




Abstract
To determine whether dietary fat intake during childhood affects the later risk of developing breast cancer, we fed prepubertal rats between post-natal days 5 and 25 a low (16% energy) or high-fat (39% energy) diet composed mainly of n -6 or n -3 polyunsaturated fatty acids (PUFAs) originating either from corn oil or menhaden oil, respectively, in the ratios of 16–17:1 ( n -6 PUFA diets) or 2–3:1 ( n -3 PUFA diets). We also examined whether changes in risk are associated with perturbations in biological processes previously linked to fatty acid intake and breast cancer. Mammary tumorigenesis was induced by treating 50-day-old rats with the carcinogen 7,12-dimethylbenz[ a ]anthracene. When compared with the reference low-fat n -6 PUFA diet, prepubertal exposure to the low-fat n -3 PUFA diet decreased, whereas a high-fat n -3 PUFA diet increased mammary tumor incidence; the high-fat n -6 PUFA diet had no effect. Both the low and high-fat n -3 PUFA diets induced mammary epithelial differentiation by reducing the number of terminal end buds (TEBs) and increasing the presence of lobulo-alveolar structures. They also increased lipid peroxidation and reduced cyclooxygenase-2 activity. Prepubertal exposure to the low-fat n -3 PUFA diet increased apoptosis, determined using TUNEL assay, and reduced cell proliferation, determined using PCNA staining. In marked contrast, prepubertal exposure to the high-fat n -3 PUFA diet induced cell proliferation and inhibited apoptosis in the TEBs and lobular structures. The latter is consistent with the finding that pAkt, a survival factor that inhibits apoptosis, was elevated in their mammary glands. In summary, although prepubertal exposure to a low-fat n -3 PUFA diet reduced later mammary tumorigenesis in rats, high levels of this fatty acid can have adverse effects on the prepubertal mammary gland and increase subsequent breast cancer risk.
Introduction
Cohort studies in humans do not consistently support a link between breast cancer and total fat intake ( 1 , 2 ). However, as people emigrate from countries of low-fat consumption and low breast cancer risk to countries of high-fat consumption and high breast cancer risk, their risk to breast cancer increases. The risk of children born to immigrants in the high-fat consumption countries is as high as that of the natives ( 3 ), suggesting that exposures early in life play a critical role in affecting breast cancer susceptibility. This interpretation is in agreement with the proposal of Colditz and Frazier ( 4 ) that the risk of developing breast cancer is determined before the birth of the first child. Their proposal is based on a mathematical model that builds on the Pike model ( 5 ) and the Nurses Cohort ( 6 ), and is supported by the fact that in humans as well as in laboratory animals the mammary glands develop during the adolescent growth spurt ( 7 , 8 ). Mammary gland development is completed during the first full-term pregnancy and nursing. However, only a handful of studies have examined whether childhood dietary exposures modify later breast cancer risk. Difficulties in obtaining accurate dietary information retrospectively, particularly regarding childhood dietary exposures, is a major problem for these studies.
There are a few human studies that investigated the effects of dietary fat intake during childhood on breast cancer risk. The results of these studies have shown that higher consumption of eggs and vegetable fat is inversely associated with breast cancer risk or proliferative benign breast disease ( 9 , 10 ). In addition, an inverse relationship between adolescent fat intake from milk, cheese and yogurt, and breast cancer risk has been reported ( 11 ). In one case–control study, however, no associations between childhood diet and breast cancer risk were found ( 12 ).
The type of fat most likely to reduce breast cancer risk is n -3 polyunsaturated fatty acid (PUFA) present in fish and other marine products as well as some vegetable oils (e.g. canola and linseed oils). Some human studies have provided evidence suggesting that diets high in n -3 PUFAs are protective against breast cancer ( 13 , 14 ), including the findings in Japanese and Norwegian women who consumed five or more servings of fish per week ( 15 , 16 ). These data are consistent with the findings showing that n -3 PUFA levels in adipose tissue are inversely linked to breast cancer risk ( 13 , 17 ). However, some studies indicate no change or a significant increase in breast cancer risk among women who consumed high levels of n -3 PUFAs ( 18 – 21 ). Data from animal studies have also generated conflicting data: some studies show that dietary intake of n -3 PUFAs inhibits tumorigenesis ( 22 ), whereas others do not support these data ( 23 , 24 ). These inconsistent findings may reflect differences in the levels of n -3 PUFAs fed to the animals, i.e. the exposures ranged from physiological to pharmacological. Further, it is not known whether prepubertal dietary exposure to n -3 PUFAs affects later susceptibility to breast cancer.
We report here the findings of an animal study that investigated the effects on breast cancer risk of a prepubertal exposure to n -3 PUFA diets at a n -6: n -3 PUFA ratio of 2–3:1. The n -6: n -3 PUFA ratio in the n -6 PUFA diets was 16–17:1, mimicking Western diets that contain very low levels of n -3 PUFAs. Our ancestors' diet, in contrast, was composed of these fatty acids in the ratio of 1:1. Two different n -3 and n -6 PUFA exposure levels were used, but to keep the n -6: n -3 PUFA ratios constant, the diets were either low or high in fat. The results indicated that a low-fat n -3 PUFA diet reduced, whereas a high-fat n -3 PUFA diet increased mammary tumorigenesis, when compared with a low-fat n -6 reference diet or prepubertal high-fat n -6 PUFA diet. Since both n -3 PUFA diets reduced the number of epithelial targets for malignant transformation, i.e. terminal end buds (TEBs), the expression of cyclooxygenase (COX)-2 and increased lipid peroxidation, these changes are unlikely to explain the opposing effects of the low- and high-fat diets on breast cancer risk. The protective effects of a low-fat n -3 PUFA might have been caused by decreased cell proliferation and increased apoptosis in the TEBs. Prepubertal exposure to a high-fat n -3 PUFA diet, in contrast, increased cell proliferation and inhibited apoptosis in the TEBs. Since prepubertal exposure to a high-fat n -6 PUFA did not affect mammary tumorigenesis, high levels of n -3 PUFAs rather than a high-fat diet itself probably led to an increase in susceptibility to breast cancer.
Materials and methods
Dietary exposures
Timed–pregnant Sprague–Dawley rats were obtained from Charles River (Wilmington, MA) on gestation day 10 and fed semipurified AIN93 (American Institute for Nutrition) diet upon arrival. Animals were housed individually in a temperature- and humidity-controlled room at the Georgetown University Resource Animal Facility under a 12-h light–dark cycle. All animal procedures were approved by the Georgetown University Animal Care and Use Committee, and the experiments were performed following the guidelines of National Institutes of Health for the proper and humane use of animals in biomedical research.
One day after the animals gave birth, male rat pups were killed. The female pups from 3–4 litters were pooled and divided into 2–3 groups of 10 pups each per nursing dam. Nursing dams were either kept on an AIN93 diet or switched to one of the three experimental diets when the offspring were 5 days old. The dietary groups were: (i) a low-fat n -6 PUFA reference diet (AIN93 diet); (ii) a high-fat n -6 PUFA diet; (iii) a low-fat n -3 PUFA diet; and (iv) a high-fat n -3 PUFA diet. Two independent studies were performed, each with ∼35–40 rat pups per group. The studies were identical except that in the second study, one of the four dietary exposures was eliminated (high-fat n -6 PUFA), because it did not cause significant difference in any of the endpoints studied when compared with low-fat n -6 PUFA reference group.
The diets were prepared commercially by Bioserv (Frenchtown, NJ). Low-fat diets contained 16% energy from fat and high-fat diets contained 39% energy from fat. The n -6 and n -3 PUFAs were derived from corn oil or menhaden oil, respectively. Both corn oil and menhaden contain small amounts of saturated (13–30% of total feed weight) and monounsaturated fats (27%), but corn oil is high in n -6 PUFAs (54% of total weight) and menhaden oil in n -3 PUFAs (27%). Menhaden oil contains 9% docosahexanoic acid (DHA) and 13% eicosapentanoic acid (EPA), and humans can be exposed to it when they consume, e.g. poultry, live-stock or farmed fish (the poultry industry is currently the largest user of menhaden meal, followed by the turkey, swine and ruminant industries). There are some concerns that very high levels of DHA can have adverse effects, such as slow down of blood coagulation, and thus an upper recommended daily DHA limit is set to 3 g.
The AIN-93 rodent laboratory diet; i.e. the low-fat n -6 PUFA reference diet, contained 65 g of corn oil and 5 g of menhaden oil per kg feed. The high-fat n -6 PUFA diet contained 175 g of corn oil and 15 g of menhaden oil per kg feed, the low-fat n -3 PUFA diet 35 g of corn oil and 35 g of menhaden oil per kg feed, and the high-fat n -3 PUFA diet 120 g of corn oil and 70 g of menhaden oil per kg feed. The estimated daily DHA exposure in the high-fat n -3 PUFA group was 20 mg. Absolute levels of menhaden and corn oil varied in the low and high-fat diets, but the ratio of n -6: n -3 PUFAs remained constant (16–17:1 in n -6 PUFA diets and 2–3:1 in n -3 PUFA diets). Protein, mineral and vitamin contents of the low- and high-fat n -3 and n -6 PUFA diets were similar, and the diets were made isocaloric by adjusting the energy content with fiber (contains no energy). The diets have been described in more detail in our previous paper ( 25 ).
Between days 5 and 15, dietary exposures occurred via the milk from the dams that closely reflects the dietary intake of dams; i.e. fatty acid composition of the milk is similar to the diet a dam consumes ( 26 ). Between days 15 and 25, the pups were also exposed to the diets through the consumption of food pellets. All animals were switched to the low-fat n -6 PUFA reference diet on post-natal day 26, when the offspring were weaned.
Carcinogen exposure
When the rats were 50 days of age, they were administered 10 mg of the mammary carcinogen 7,12-dimethylbenz[ a ]anthracene (DMBA) (Sigma Chemical, St Louis, MO) by oral gavage. The low-fat n -6 PUFA control group was composed of 52 rats, the high-fat n -6 PUFA group had 25 rats, the low-fat n -3 PUFA group 55 rats and the high-fat n -3 PUFA group 52 rats. This DMBA dose produces a sufficient number of tumors to facilitate detection of either an increase or decrease in tumorigenicity ( 27 ). Carcinogen was dissolved in peanut oil and given in a volume of 1 ml. Animals were checked weekly for mammary tumors by palpation. Tumor growth was measured using a caliper, and the length, width and height of each tumor were recorded. Animals were killed when the tumor burden was ∼10% of total body weight. All remaining animals, including those that did not develop tumors, were killed 18 weeks after DMBA administration.
Endpoints for data analysis were time to tumor appearance (tumor latency), the number of tumors per animal (tumor multiplicity) and proportion of animals that developed at least one tumor per group (tumor incidence).
Mechanisms mediating the effects of prepubertal PUFA diets on mammary tumorigenesis
To identify the changes induced by prepubertal dietary PUFA exposures that might have mediated the effects of these diets on mammary tumorigenesis, we obtained serum and the third thoracic and fourth abdominal mammary glands from 3- or 8-week-old rats that were fed different PUFA diets during prepuberty but not treated with a carcinogen. The glands were either processed for wholemounts (see below), snap-frozen in liquid nitrogen and stored at −80°C or fixed in formalin. For the latter, the glands were put into a histology cassette and fixed in 10% buffered formalin overnight. Fixed glands were embedded in paraffin, and 5 μm sections were cut and mounted onto slides. Ten to twelve animals per group and age were used for each assay, and they were obtained from both the independent sets of experiments.
Lipid peroxidation assay
The levels of lipid peroxidation in the serum was determined using a lipid hydroperoxide assay kit from Cayman Chemical (Ann Arbor, MI). In brief, lipid hydroperoxides were extracted from serum into chloroform and stored at −80°C; the assay was performed subsequently following the manufacturer's instructions. A standard curve was generated for each assay and used to calculate the hydroperoxide values of each sample. Each sample was run in triplicate and the mean value was calculated.
Whole mount analysis
The harvested fourth abdominal glands were stretched on a microscopic slide and placed overnight in mammary gland fixative solution which contained 25% glacial acetic acid (EM Science, Gibbstown, NJ) and 75% ethanol (Fisher Scientific). Mammary glands were then washed in 70% ethanol for 1 day and stained in carmine aluminum overnight. The next day, glands were again washed in 70% ethanol overnight, followed by washing them in 95 and 100% ethanol, and then placed in xylene (Fisher Scientific). Glands were covered with Permount (Fisher Scientific) and coverslips were placed on top.
Mammary wholemounts were analyzed blindly by two independent investigators using an Olympus Vanox-S microscope (Olympus America). The structures of the mammary glands were evaluated in a double-blind manner. The presence of alveolar buds (ABs) in 3-week-old pups and lobulo-alveolar units (LAUs) in 8-week-old rats was evaluated using a 5-point scale (0, absent; 1, low; 2, low–moderate; 3, moderate; 4, moderate–high; 5, high). We have validated and successfully used this scale in our earlier studies ( 27 ). The number of TEBs per total mammary gland was also determined.
TUNEL staining
The paraffin embedded sections were used to measure apoptosis with the ApopTag Kit (Serologicals, Norcross, GA) following the manufacturer's protocol. In brief, the sections were deparaffinized and rehydrated in a series of graded alcohols. The sections were pretreated with 20 μg/ml of Proteinase K for 15 min. Endogenous peroxidases were quenched with 3% H 2 O 2 for 5 min. The sections were washed with equilibration buffer (ApopTag Kit) and incubated with the terminal deoxynucleotidyl transferase TdT enzyme that adds digoxigenin-labeled nucleotides to the 3′-OH ends of the DNA. The reaction was stopped and the sections were incubated with a digoxigenin peroxidase conjugate. The sections were washed, incubated with the peroxidase substrate for 6 min, rewashed and counterstained with 0.5% methyl green (Vector Laboratories, Burlingame, CA) for 10 min, and finally washed in water followed by washes in 100% butanol and dehydrated. Staining in 1000 ductal, 1000 LA and 1000 TEB cells were counted. Apoptotic index was determined by calculating the percentage of cells that were apoptotic through both positive staining and histological evaluation. All slides were blindly evaluated by two independent investigators.
Immunohistochemistry
COX-2 and proliferating cell nuclear antigen (PCNA) protein expression were measured by immunohistochemistry in the paraffin embedded sections. The sections were deparaffinized in two 5 min changes of xylene and rehydrated through graded alcohols to distilled water. The sections were microwave heated for antigen retrieval in antigen retrieval solution (Vector Laboratories) for 20 min, incubated with 3% H 2 O 2 for 15 min to block endogenous peroxidases and washed in phosphate-buffered saline containing 0.1% Triton X-100 for 20 min to reduce non-specific binding. Next, the sections were blocked with Vectastain blocking serum from the Vectastain Elite ABC Kit (Vector Laboratories) for 20 min and incubated with the primary antibody [1:250, COX-2 (M-19)], or PCNA of dilution 1:500 (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. After several washes, the sections were treated with a biotinylated anti-goat IgG from the Vectastain Elite ABC Kit (Vector Laboratories) for 30 min at room temperature. After three washes, the sections were treated with an avidin and biotinylated horseradish peroxidase (HRP) complex from the Vectastain Elite ABC Kit (Vector Laboratories) for 30 min at room temperature. The sections were then washed and stained with the chromogen, 3,3′-diaminobenzidine (DAB) (Vector Laboratories) for 1 min, washed and counterstained with Vector's hematoxylin QS nuclear counterstain (Vector Laboratories) for 45 s.
The slides were analyzed blindly by two independent investigators. Cell types were divided into ductal, LA or TEB cells. One thousand of each cell type were counted per slide and the number of cells that stained positive was recorded. The percentages of COX-2 or PCNA expressing cells per total number of cells counted per structure were used when analyzing the data.
Western blots
Akt phosphorylation in the mammary glands was assessed using western blot assays. The mammary glands were homogenized in buffer containing 25 mM Tris–HCl, 2 mM MgCl 2 , 1 mM EDTA, 1% Triton X-100, 10% glycerol, 1 mM DTT, 1 mM PMSF, 1 μM leupeptin, 1 μM pepstatin and 1 μg/ml aprotinin. Homogenates were centrifuged and the protein extract collected from the supernatant. NuPAGE sample buffer with 10% β-3 mercaptoethanol was added to 30 μg of protein extract, boiled for 5 min at 100°C and loaded onto a NuPAGE 12% Bis–Tris gel (Invitrogen Life Technologies, Carlsbad, CA). The gels were run at 150 V for 1 h and transferred for 2 h. Membranes were washed with TBST (0.1 M Tris-HCl, 1.5 M NaCl and 0.5% Tween-20) and blocked in 5% bovine serum albumin (BSA) in TBST for 2 h at room temperature. After blocking, membranes were incubated with the phospho-Akt antibody Ser473 (1:1000, Cell Signaling Technology, Beverly, MA) overnight at 4°C. Next, membranes were washed with TBST and incubated with the secondary anti-rabbit IgG HRP antibody (1:5000, Amersham Pharmacia Biotech, Piscataway, NJ) for 1 h at room temperature. Membranes were washed with TBST and developed using ECL (Amersham Pharmacia Biotech). For total Akt levels, blots were stripped in a solution containing 2% SDS, 6% 1 M Tris (pH 6.8) and 0.7% β-mercaptoethanol for 20 min. Blots were washed several times with TBST and reblocked in 5% milk in TBST for 1 h. Stripping efficiency was checked by incubating the membranes with fresh secondary antibody for 1 h and developing the blot to check for the presence of bands. Membranes were then washed and incubated with primary antibody for total Akt (1:1000, Cell Signaling) overnight at 4°C. Membranes were washed again with TBST, incubated with secondary anti-rabbit HRP antibody (1:5000), washed and developed. Beta-actin was used to check for equivalency of protein loading (1:5000, Sigma Chemical). Fold differences were calculated by normalizing phophorylated Akt to total Akt and then β-actin.
Statistical analysis
Results for the data obtained on food consumption, body weight, some mammary tumor end-points (latency and multiplicity), lipid peroxidation, mammary gland morphology, TUNEL staining, immunohistochemistry and western blots were analyzed with SigmaStat software using one- or two-way analysis of variance (ANOVA), separately at 3 and 8 weeks of age ( 28 ). Where appropriate, between-group comparisons were done using Tukey's multiple comparisons test. To determine tumor incidence in each group, the time to tumor presentation was measured as the number of weeks from DMBA exposure to the time the first tumor per animal could be palpated. Estimations of tumor presentation were calculated by the methods developed by Kaplan and Meijer ( 28 ). Differences among the treatment arms were tested using the log rank test ( 29 ). We also determined differences in final tumor incidence among the groups using χ 2 test. The differences were considered significant if the P -value was <0.05. All probabilities are two-tailed.
Results
Effects of diet on mammary tumorigenesis
Mammary tumor incidence was significantly lower in the rats fed the low-fat n -3 PUFA diet during prepuberty, compared with the rats fed the reference diet ( P = 0.03) ( Figure 1A ). In contrast, mammary tumor incidence was significantly higher in the rats fed the high-fat n -3 PUFA diet, compared with the rats fed the reference diet ( P = 0.0008). Thus, 18 weeks after DMBA administration, 41% of the animals fed the low-fat n -3 PUFA diet had developed mammary tumors; also, 89% of the animals fed the high-fat n -3 PUFA diet, 67% of the animals fed the low-fat n -6 PUFA reference diet and 75% of the animals fed the high-fat n -6 PUFA diet (χ 2 = 27.14, P < 0.0148) showed incidence of tumor. No differences in tumor incidence or other tumorigenesis end-points were noted between the low-and high-fat n -6 PUFA diets.
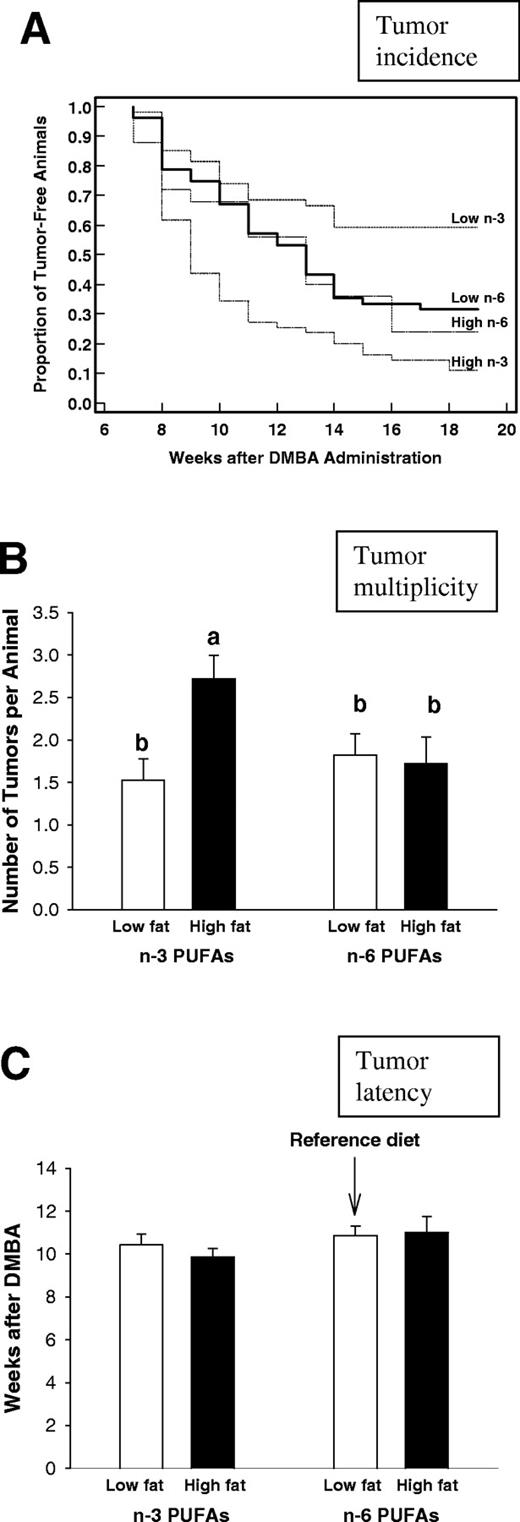
Mammary tumorigenesis in rats exposed to low- or high-fat n -3 or n -6 PUFA diets from post-natal day 5 until day 25. A dose of 10 mg DMBA was administered on day 50 and mammary tumors were monitored weekly by palpation for 18 weeks. ( A ) Mammary tumor incidence was significantly reduced in the prepubertally low-fat n -3 PUFA fed rats ( P < 0.03) and increased in the high-fat n -3 PUFA fed rats ( P < 0.0008), when compared with the reference group (Kaplan–Meier survival analysis and log rank test). ( B ) Tumor multiplicity, measured as the number of tumors per animal at the time of killing, was significantly lower in the low-fat n -3 PUFA fed rats than in the high-fat n -3 PUFA fed rats (one-way ANOVA, P = 0.007; bars markerd with different letters are significantly different from each other at the level P < 0.05 by using a Tukey test). ( C ) Tumor latency, measured as the number of weeks after DMBA administration until the appearance of the first tumor, was not different among the groups. The mean + SEM of all tumors included to the analysis per dietary treatment are shown. The numbers of animals per group were: low-fat n -6 PUFA group: n = 52; high-fat n -6 PUFA group: n = 25; low-fat n -3 PUFA group: n = 55; and high-fat n -3 PUFA group: n = 52.
Tumor multiplicity was significantly different among the different dietary treatments ( P = 0.007) with the rats fed the high-fat n -3 PUFA diet developing significantly more tumors per animal than the rats fed the low-fat n -3 PUFA diet ( P < 0.05) ( Figure 1B ). Tumor latency was not affected by the prepubertal dietary exposures ( Figure 1C ).
Mammary gland morphology
Mammary gland morphology was studied in the wholemounts to determine whether prepubertal n -3 PUFA exposures alter the number of targets for malignant transformation (TEBs) or their differentiation to ABs and further to LAUs. At 3 weeks of age, animals fed the low- or high-fat n -3 PUFA diet had lower numbers of TEBs, compared with the reference or the high-fat n -6 PUFA fed animals ( P < 0.004) ( Figure 2A ). No differences were seen in the density of ABs ( Figure 2B ); these are the predominant epithelial structures in the mammary glands at this age.
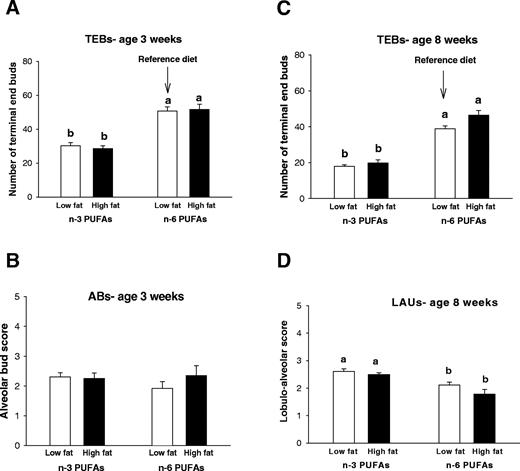
Changes in mammary gland morphology in 3- and 8-week-old rats that were exposed to a low-fat or high-fat n -3 or n -6 PUFA diets from post-natal day 5 until day 25. The number of terminal end buds (TEBs) ( A ) was counted and the density of ABs was scored on a scale from 0–5 at 3 weeks of age ( B ). TEBs and LAUs at 8 weeks of age ( C and D ) were counted or scored. The mean + SEM of 9–12 animals per dietary treatment are shown (except n = 5 in the high-fat n -6 PUFA group). Bars marked with different letters are significantly different from each other ( P < 0.05) using a Tukey test.
At 8 weeks of age, the animals exposed to low- and high-fat n -3 PUFA diets had less TEBs than the reference low-fat n -6 PUFA diet or the high-fat n -6 PUFA diet ( P < 0.001) ( Figure 2C ). In addition, the glands of animals fed the low-fat or high-fat n -3 PUFA diets during prepuberty contained a higher number of LAUs, compared with the animals fed the reference diet ( P < 0.001) ( Figure 2D ). To summarize, the mammary glands of the animals exposed to the n -3 PUFA diets during prepuberty were more differentiated when compared with the reference diet and the high-fat n -6 PUFA diet.
Lipid peroxidation levels in PUFA-fed animals
One of the changes induced by n -3 PUFAs is an increase in lipid peroxidation, which can result in an increase in apoptosis ( 30 ). Lipid peroxidation was examined by measuring lipid hydroperoxide levels in serum. As expected, the animals exposed to the high-fat n -3 PUFA diet had the highest levels of lipid hydroperoxides, whereas the reference diet fed rats had the lowest levels ( P < 0.001) ( Figure 3A ). It is of interest that the increased levels persisted for at least 5 weeks after the dietary exposure. Low-fat n -3 PUFA diet also modestly increased lipid peroxidation, but only at 8 weeks of age ( P < 0.05) ( Figure 3B ). Prepubertally high-fat n -6 PUFA fed rats were not included to measuring this end point.
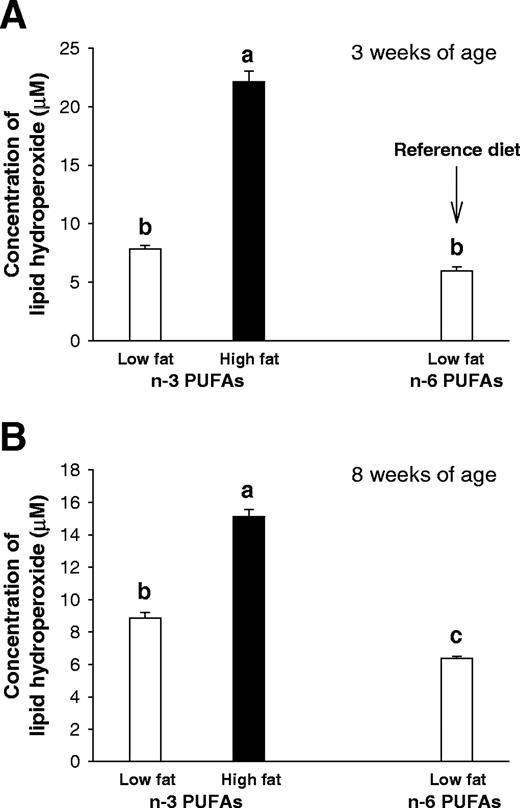
Lipid hydroperoxide levels in rats exposed prepubertally to a low- or high-fat n -3 PUFA or the reference low-fat n -6 PUFA diets. Lipid hydroperoxides, analyzed in the serum at 3 weeks ( A ) and 8 weeks ( B ) were significantly higher in the high-fat n -3 PUFA group than in the low-fat n -3 PUFA or reference group (one-way ANOVA for both 3 and 8 weeks: P < 0.001). Bars marked with different letters are significantly different from each other ( P < 0.05) using a Tukey test. Values represent the mean of 12 animals per group + SEM.
Apoptosis
We used a standard TUNEL assay to measure apoptosis in the mammary glands of 3- and 8-week-old rats. The level of apoptosis was significantly higher in the animals fed the low-fat n -3 PUFA diet compared with the reference low-fat n -6 PUFA diet at both 3 (data not shown) and 8 weeks of age ( P < 0.05) ( Figure 4A, C and E ). In contrast, animals exposed to a high-fat n -3 PUFA diet had a significantly lower level of apoptosis in both the LA structures and the TEBs, compared with the reference group ( P < 0.05).
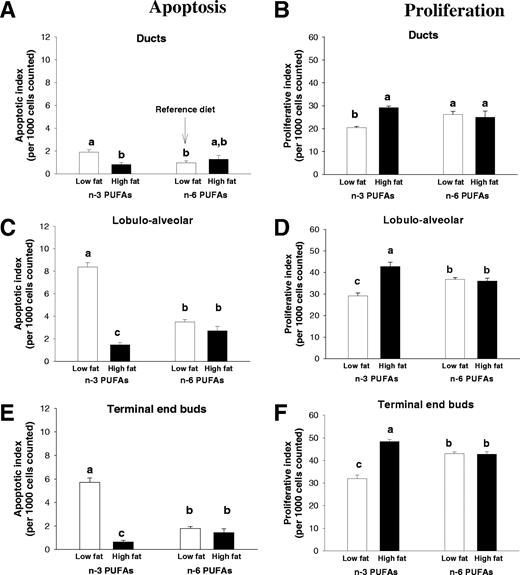
Apoptosis and proliferation in the mammary glands of 8-week-old rats exposed prepubertally to the low- or high-fat n -3 and n -6 PUFA diets. Apoptosis was evaluated via a TUNEL assay and confirmed by histological examination in the ductal cells ( A ), the LA cells ( C ) and the TEB cells ( E ). Data are illustrated as a percentage of apoptotic cells per 1000 cells counted. Proliferation was evaluated via immunohistochemistry for PCNA in the ductal cells ( B ), the LA cells ( D ) and the TEB cells ( F ). Data are illustrated as a percentage of proliferating cells per 1000 cells counted. Values represent the mean of 6–12 animals per group + SEM. Significantly different at the level of P < 0.001 in all three cells types for both apoptosis and proliferation; bars marked with different letters are significantly different from each other ( P < 0.05) using a Tukey test.
Cellular proliferation
Cell proliferation was assessed by measuring PCNA protein expression by immunohistochemistry. We observed significantly higher levels of proliferating cells in the LA structures and TEBs at 3 ( P < 0.001) (data not shown) and 8 ( P < 0.001) ( Figure 4B, D and F ) weeks of age in rats fed the high-fat n -3 PUFA diet compared with animals fed the reference diet. In the rats fed the low-fat n -3 PUFA diet, cell proliferation at week 8 was lower in the ducts, LA structures and TEBs ( P < 0.05) than in the rats fed the reference diet.
Akt expression
In addition to assessing apoptosis using the TUNEL assay, we also determined the levels of phosporylated Akt by western blot. Akt is a survival factor that inhibits apoptosis ( 31 ). Furthermore, previous studies indicate that n -3 PUFAs modulate Akt expression ( 32 ). The results indicated that phospho-Akt levels in the mammary glands of animals exposed to a high-fat n -3 PUFA diet were significantly higher than those exposed to the low-fat n -3 PUFA diet during prepuberty ( P = 0.03) ( Figure 5 ).
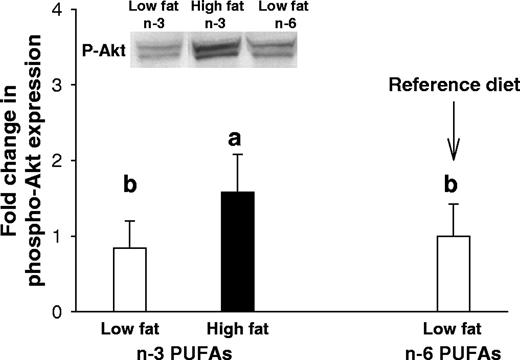
Akt phosphorylation in the mammary glands of 8-week-old rats exposed prepubertally to low- or high-fat n -3 PUFA or the reference low-fat n -6 PUFA diet. Phosphorylated Akt was examined by western blot and data of total Akt and β-actin were normalized for differences in protein loading. Values represent the mean of 12 animals per group + SEM. Akt phosphorylation was significantly higher in the high-fat n -3 PUFA than low-fat n -3 PUFA group; one-way ANOVA: P < 0.028. Bars marked with different letters are significantly different from one another ( P < 0.05).
COX-2 expression
COX-2 is involved in the metabolism of both n -3 and n -6 PUFAs. Consequently, the presence of n -3 PUFAs in the diet reduces the activity of COX-2 by creating a competition for this enzyme. We determined the COX-2 enzyme levels using immunohistochemistry. Both at 3 and 8 weeks of age, COX-2 protein levels were lower in the mammary glands of rats fed either the low- or high-fat n -3 PUFA diets, compared with the rats fed low-fat n -6 PUFA reference diet (week 3: P < 0.05 and week 8: P < 0.05) ( Figure 6 , data obtained at 3 weeks not shown). In particular, in the mammary glands of prepubertally n -3 PUFA fed 8-week-old rats, COX-2 expression was reduced in the TEBs ( Figure 6A ) and LA structures ( Figure 6B ), but not in the ducts ( Figure 6C ).
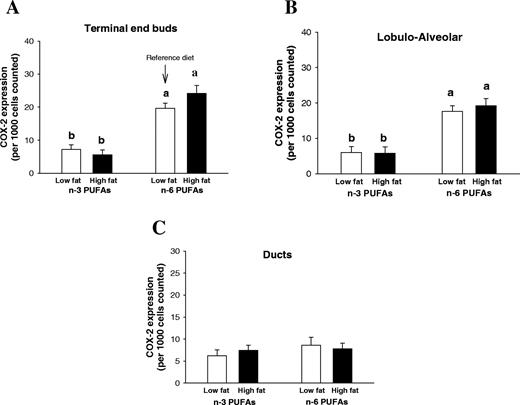
Cox-2 expression in the terminal end buds, TEBs (A) , LAUs ( B ) and ducts ( C ) at the age of 8 weeks in the animals exposed prepubertally to low- or high-fat n -3 PUFA or the reference low-fat n -6 PUFA diets. Data are illustrated as a mean + SEM percentage of cells expressing Cox-2 staining per 1000 cells counted, in 12 rats per group. Cox-2 expression was significantly lower in the n -3 PUFA fed groups than in the reference or high-fat n -6 PUFA fed rats (one-way ANOVA: P < 0.05). Bars marked with different letters are significantly different from each other ( P < 0.05) using a Tukey test.
Discussion
In this study, the effects on mammary tumorigenesis of a prepubertal exposure to a low- or high-fat, n -3 or n -6 PUFA diet, were investigated. Findings in human populations have generated conflicting data as to whether an adult exposure to dietary n -3 PUFAs affects breast cancer risk ( 20 , 21 ). We anticipated that an exposure to n -3 PUFAs early in life would affect later breast cancer risk, since the mammary gland is then undergoing extensive changes and shown to be vulnerable, e.g. to radiation ( 33 , 34 ). The results indicated that rats fed a low-fat n -3 PUFA diet during prepuberty were at a significantly lower risk of developing DMBA-induced mammary tumors than the rats fed a low-fat n -6 PUFA reference AIN93 diet. Rats fed prepubertally a high-fat n -3 PUFA diet, in contrast, exhibited a significantly higher tumor incidence (proportion of rats per group that develop tumors) and multiplicity (number of tumors per animal) than either the control low- or high-fat n -6 PUFA or low-fat n -3 PUFA fed rats.
Mammary glands of the rats fed the low-fat n -3 PUFA diet during prepuberty exhibited several biological changes that may provide protection against breast carcinogenesis. The structure of the mammary epithelial tree was altered in these rats. The number of TEBs was reduced and the density of differentiated LAUs was increased, when compared with a low-fat n -6 PUFA AIN93 laboratory chow. These findings are supportive of the earlier studies showing that a reduced risk of developing breast cancer is associated with a reduction in targets for malignant transformation ( 8 ).
The increase in lipid peroxidation in animals fed the low-fat n -3 PUFA diet is consistent with earlier studies showing that n -3 PUFAs promote lipid peroxidation ( 30 , 35 ). Since this was seen in the 8-week-old rats, i.e. 5 weeks after the diets were switched to semi-purified control AIN93 diet, the early dietary exposures probably altered cell membrane composition permanently. Lipid peroxidation is a process by which PUFAs are oxidized and reactive oxygen species are produced that can then damage DNA ( 36 , 37 ). An increase in lipid peroxidation may have signaled the damaged cells to undergo apoptosis, since the mammary glands of low-fat n -3 PUFA contained more apoptotic cells than the glands of the control rats. Increased apoptosis in their mammary glands is consistent with previous studies showing that n -3 PUFAs induce apoptosis in human colon cancer and leukemia cells lines ( 38 , 39 ). However, since lipid peroxidation was not elevated at 3 weeks, but apoptosis was, other pathways are clearly involved in causing cell death in rats exposed to a low-fat n -3 PUFA diet during prepuberty.
An additional beneficial effect was reduced proliferation of cells in the mammary epithelial structures in the prepubertally low-fat n -3 PUFA fed rats, including in the TEBs. This is in line with findings obtained in vitro and in vivo indicating that n -3 PUFAs reduce cell proliferation ( 40 ). Since the balance between proliferation and apoptosis determines whether preinitiated cells will form a palpable tumor, the reduced proliferation and increased apoptosis in different mammary epithelial structures of the prepubertally low-fat n -3 PUFA fed rats as well as lower number of TEBs could have contributed to their reduced mammary tumorigenesis.
We found that rats fed a high-fat n -3 PUFA diet during prepuberty were at an increased risk of developing mammary tumors. Previous animal studies have generated conflicting data regarding n -3 PUFA intake and mammary tumorigenesis. Findings in athymic mice have shown that n -3 PUFAs inhibit the growth of human breast cancer cells ( 22 ). However, in rats a diet containing menhaden oil did not modify N -nitrosomethylurea-induced mammary tumor growth ( 23 ). Furthermore, it has been reported that when the amount of n -3 PUFAs relative to n -6 PUFAs increases above 7, DMBA-induced mammary tumor incidence and tumor weights also increase in rats ( 24 ). In our study, n -6: n -3 PUFA ratio was similar in the low- and high-fat n -3 PUFA diets (2–3:1), but high-fat diet contained more total n -3 PUFAs than the low-fat diet. It is probable that high absolute levels of n -3 PUFAs contributed to the increased mammary tumorigenesis in the high-fat n -3 PUFA group, since high-fat diet itself (i.e. high-fat n -6 PUFA) did not affect breast cancer risk. In addition, a previous study has reported that high (pharmacological) n -3 PUFA levels in a low-fat diet promoted mammary tumorigenesis in rats ( 24 ).
Some of the biological changes seen in the mammary glands of the rats prepubertally exposed to a high-fat n -3 PUFA diet were similar to those seen in the glands of rats exposed prepubertally to a low-fat n -3 PUFA diet, but some were opposite. Mammary gland morphology was similarly altered in the low- and high-fat n -3 PUFA fed groups; i.e. both the low- and high-fat n -3 PUFA glands contained less TEBs and more differentiated LAUs than the reference group. Since mammary tumorigenesis was reduced in the prepubertally low-fat n -3 PUFA fed rats and increased in the high-fat n -3 PUFA fed rats, this could be interpreted to argue against the commonly held view that morphological changes are predictive of altered breast cancer risk ( 8 , 27 ). However, our results indicated that cell proliferation and apoptosis within the TEBs and LAUs were different in the low- and high-fat n -3 PUFA groups. TEBs contain higher levels of proliferating cells than the other mammary epithelial structures ( 8 ), as also found in our study. When compared with the reference group, the TEBs of the prepubertally high-fat n -3 PUFA fed animals contained even a higher number of proliferating cells. As discussed above, cell proliferation in the TEBs of the low-fat n -3 PUFA fed animals was significantly reduced. Furthermore, the level of apoptosis was different in the two n -3 PUFA groups, with the prepubertally high-fat n -3 PUFA exposed rats showing a significantly reduced rate of apoptosis in the TEBs and LAUs than the control or low-fat n -3 PUFA fed rats. The levels of proliferation and apoptosis were not altered in the ducts, suggesting that high levels of n -3 PUFAs specifically targeted TEBs and LAUs. To summarize, although the number of TEBs at the time the carcinogen is administered is closely associated with the gland's susceptibility to malignant transformation ( 8 ), the present results suggest that even more important is the level of cell proliferation taking place in these structures. Thus, an animal with low levels of TEBs can be at an increased breast cancer risk, if their TEBs contain elevated levels of proliferating cells.
Several genes regulate apoptosis, including Akt. Akt is a survival factor, and its upregulation is linked to resistance to apoptosis ( 31 ). Earlier studies have found that n -3 PUFAs interact with Akt to downregulate its phosphorylation ( 32 ), and this could at least partly explain how n -3 PUFAs induce apoptosis. We noted increased levels of pAkt in the high-fat n -3 PUFA fed rats and no changes in this gene in the low-fat n -3 PUFA fed rats. It is possible that the reduced levels of apoptosis in the TEBs and LAUs of the prepubertally high-fat n -3 PUFA fed rats may be related to the finding that phosphorylated Akt expression was increased in the animals fed the high-fat n -3 PUFA diet during prepuberty. However, an increase in apoptosis in the low-fat n -3 fed rats cannot be because of pAkt, since no changes in its expression were seen in these rats.
Non-steroidal antiinflammatory drugs inhibit COX activity and have been shown to inhibit tumorigenesis ( 41 ). Conversely, COX-2 overexpression in mice is sufficient to induce mammary tumors ( 42 ). Earlier findings in rats fed n -3 PUFAs indicate increased incorporation of n -3 PUFAs into the membrane phospholipids and a subsequent inhibition of COX-2 expression ( 43 , 44 ). In agreement with these observations, we found that both the low- and the high-fat n -3 PUFA diets decreased the expression of COX-2, perhaps reflecting an optimal n -6: n -3 PUFA ratio in these diets. Since the high-fat n -3 PUFA diet increased mammary tumorigenesis, COX-2 expression is unlikely to be correlated with this finding. Whether the reduced expression contributed to the reduced tumorigenesis in the prepubertally low-fat n -3 PUFA fed rats, remains to be established.
In conclusion, we report here that rats fed prepubertally a low-fat n -3 PUFA diet are at a reduced risk of developing carcinogen-induced breast cancer. In marked contrast, rats fed the high-fat n -3 PUFA diet during prepuberty are at a significantly increased risk of mammary tumorigenesis. Mechanisms mediating the effects of these opposite findings were examined, and many changes previously associated with n -3 PUFA functions were identified. We propose that the reduced breast cancer risk in rats fed the low n -3 PUFA diet during prepuberty may reflect increased epithelial differentiation and apoptosis, and reduced cell proliferation and COX-2 activity. Thus, less DNA damage is inflicted, because the cells exhibit low levels of proliferation and the glands can eliminate damaged cells through apoptosis. In the high-fat n -3 PUFA fed rats, interrelated pathways/activities are strongly implicated in the increase in breast cancer risk. These rats show elevated levels of lipid peroxidation and proliferating mammary cells, and reduced apoptosis. The latter finding may be caused by an activation of Akt signaling. Thus, the mammary glands of rats fed the high-fat n -3 PUFA diet during prepuberty may acquire higher levels of DNA damage, but are less effective in eliminating the damaged cells through apoptosis.
The work was supported by a predoctoral fellowship grant from Department of Defense to S.E.O. and grants to L.H.C. from NCI (5 RO1 CA89950 and 1 U54 CA100970), the Susan G.Komen Breast Cancer Research Foundation and American Institute for Cancer Research.
Conflict of Interest Statement : None declared.
References
Boyd,N.F., Stone,J., Vogt,K.N., Connelly,B.S., Martin,L.J. and Minkin,S. (
Smith-Warner,S.A., Spiegelman,D., Adami,H.-O. et al . (
Ziegler,R.G., Hoover,R.N., Pike,M.C., Hildesheim,A., Nomura,A.M.Y., West,D.W., Wu-Williams,A.H., Kolonel,L.N., Horn-Ross,P.L., Rosenthal,J.F. and Hyer,M.B. (
Colditz,G.A. and Frazier,A.L. (
Pike,M.C., Krailo,M.D., Henderson,B.E., Casagrande,J.T. and Hoel,D.G. (
Rosner,B., Colditz,G. and Willett,W. (
Marshall,W.A. and Tanner,J.M. (
Russo,J. and Russo,I.H. (
Frazier,A.L., Ryan,C.T., Rockett,H., Willett,W.C. and Colditz,G.A. (
Baer,H.J., Schnitt,S.J., Connolly,J.L., Byrne,C., Cho,E., Willett,W.C. and Colditz,G.A. (
Pryor,M., Slattery,M.L., Robinson,L.M. and Egger,M. (
Potischman,N., Weiss,H.A., Swanson,C.A., Coates,R.J., Gammon,M.D., Malone,K.E., Brogan,D., Stanford,J.L., Hoover,R.N. and Brinton,L.A. (
Maillard,V., Bougnoux,P., Ferrari,P., Jourdan,M.L., Pinault,M., Lavillonniere,F., Body,G., Le,F.O. and Chajes,V. (
Kaizer,L., Boyd,N.F., Kriukov,V. and Tritchler,D. (
Lund,E. and Bonaa,K.H. (
Key,T.J., Sharp,G.B. and Appleby,P.N. (
Klein,V., Chajes,V., Germain,E., Schulgen,G., Pinault,M., Malvy,D., Lefrancq,T., Fignon,A., Le Floch,O., Lhuillery,C. and Bougnoux,P. (
Zhu,Z.R., Agren,J., Mannisto,S., Pietinen,P., Eskelinen,M., Syrjanen,K. and Uusitupa,M. (
Stripp,C., Overvad,K., Christensen,J., Thomsen,B.L., Olsen,A., Moller,S. and Tjonneland,A. (
Goodstine,S.L., Zheng,T., Holford,T.R., Ward,B.A., Carter,D., Owens,P.H. and Mayne,S.T. (
Caygill,C.P.J. and Hill,M.J. (
Rose,D.P. and Connolly,J.M. (
Cohen,L.A., Chen-Backlund,J.Y., Sepkovic,D.W. and Sugie,S. (
Sasaki,T., Kobayashi,Y., Shimizu,J., Wada,M., In'nami,S., Kanke,Y. and Takita,T. (
Hilakivi-Clarke,L., Cho,E., Cabanes,A., DeAssis,S., Olivo,S., Helferich,W., Lippman,M.E. and Clarke,R. (
Saste,M.D., Carver,J.D., Stockard,J.E., Benford,V.J., Chen,L.T. and Phelps,C.P. (
Hilakivi-Clarke,L., Clarke,R., Onojafe,I., Raygada,M., Cho,E. and Lippman,M.E. (
Kaplan,E.L. and Meijer,P. (
Das,U.N. (
Franke,T.F., Hornik,C.P., Segev,L., Shostak,G.A. and Sugimoto,C. (
DeGraffenried,L.A., Friedrichs,W.E., Fulcher,L., Fernandes,G., Silva,J.M., Peralba,J.M. and Hidalgo,M. (
Hildreth,N.G., Shore,R.E. and Dvoretsky,P.M. (
Bhatia,S., Robinson,L.L., Oberlin,O., Greenberg,M., Bunin,G., Fossati-Bellani,F. and Meadows,A.T. (
Ando,K., Nagata,K., Yoshida,R., Kikugawa,K. and Suzuki,M. (
Marnett,L.J. (
Anderson,D. (
Arita,K., Kobuchi,H., Utsumi,T., Takehara,Y., Akiyama,J., Horton,A.A. and Utsumi,K. (
Igarashi,M. and Miyazawa,T. (
Rose,D.P., Connolly,J.M., Rayburn,J. and Coleman,M. (
Harris,R.E., Alshafie,G.A., bou-Issa,H. and Seibert,K. (
Liu,C.H., Chang,S.H., Narko,K., Trifan,O.C., Wu,M.-T., Smith,E., Haudenschild,C., Lane,T.F. and Hla,T. (
Singh,J., Hamid,R. and Reddy,B.S. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}