the overall cell area by a blinded investigator (six cells per condition were analysed).
2.16 Western-blotting analysis
Whole cell lysate was prepared from cultured RBMECs using 120 µL of lysis buffer from the G-LISA kit (Cat# BK124; Cytoskeleton, Inc.) and precleared by centrifugation at 10 000×g for 1 min at 4°C. Relative protein concentration was measured using Precision Red reagent (Cat# ADV02; Cytoskeleton, Inc.), and samples were diluted to equal amounts of protein and stored at −80°C. Prior to electrophoresis, samples were mixed with 6× sample buffer (Cat# BP111R; Boston BioProducts) and heated to 95°C for 5 min. Following separation by SDS–PAGE, proteins were transferred to a nitrocellulose membrane and assessed by western-blotting with primary antibodies against Poldip2 (Cat# ab181841; Abcam), RhoA (Cat# 2117S; Cell Signalling), or Vinculin (Cat# V4505-100UL; Sigma). Blots were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies depending on the species of the primary antibody [anti-mouse (Cat# AP181P; Millipore) and anti-rabbit (Cat# 711035152; Jackson Labs)] and assessed using enhanced chemiluminescence (Cat# 32106, GE). HRP-induced luminescence was detected with autoradiography film (Cat# XC59X; MidSci). Detected bands were scanned and densitometry was performed using ImageJ.
2.17 Statistical analysis
Data are expressed as mean ± SEM. Shapiro–Wilk normality test was performed. Statistical analyses were performed using a two-way ANOVA with a Tukey’s correction for multiple comparison. For comparison between two groups, two-tailed T-test was used. P < 0.05 was considered statistically significant. The survival curve (Kaplan–Meier curve) was analysed using the log-rank test.
3. Results
3.1 Endothelial-specific Poldip2 deletion reduces immune cell infiltration in the lung after LPS injection
Given the previously described beneficial effect of reduced whole-body Poldip2 in LPS-induced lung injury,16 we sought to determine if endothelial-specific Poldip2 deletion (Poldip2 EC−/−) would exhibit similar effects. Indeed, while LPS injection induced a significant increase in cell counts in the bronchoalveolar lavage (BAL) from Poldip2 EC+/+ mice, this effect was fully abolished in Poldip2 EC−/− mice (1.29 ± 1.8 ×106 vs. 0.21 ± 0.9 ×106 cells/ml) (Supplementary material online, Figure S1A). This reduction in lung oedema was confirmed by measurement of protein concentration in BAL and Evans blue extravasation (Figure 1C and D). Histological analysis was also consistent with decreased lung oedema in Poldip2 EC−/− mice (Figure 1B). Further analysis of BAL fluid revealed that the immune cell BAL composition was not different between the genotypes after LPS injection (Supplementary material online, Figure S2) indicating that in the BAL of Poldip2 EC−/− mice the extent of infiltration was reduced universally without favouring any particular cell-type. Neutrophil-specific staining of lung sections, however, revealed a reduction in infiltrating neutrophils in the lungs of Poldip2 EC−/− mice after LPS injection (7.2 ± 0.8-fold vs. 31.5 ± 4-fold increase in Poldip2 EC+/+ mice) (Figure 1D and E). These data suggest that at this stage of sepsis, neutrophils are still retained in tissue and indicate that loss of EC Poldip2 might also affect neutrophil adhesion or release from tissue. While we did not detect a significant difference in mortality, we did observe a significant reduction in sepsis severity as assessed by core temperature measurement30 in Poldip2 EC−/− mice (Supplementary material online, Figure S3).
![Endothelial-specific Poldip2 knock-out prevents LPS-induced lung injury. (A) Poldip2 EC−/− and Poldip2 EC+/+ littermates were injected with 18 mg/kg LPS. BAL was collected 18 h later and cell counts are represented as averages ± SEM (n = 4–6). $$$P < 0.001 compared to PBS injected Poldip2 EC+/+ mice, ####P < 0.0001 compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction). (B) Lungs harvested from mice treated as in (A) were stained with haematoxylin eosin, and show increased lung oedema in Poldip2 EC+/+ (arrows), but not in Poldip2 EC−/− mice after LPS injection. (C) Protein concentration measurement in BAL showing a significant increase after LPS injection. This response was significantly blunted in Poldip2 EC−/− mice (n = 5 in PBS group, n = 6–7 in LPS treated groups). $$P < 0.01 compared to PBS injected Poldip2 EC+/+ mice, #P < 0.05 compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction). (D) Lung permeability to Evans blue dye increased after LPS injection in Poldip2 EC+/+ with a significantly lower increase in Poldip2 EC−/− mice (n = 9 for PBS injected groups, n = 11–12 for LPS injected mice, $P < 0.05 compared to PBS injected Poldip2 EC+/+ mice, #P < 0.05 compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction)]. (E) Neutrophil-specific staining of LY6G and LY6C showed a significantly increased lung parenchyma infiltration with neutrophils in Poldip2 EC+/+ mice after LPS injection, which was significantly lower in Poldip2 EC−/− mice. Data quantified in (F), represent averages ± SEM (n = 5) $$$P < 0.001 compared to PBS injected Poldip2 EC+/+ mice, #P < 0.05 compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/118/11/10.1093_cvr_cvab295/2/m_cvab295f1.jpeg?Expires=1716448650&Signature=NLcsBAH0p08uyjLRMv9x97mphwvbS11HRtxRUGnC~E52SOtpEdXz4MqlrRilcz7QMfiDuqAuocABGVBqxEzVBmOK~ARbyPsxAFgADbZCrvUdVdZ7fSvRedNY7tGrfWRfPP0zrCmzd6SUMLkeUiMCAyFvzZYCLFRooCYTWEhb-YqpbmeLxQvB6d4NWv3qIS7y01ORYiXb9l1g7ye~~l7Fopj4fjkNSRDMi1L1MY-JSJGMbR6qEF4AqDOdzkYqoxqRNAM9rA233ExJzNZiY1SDCJsHWhkBk7noIDGgKh7Fqt7ihaie55ggx35WLsX-2HMFcOy5AVMP159LG~9q6nL4aQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Endothelial-specific Poldip2 knock-out prevents LPS-induced lung injury. (A) Poldip2 EC−/− and Poldip2 EC+/+ littermates were injected with 18 mg/kg LPS. BAL was collected 18 h later and cell counts are represented as averages ± SEM (n = 4–6). $$$P < 0.001 compared to PBS injected Poldip2 EC+/+ mice, ####P < 0.0001 compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction). (B) Lungs harvested from mice treated as in (A) were stained with haematoxylin eosin, and show increased lung oedema in Poldip2 EC+/+ (arrows), but not in Poldip2 EC−/− mice after LPS injection. (C) Protein concentration measurement in BAL showing a significant increase after LPS injection. This response was significantly blunted in Poldip2 EC−/− mice (n = 5 in PBS group, n = 6–7 in LPS treated groups). $$P < 0.01 compared to PBS injected Poldip2 EC+/+ mice, #P < 0.05 compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction). (D) Lung permeability to Evans blue dye increased after LPS injection in Poldip2 EC+/+ with a significantly lower increase in Poldip2 EC−/− mice (n = 9 for PBS injected groups, n = 11–12 for LPS injected mice, $P < 0.05 compared to PBS injected Poldip2 EC+/+ mice, #P < 0.05 compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction)]. (E) Neutrophil-specific staining of LY6G and LY6C showed a significantly increased lung parenchyma infiltration with neutrophils in Poldip2 EC+/+ mice after LPS injection, which was significantly lower in Poldip2 EC−/− mice. Data quantified in (F), represent averages ± SEM (n = 5) $$$P < 0.001 compared to PBS injected Poldip2 EC+/+ mice, #P < 0.05 compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction).
3.2 Endothelial-specific Poldip2 knock-out reduces the expression of markers of inflammation and chemokines in the lung after LPS injection
ALI-induced immune cell activation and infiltration are associated with increased expression of a number of inflammatory cytokines,31 chemokines,32 and adhesion molecules.32 We therefore examined mRNA expression levels of Tnfα, Iκbα, Cxcl1, Cxcl2, and Vcam1 and found they were dramatically increased after LPS injection in Poldip2 EC+/+ mice, but the increase was blunted in the lungs Poldip2 EC−/− mice (Figure 2). We also assessed BAL concentrations of cytokines with a known role in sepsis (such as IL-1β and IL-6) along with protein levels of CXCL1 and CXCL2 and lung tissue levels of TNFα, all of which were increased after LPS injection but to a much lesser degree in Poldip2 EC−/− compared to Poldip2 EC+/+ mice (Figure 3). These data support the idea that endothelial-specific Poldip2 reduction is capable of containing the inflammatory cascade during ALI (i.e. endothelial activation, leucocyte adhesion, chemokine expression, and cytokine secretion), thus preventing the downward-spiral of sepsis-induced lung injury.
![Endothelial-specific Poldip2 knock-out abrogates inflammatory gene expression in LPS-induced lung injury. LPS injection induced a significant increase in expression of inflammatory markers [Tnfα (A), Iκbα (B)), chemoattractant molecules [Cxcl1 (C), Cxcl2 (D)], and the vascular activation marker [Vcam1 (E)] in Poldip2 EC+/+ mice. This effect was significantly blunted in Poldip2 EC−/− mice. Graphs represent averages ± SEM (n = 5). P < 0.0001, $$$P < 0.001, $$P < 0.01, compared to PBS injected Poldip2 EC+/+ mice, ##P < 0.01, #P < 0.05, compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/118/11/10.1093_cvr_cvab295/2/m_cvab295f2.jpeg?Expires=1716448650&Signature=bAAHB6c0NPZRFc5J2NQD2~R9krRIMaQjiNkcmf7QMDzer4maxF1CwbgD3KAm~dV9eO7BNdG5qER3b8MGHh5IOBMnTQr1jAMmFN-teWYwAhhGhT2mSlOoPioAdQPxhD1F7PG~O6FEjr11cDmE78UaaKOdKsipSKlIVtzIjMNCyYr6ZTGIVjbHN2lbh9tpmEtKF37jorm-gBW35Ym-x~YUJIWLKOto7OA-OKI6g8yU2yXBhsGSimD8cLsPsrGuJE272sHHJ1tr5yn5B6M8Ofu3abvFys7QBv13gNdWboWlAxYPpM7NWyNMl2ovXfmLhRmzqwBdSv29keQQGe-jBDFs7A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Endothelial-specific Poldip2 knock-out abrogates inflammatory gene expression in LPS-induced lung injury. LPS injection induced a significant increase in expression of inflammatory markers [Tnfα (A), Iκbα (B)), chemoattractant molecules [Cxcl1 (C), Cxcl2 (D)], and the vascular activation marker [Vcam1 (E)] in Poldip2 EC+/+ mice. This effect was significantly blunted in Poldip2 EC−/− mice. Graphs represent averages ± SEM (n = 5). P < 0.0001, $$$P < 0.001, $$P < 0.01, compared to PBS injected Poldip2 EC+/+ mice, ##P < 0.01, #P < 0.05, compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction).
![Endothelial-specific Poldip2 knock-out decreases cytokine and chemoattractant levels in BAL in LPS-induced lung injury. LPS injection induced a significant increase in cytokine levels [IL-1β (A), IL-6 (B)] and chemoattractant molecules [CXCL1 (C), CXCL2 (D)] in BAL in Poldip2 EC+/+ mice. LPS injection also induced a significant increase in TNFα lung tissue levels (E) in Poldip2 EC+/+ mice. These effects were significantly blunted in Poldip2 EC−/− mice. Graphs represent averages ± SEM (BAL experiments: n = 5–6 for PBS controls, n = 13 in LPS treated mice; lung tissue experiment: n = 6 for PBS controls, n = 7 and 10 in LPS treated Poldip2 EC−/−, and Poldip2 EC+/+ mice, respectively). P < 0.0001, $$$P < 0.001, $$P < 0.01, compared to PBS injected Poldip2 EC+/+ mice, ##P < 0.01, #P < 0.05, compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/118/11/10.1093_cvr_cvab295/2/m_cvab295f3.jpeg?Expires=1716448650&Signature=qIL8OSSHRWy1eGwff8eLpOvHu56BDTuxFjQt12aIdSq3JUuHpEDFCGmr-DgYlUp7TPNgIe2crunqoljgYp2L7KgxlSbJCAozMgiqP7hh5AijRCq-YBni0OaJgT0ZzHx8lua5SZqLkjrULJBSwS5uzAxHISGRiUrIsiO72rB3DdZWgO8yQ0u6WOU8DAD~crcXS0bw5127s-At0qFKhtJcGi0z2TZ8fgex4abuB01coOg35W0MS~fByncG07qhP~zy~MmGOroB8bQGMSbgEIhoS810CS6dyiYdv5aR~EIksYLMca0YzJ6McIVsFpAql1IqxNsFQJBGLMtwvUJs3oOClA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Endothelial-specific Poldip2 knock-out decreases cytokine and chemoattractant levels in BAL in LPS-induced lung injury. LPS injection induced a significant increase in cytokine levels [IL-1β (A), IL-6 (B)] and chemoattractant molecules [CXCL1 (C), CXCL2 (D)] in BAL in Poldip2 EC+/+ mice. LPS injection also induced a significant increase in TNFα lung tissue levels (E) in Poldip2 EC+/+ mice. These effects were significantly blunted in Poldip2 EC−/− mice. Graphs represent averages ± SEM (BAL experiments: n = 5–6 for PBS controls, n = 13 in LPS treated mice; lung tissue experiment: n = 6 for PBS controls, n = 7 and 10 in LPS treated Poldip2 EC−/−, and Poldip2 EC+/+ mice, respectively). P < 0.0001, $$$P < 0.001, $$P < 0.01, compared to PBS injected Poldip2 EC+/+ mice, ##P < 0.01, #P < 0.05, compared to LPS injected Poldip2 EC+/+ mice (two-way ANOVA, with Tukey’s correction).
3.3 Reduced Poldip2 levels prevent TNFα-induced increase in endothelial permeability via a RhoA-associated pathway
Leucocyte extravasation into the inflamed lung tissue is a complex process that starts with attachment of leucocytes to the endothelium.33 However, for the leucocytes to enter the extravascular space, the endothelium must become permeable, allowing transmigration.33 We therefore sought to determine if decreased Poldip2 also affected endothelial permeability using an in vitro collagen-biotin permeability assay. Treatment with TNFα resulted in formation of intercellular gaps in HLMEC monolayers, as can be seen with increased binding of FITC-avidin to the exposed bottom of the dish coated with collagen-biotin (Figure 4A, top). In the case of HLMECs pre-treated with siPoldip2, however, the extent of FITC-avidin binding was significantly lower (5 ± 0.5-fold in siPoldip2 vs. 17.5 ± 3-fold in siControl treated cells after TNFα), an indication of decreased gap formation and reduced endothelial permeability (Figure 4A, bottom), as quantified in Figure 4B. We confirmed these findings using TEER measurements, which demonstrated a significant drop in resistance after TNFα treatment that was not present in siPoldip2 pre-treated cells (Figure 4C). These results indicate a potential role of decreased Poldip2 expression in the maintenance of intercellular junctions.
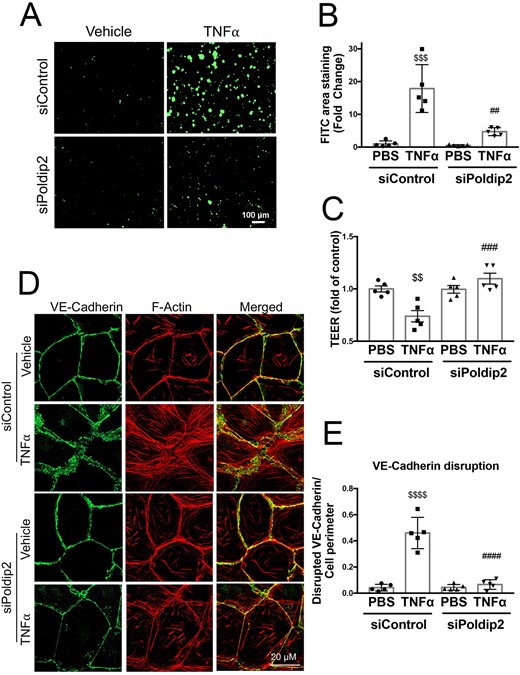
Poldip2 knock-down reduces endothelial permeability and preserves junctional organization in response to inflammation. (A) TNFα treatment induced intercellular gap formation in HLMECs, as assessed by the increased binding of FITC-avidin to collagen-biotin coated dishes. Poldip2 knock-down diminished gap formation in response to TNFα. Images quantified in (B) represent averages ± SEM (n = 5). $$$P < 0.001 compared to siControl PBS, ##P < 0.01 compared to siControl TNFα (two-way ANOVA, with Tukey’s correction). (C) TEER results showing significant decrease in endothelial resistance in siControl treated cells after TNFα treatment compared to siControl and PBS-treated cells. This decrease was not observed in siPoldip2 treated cells (n = 5), $$P < 0.01 compared to PBS siControl treated cells, ###P < 0.001 compared to TNFα and siControl treated cells (two-way ANOVA, with Tukey’s correction). TNFα induced disruption of VE-cadherin at the cell–cell junction along with actin disorganization and stress-fibre formation in HLMECs, while knocking down Poldip2 prevented these effects (D). Images quantified in (E) represent averages ± SEM (n = 5). P < 0.0001 compared to PBS siControl treated cells, ####P < 0.0001 compared to TNFα siControl treated cell (two-way ANOVA, with Tukey’s correction).
VE-cadherin is one of the main junctional proteins in endothelial cells with an established role in the maintenance of the endothelial barrier in the lung.34 To assess junctional integrity, we examined VE-cadherin distribution in the presence and absence of Poldip2 knock-down.35 As is evident in Figure 4D and E, after TNFα treatment, VE-cadherin staining at the cell–cell junctions was dramatically disrupted and depletion of Poldip2 nearly abolished this disruption. Of interest, siPoldip2 also resulted in redistribution of actin fibres along the border of the cell and prevented TNFα-induced stress-fibre formation (Figure 4D).
To investigate potential mechanisms responsible for the preservation of the endothelial barrier upon Poldip2 depletion, we performed an RNASeq experiment followed by Reactome analysis in HLMECs stimulated with TNFα after transfection with siControl or siPoldip2. Interestingly, among all the pathways identified in the analysis, the most significantly affected by Poldip2 knock-down after TNFα treatment were pathways involving Rho GTPases (Figure 5A and Supplementary material online, Table S2). Rho GTPases and RhoA activity, in particular, have been previously linked to increased endothelial permeability.34 To determine if Poldip2 knock-down affected RhoA activity, we performed a G-LISA assay in RBMECs. TNFα treatment caused a significant increase in activation of RhoA in siControl treated cells, but not in siPoldip2 treated cells (1.29 ± 0.01 vs. 0.94 ± 0.05 relative RhoA activity after TNFα) (Figure 5B), while total RhoA levels remained unchanged (Figure 5B and Supplementary material online, Figure S4). Furthermore, treatment with siPoldip2 resulted in a redistribution of active-RhoA staining away from the cell periphery with a distinct perinuclear localization (Figure 4C and D) both in the presence and absence of TNFα. These results suggest that Poldip2 knock-down affects both activity and availability of active-RhoA near cell junctions.
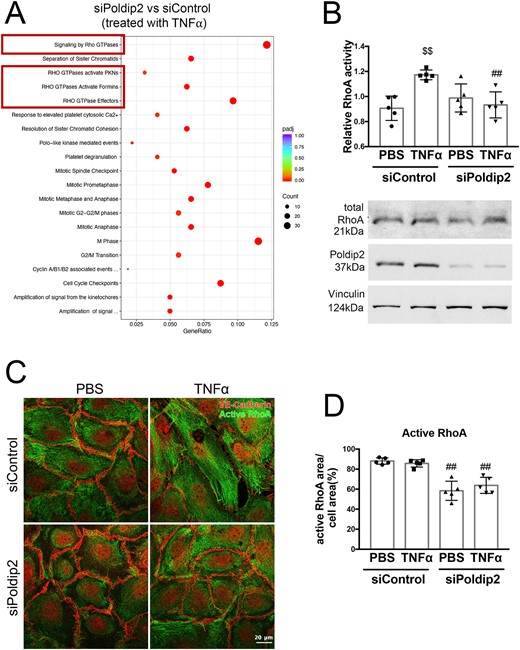
Poldip2 knock-down alters RhoA activity and localization. (A) Reactome analysis of RNASeq data showing the most highly significantly affected pathways when comparing siPoldip2 vs. siControl treated HLMECs after TNFα treatment (Rho GTPase pathways are highlighted in the red boxes) (n = 3). (B) TNFα treatment induced RhoA activity in siControl but not in siPoldip2 treated RBMECs, as measured in G-LISA assays. The graph represents averages ± SEM (n = 5), $$P < 0.01 compared to PBS siControl treated cells, ##P < 0.01 compared to TNFα and siControl treated cells (two-way ANOVA, with Tukey’s correction). Representative western blot showing unchanged total RhoA protein expression and efficiency of Poldip2 knock-down. Vinculin served as a loading control (see Supplementary material online, Figure S4 for Western blot quantification). (C) Immunofluorescence assay of active-RhoA, shown as green staining. Active-RhoA was localized to the entire cell area in siControl treated HLMECs with siPoldip2 shifting active-RhoA staining centrally. VE-cadherin staining (red) detects cell junctions. Images quantified in (D) represent averages ± SEM (n = 5), ##P < 0.01 compared to corresponding siControl treated cells (two-way ANOVA, with Tukey’s correction).
4. Discussion
In this study, we identify endothelial Poldip2 as a previously unrecognized regulator of the vascular response to inflammatory stimuli. We show that loss of Poldip2 prevents VE-cadherin disruption at the endothelial cell border and the subsequent increase in permeability in response to TNFα, likely in a Rho pathway-dependent manner. Preservation of the endothelial barrier is also evident in vivo, because endothelial-specific deletion of Poldip2 prevented the infiltration of all major types of inflammatory cells into the BAL and lung tissue following a challenge with LPS. This led to an overall lower inflammatory response, as reflected in a reduction in cytokine induction and a partial preservation of normal body temperature. Our data suggest that Poldip2 may be an important novel player in diseases in which the endothelial barrier is compromised, such as ALI or sepsis.
Sepsis is a life-threatening condition that develops due to a dysregulated host response to infection.1 Sepsis starts from a pathogen, with bacterial components (i.e. LPS) and products of cellular breakdown entering the bloodstream and endothelial cells being one of the first cell-types to encounter and respond to the insult. Upon stimulation, endothelial cells express adhesion molecules, vasoactive compounds, inflammatory cytokines, and chemoattractants, thus promoting thrombus formation and immune cell recruitment at the site of infection.36 Locally, endothelial activation aids in isolating and eliminating the source of infection. Systemic endothelial activation, however, may result in microvascular thrombosis and increased capillary permeability, leading to tissue hypoxia and eventually to tissue damage. Endothelial activation may be further enhanced by circulating cytokines produced by immune cells (such as TNFα, IL-1β, and IL-6).37 It has been suggested that endothelial activation and damage can facilitate the disruption of the alveolar-capillary barrier, leading to excess fluid extravasation, an important part of sepsis-induced ALI.38
Our previous studies indicated an important role of Poldip2 in endothelial permeability and activation in stroke39 and sepsis models.40 Heterozygous deletion of Poldip2 was protective against increased blood–brain barrier permeability in stroke39 and sepsis40 as well as against LPS-induced lung oedema.16 The whole-body nature of the knock-out, however, did not allow us to discern the role of the endothelium in the observed phenomena. In this study, we now provide concrete evidence that endothelial Poldip2 plays a critical role in barrier function and that endothelial-specific reduction in Poldip2 is protective against LPS-induced ALI. The lack of significant improvement in mortality with endothelial-specific Poldip2 knock-out as opposed to that seen in the whole-body heterozygotes could be potentially explained by the additive effect of the other cell-types (i.e. immune cells) in the heterozygotes.16 The contribution of other cell-types to the protective phenotype of whole-body heterozygous knock-out is the subject of ongoing studies. It is intriguing, however, that we still observe changes in parameters associated with sepsis severity (such as body temperature drop,30 TNFα expression, and protein levels of IL-1β and IL-641) indicating the crucial role endothelial activation plays in the downward-spiral of sepsis. It is also possible that the dose of LPS in our study was too high to discern more subtle differences in mortality potentially present in the cell-type-specific knock-out as compared to the whole-body knock-out.
In our previous work, we demonstrated that siRNA-mediated Poldip2 knock-down reduced endothelial activation in response to LPS via inhibition of NF-κB40 nuclear translocation and resulted in a decrease in VCAM1 expression.16 Those changes are likely responsible for decreased cytokine expression by the endothelial cells as well. We also showed that Poldip2 knock-down preserved brain endothelial cell monolayer integrity after LPS treatment;40 however, the exact mechanism responsible for this effect remained unclear. The RhoA/ROCK pathway, a well-established player in intercellular permeability,42 has been previously connected to NF-κB activation43 and could provide a potential link between generalized inflammation (characterized among other things by the elevated levels of TNFα)41 and the cytoskeletal changes responsible for intercellular gap formation and increased endothelial permeability. Here, we demonstrate in vitro that decreased Poldip2 results in accumulation of active-RhoA in the centre of the cell and decreased overall RhoA activity, reducing its availability for cellular contraction and junctional disassembly upon TNFα treatment. This leads to preservation of adherens junctions, reduction of stress-fibre formation, and reorganization of actin fibres during the inflammatory state caused by sepsis, as modelled in this study by the addition of TNFα in vitro. These findings are consistent with previous work showing that Poldip2 overexpression in vascular smooth muscle cells activates RhoA and that Poldip2 can activate the Rho-GEF, Ect2.44,45 Of interest, Ect2-regulated RhoA activity has been linked to leucocyte diapedesis;46 however, further work will be necessary to fully delineate the pathways by which Poldip2 regulates Rho-dependent signalling.
Other possible mechanistic pathways deserve consideration as well. While our RNAseq data (confirmed by PCR) suggest that knocking down Poldip2 has only a slight effect on TNF receptor expression (Supplementary material online, Figure S5), possibilities such as this also require additional investigation. Additionally, Nox4 (which is known to be regulated by Poldip244) could play a role in downstream effects of decreased Poldip2 and requires further studies.
Endothelial-specific knock-down of Poldip2 opens new possibilities for therapeutic interventions in patients with sepsis. Due to the molecular consequences of Poldip2 knock-down, targeting endothelial Poldip2 could be useful not only in sepsis caused by gram-negative bacteria, but also in other conditions characterized by excessive endothelial activation, such as atherosclerosis47 or COVID-19 infection.48 The direct proximity of the endothelium to the bloodstream makes it easily accessible for viral particles, siRNA, and various circulating compounds. A reduction of Poldip2 levels primarily in the endothelium, would therefore avoid potential off-target effects.
One of the main limitations of our model is the presence of Poldip2 knock-down or deletion prior to the insult, which would be impossible in the clinical setting of sepsis. Whether or not Poldip2 knock-down would also be able to offer similar protection after the onset of sepsis requires further investigation. Nevertheless, this study offers a proof-of-principle and adds to our understanding of the molecular mechanisms involved in the beneficial effects of reduced Poldip2 in endothelium during the LPS-induced lung injury.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
E.V.D., S.J.F. and K.K.G. conceived and designed the study. E.V.D. and S.J.F. performed the majority of experiments and analysed data. E.V.D, S.K., K.K.G. wrote and edited the manuscript. G.J. helped with histology and immunohistochemistry. S.K and H.J. performed endothelial and smooth muscle expression profile analysis. M.S.H. and H.Q. performed bronchoalveolar lavage experiments. H.C.W. and Z.O. performed FACS experiments. K.W. performed TEER experiments. B.L. helped with mouse breeding colony maintenance and strategies, provided technical assistance on assays, and helped edit the manuscript. A.V. provided guidance and assistance on RhoA activity assays and edited the manuscript. A.K. provided guidance on the VE-cadherin experiments. M.S.H. helped with conceptual design, and helped to edit the manuscript. K.K.G. provided expertise, helped with conceptual design, and helped to edit the manuscript.
Acknowledgement
Experiments including immunocytochemistry and immunofluorescence were performed in part through the use of the Microscopy in Medicine (MiM) Core in the Division of Cardiology at Emory University.
Funding
E.V.D. is supported by National Institute of Health [grant number F32-HL151133-504 01]. M.S.H. is supported by American Heart Association [grant number 17SDG33410777]. K.K.G., B.L. and M.S.H are supported by National Institute of Health [grant number HL152167].
Data Availability
The data underlying this article are available in GenBank at GSE165410 and GSE165939.
Translational perspective
Sepsis is a life-threatening condition that develops due to a dysregulated host response to infection and is often accompanied by sepsis-induced lung injury. In this study, we identify endothelial Poldip2 as a novel regulator of the vascular response to inflammatory stimuli. We show that endothelial-specific loss of Poldip2 preserves endothelial barrier in in vivo sepsis model preventing neutrophil infiltration into the lungs. This in turn leads to an overall lower inflammatory response and decreased sepsis severity. Therefore, endothelial-specific knock-down of Poldip2 opens new possibilities for therapeutic interventions in patients with sepsis.
References
Author notes
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}