





Abstract
Motivation: Identification of conserved motifs in biological sequences is crucial to unveil common shared functions. Many tools exist for motif identification, including some that allow degenerate positions with multiple possible nucleotides or amino acids. Most efficient methods available today search conserved motifs in a set of sequences, but do not check for their specificity regarding to a set of negative sequences.
Results: We present a tool to identify degenerate motifs, based on a given classification of amino acids according to their physico-chemical properties. It returns the top K motifs that are most frequent in a positive set of sequences involved in a biological process of interest, and absent from a negative set. Thus, our method discovers discriminative motifs in biological sequences that may be used to identify new sequences involved in the same process. We used this tool to identify candidate effector proteins secreted into plant tissues by the root knot nematode Meloidogyne incognita. Our tool identified a series of motifs specifically present in a positive set of known effectors while totally absent from a negative set of evolutionarily conserved housekeeping proteins. Scanning the proteome of M.incognita, we detected 2579 proteins that contain these specific motifs and can be considered as new putative effectors.
Availability and Implementation: The motif discovery tool and the proteins used in the experiments are available at http://dtai.cs.kuleuven.be/ml/systems/merci.
Contact: celine.vens@cs.kuleuven.be
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Conserved motifs in biological sequences reflect functionally important shared features. In genome sequences, conserved motifs can point to promoters or regulatory elements, regions of splice junctions between protein-coding exons or regions affecting the shape of the chromatin. In protein sequences, such conserved motifs can highlight signals that are important for controlling the cellular localization (e.g. nucleus, cytoplasm, extracellular compartment), regions shared between proteins that interact with a same partner or regions important for the biochemical function itself.
Physico-chemical properties and 3D structures of proteins are more conserved than the suite of amino acids itself. Thus, at a given position in a protein sequence, different amino acids may have similar structural or physico-chemical roles. Degenerate motifs allowing multiple possible amino acids at one position are necessary to comply with this variability. Several methods allow for discovery of degenerate motifs (Bailey and Elkan, 1994; Ji and Bailey, 2007), but few of them take into account similarity in terms of physico-chemical properties of amino acids at a given position (Jonassen, 1997; Rigoutsos and Floratos, 1998).
When the purpose of the obtained motifs is to scan large datasets (e.g. genomes, proteomes) in order to find new sequences potentially involved in the same biological process, another relevant point in the motif discovery is the specificity of the identified motifs regarding the biological process. Some systems make use of statistics to attach a measure of significance to each of the discovered patterns, as deduced from a model based on the input sequences or a public sequence database (Bailey and Elkan, 1994; Jonassen, 1997; Rigoutsos and Floratos, 1998). For many biological applications, however, a negative set of sequences not involved in the process of interest can be compiled, and this set can be used as a more direct way to evaluate the relevance of the motifs. While several motif discovery processes take into consideration a negative sequence set (Bailey et al., 2010; Redhead and Bailey, 2007), this set is often used to guide the search toward motifs overrepresented in the positive sequences, rather than discriminating motifs.
In this article, we propose a method that identifies motifs consisting of specific amino acids and physico-chemical properties, that can be used as discriminators to identify new sequences involved in a biological process of interest. To our knowledge, no motif discovery method exists that combines these two features. Our method outputs the top K motifs that are most frequent in a positive set of proteins and are absent from a negative set of proteins.
We applied this method to find motifs in root-knot nematode effectors. Root-knot nematodes are the most damaging plant-parasitic animals to the agriculture worldwide, causing billions of euro losses every year (Agrios, 1997). They have sophisticated interactions with plants that include penetration of root tissue and establishment of a feeding site. A set of effector proteins that are secreted by the nematode into plant tissue is believed to be crucial for these processes. Most known effectors to date are expressed in nematode secretory glands and delivered to plant tissue through a syringe-like stylet. With the availability of two annotated genome sequences for root-knot nematodes (Abad et al., 2008; Opperman et al., 2008), identifying the whole set of candidate secreted effectors can now be envisioned. Although many effectors possess a signal peptide for secretion, others clearly present in secretory glands and/or nematode secretions have no predicted signal peptide (Bellafiore et al., 2008; Jaubert et al., 2002). Similarly, many root-knot nematode proteins bearing a signal peptide are not delivered to the plant but have conserved functions in different species. Hence, the presence of a signal peptide cannot be used as a discriminator to identify new effectors, and no reliable motif to predict secretion of a nematode protein in plants is currently available. We constructed a positive set of proteins known to be secreted by root-knot nematodes into plant tissue and a negative set of evolutionarily conserved proteins, in order to identify specific motifs in positive proteins. Using these datasets, our method identified a set of effector-specific motifs at the N-terminal region of the positive proteins.
2 METHODS
2.1 Classification schemes
Several amino acid classifications group amino acids according to their physico-chemical properties. In this work, we consider two classification schemes. The first one was proposed by Koolman and Rohm (1996). It contains seven non-overlapping properties, see Table 1. The second one is RasMol's classification (Sayle and Milner-White, 1995), which is much larger, see Table 2. It contains 15 classes,1 with a lot of overlap.
Koolman and Rohm (1996) amino acid classification
| Property | Amino acids | Property | Amino acids |
|---|---|---|---|
| Aliphatic | A,G,I,L,V | Neutral | S,T,N,Q |
| Sulfur containing | C,M | Acidic | D,E |
| Aromatic | F,Y,W | Basic | R,H,K |
| Cyclic | P |
| Property | Amino acids | Property | Amino acids |
|---|---|---|---|
| Aliphatic | A,G,I,L,V | Neutral | S,T,N,Q |
| Sulfur containing | C,M | Acidic | D,E |
| Aromatic | F,Y,W | Basic | R,H,K |
| Cyclic | P |
Koolman and Rohm (1996) amino acid classification
| Property | Amino acids | Property | Amino acids |
|---|---|---|---|
| Aliphatic | A,G,I,L,V | Neutral | S,T,N,Q |
| Sulfur containing | C,M | Acidic | D,E |
| Aromatic | F,Y,W | Basic | R,H,K |
| Cyclic | P |
| Property | Amino acids | Property | Amino acids |
|---|---|---|---|
| Aliphatic | A,G,I,L,V | Neutral | S,T,N,Q |
| Sulfur containing | C,M | Acidic | D,E |
| Aromatic | F,Y,W | Basic | R,H,K |
| Cyclic | P |
RasMol's classification of amino acids (Sayle and Milner-White, 1995)
| Property | Amino acids |
|---|---|
| Acidic | D,E |
| Acyclic | A,R,N,D,C,E,Q,G,I,L,K,M,S,T,V |
| Aliphatic | A,G,I,L,V |
| Aromatic | H,F,W,Y |
| Basic | R,H,K |
| Buried | A,C,I,L,M,F,W,V |
| Charged | D,E,R,H,K |
| Cyclic | H,F,P,W,Y |
| Hydrophobic | A,G,I,L,M,F,P,W,Y,V |
| Large | R,E,Q,H,I,L,K,M,F,W,Y |
| Medium | N,D,C,P,T,V |
| Neutral | A,N,C,Q,G,I,L,M,F,P,S,T,W,Y,V |
| Polar | R,N,D,C,E,Q,H,K,S,T |
| Small | A,G,S |
| Surface | R,N,D,E,Q,G,H,K,P,S,T,Y |
| Property | Amino acids |
|---|---|
| Acidic | D,E |
| Acyclic | A,R,N,D,C,E,Q,G,I,L,K,M,S,T,V |
| Aliphatic | A,G,I,L,V |
| Aromatic | H,F,W,Y |
| Basic | R,H,K |
| Buried | A,C,I,L,M,F,W,V |
| Charged | D,E,R,H,K |
| Cyclic | H,F,P,W,Y |
| Hydrophobic | A,G,I,L,M,F,P,W,Y,V |
| Large | R,E,Q,H,I,L,K,M,F,W,Y |
| Medium | N,D,C,P,T,V |
| Neutral | A,N,C,Q,G,I,L,M,F,P,S,T,W,Y,V |
| Polar | R,N,D,C,E,Q,H,K,S,T |
| Small | A,G,S |
| Surface | R,N,D,E,Q,G,H,K,P,S,T,Y |
RasMol's classification of amino acids (Sayle and Milner-White, 1995)
| Property | Amino acids |
|---|---|
| Acidic | D,E |
| Acyclic | A,R,N,D,C,E,Q,G,I,L,K,M,S,T,V |
| Aliphatic | A,G,I,L,V |
| Aromatic | H,F,W,Y |
| Basic | R,H,K |
| Buried | A,C,I,L,M,F,W,V |
| Charged | D,E,R,H,K |
| Cyclic | H,F,P,W,Y |
| Hydrophobic | A,G,I,L,M,F,P,W,Y,V |
| Large | R,E,Q,H,I,L,K,M,F,W,Y |
| Medium | N,D,C,P,T,V |
| Neutral | A,N,C,Q,G,I,L,M,F,P,S,T,W,Y,V |
| Polar | R,N,D,C,E,Q,H,K,S,T |
| Small | A,G,S |
| Surface | R,N,D,E,Q,G,H,K,P,S,T,Y |
| Property | Amino acids |
|---|---|
| Acidic | D,E |
| Acyclic | A,R,N,D,C,E,Q,G,I,L,K,M,S,T,V |
| Aliphatic | A,G,I,L,V |
| Aromatic | H,F,W,Y |
| Basic | R,H,K |
| Buried | A,C,I,L,M,F,W,V |
| Charged | D,E,R,H,K |
| Cyclic | H,F,P,W,Y |
| Hydrophobic | A,G,I,L,M,F,P,W,Y,V |
| Large | R,E,Q,H,I,L,K,M,F,W,Y |
| Medium | N,D,C,P,T,V |
| Neutral | A,N,C,Q,G,I,L,M,F,P,S,T,W,Y,V |
| Polar | R,N,D,C,E,Q,H,K,S,T |
| Small | A,G,S |
| Surface | R,N,D,E,Q,G,H,K,P,S,T,Y |
We can view the classification schemes as directed acyclic graphs (DAGs) representing the superclass relationship. In its simplest way, the DAG contains two levels: the classes and the amino acids. However, a closer look at the latter classification scheme can introduce more structure and levels. For instance, all basic amino acids are charged and large, and all charged amino acids are polar and surface.
2.2 Formal task description
We define the task of identifying the top K discriminative protein motifs with amino acid properties as follows:
Given: (i) a set of positive proteins P and a set of negative proteins N, (ii) a parameter K, and (iii) a set of amino acid properties C and a partial order  (structured as a DAG) defined on the union of C and the amino acid alphabet A. For all c1, c2 ∈ C ∪ A:
(structured as a DAG) defined on the union of C and the amino acid alphabet A. For all c1, c2 ∈ C ∪ A: 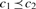 if and only if there is a directed path from c1 to c2 in the DAG.
if and only if there is a directed path from c1 to c2 in the DAG.
Find: the set of K motifs, using symbols in C ∪ A, that are maximally frequent in P and are absent from (or infrequent in, see Section 2.3.2) N.
2.3 Algorithm
The algorithm we propose is based on the well-known candidate generation principle, introduced in sequential pattern mining by the AprioriAll algorithm (Agrawal and Srikant, 1995). At each iteration, a set of candidate patterns is generated, whose frequency is tested. In order to search for discriminative patterns, we change the basic algorithm, such that it essentially looks for those patterns that are frequent in the positive sequences, and meanwhile tests if they are absent from the negative sequences. In order to include physico-chemical properties of the amino acid residues, we extend the candidate generation step. In the next sections, we describe the different steps of the algorithm.
2.3.1 Candidate generation
In order to perform a complete and efficient search, it is important that each relevant candidate is generated, and that no candidate is generated more than once. To achieve this, most candidate generation algorithms structure the search space as a lattice representing a general-to-specific structure. Generating candidates then comes down to traversing the lattice, thereby pruning as much as possible.
We follow the same approach and consider a pattern <p1, p2, p3,…, pn> more general than another pattern <q1, q2, q3,…, qm>, if and only if n ≤ m and for each pair (pi, qi) it holds that 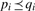 . Our candidate generation method traverses the lattice from general to specific, which is done by starting with an artificial root element that denotes the empty pattern, and at each step performing two basic operations to generate minimally specialized new candidates given a pattern: (i) add a top-level element of the DAG (extension), and (ii) minimally specialize an element of the pattern (specialization).
. Our candidate generation method traverses the lattice from general to specific, which is done by starting with an artificial root element that denotes the empty pattern, and at each step performing two basic operations to generate minimally specialized new candidates given a pattern: (i) add a top-level element of the DAG (extension), and (ii) minimally specialize an element of the pattern (specialization).
In order to ensure that no pattern is considered more than once, we only add elements to the end of the pattern, and only specialize the last element in the pattern. We transform the DAG into a tree in a preprocessing step, such that only one path leads to each amino acid. This is trivially fulfilled for the Koolman and Rohm classification. A possible spanning tree for the hierarchy by Sayle and Milner-White is shown in Figure 1. The specialization operator is performed by replacing the last element in the pattern by each of its children in this tree. The candidate generation process is illustrated in Figure 2.
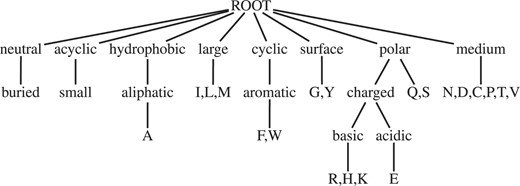
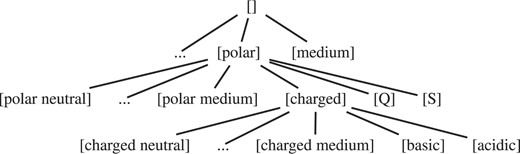
Candidate generation using the amino acid classification of Sayle and Milner-White (1995) and the spanning tree of Figure 1.
2.3.2 Finding the top K motifs
We introduce two parameters, FP and FN, which denote the minimal frequency threshold for the positive sequences, and the maximal frequency threshold for the negative sequences, respectively. By default, FN is set to zero, and motifs are searched that are absent from the negative set. However, since for some applications, constructing a pure negative set can be difficult, setting FN to a higher value can be useful. The parameter FP defines the threshold above which motifs are retained. Initially, it is set to one or to a user-defined value, and it increases throughout the execution of the algorithm: It is possible that more than K motifs have a frequency above FP at the end. In this case, one can either randomly pick motifs with frequency FP until K motifs are obtained, or one can output all of them. We opted for the latter approach, so that the user has maximal control over the output.
Initially, after finding K motifs, FP is updated to the minimum of the frequencies of those K motifs in the positive sequences.
In later stages, if a valid motif is found, it is inserted into the current list of motifs, which is sorted according to their frequency in the positive sequences. If the first K motifs have a frequency higher than FP, then FP is updated to the minimum of these K frequencies.
2.3.3 Candidate pruning and testing
In order to conduct the search efficiently, the algorithm exploits the antimonotonicity properties of the frequency constraints. This results in the following rules (freq(X, Y) returns the number of proteins in set Y that contain the motif X):
If a pattern M has freq(M, P) ≤ FP, then we should not consider new candidates C that are more specific than M, since they will have freq(C, P) ≤ FP, and thus can be discarded.
If a pattern M has freq(M, N) ≤ FN, then all new candidates C that are more specific than M will have freq(C, N) ≤ FN, hence we do not need to count their frequency in the negative set.
When checking a candidate's frequency in the positive set, we make the following two observations. First, we only have to check the frequency of a candidate in case all the candidate's parents in the lattice have passed the minimal frequency threshold FP. Secondly, it is not necessary to check the complete set of positive sequences, it suffices to check the sequences in which the parents are present. Therefore, we adopt a vertical ID-list dataset format (Zaki, 1998), where we associate to each pattern a list of (positive) sequence IDs in which it occurs. Before testing the frequency of a candidate, we check whether its parents are frequent, and if yes, intersect their sequence ID lists. Only the sequences in the intersection are checked for the presence of the pattern. If the pattern passes the minimal frequency constraint, we store it together with the sequences in which it occurs.
When a candidate has been found frequent in the positive set, we test its frequency in the negative set. As mentioned above, if a candidate's parent was infrequent in the negative set, we can output this candidate as a valid motif without any testing. In the other case, we iterate over the negatives, counting hits and stop searching if the maximal frequency FN is reached. Note that, if the maximal frequency condition is not met, we still have to keep the candidate for further processing, because the maximal frequency condition can become true for any of the new candidates generated from it.
2.3.4 Organization of the search
The search space lattice can be traversed using several search strategies, a.o. depth-first and breadth-first search. We opt for the former, since it requires less main memory: it only needs to keep sequence ID lists for candidates along a single path.2 This implies that pruning (checking whether the parents are frequent and intersecting their sequence ID lists) cannot be performed using all parents of a candidate. Therefore, we perform pruning only using the minimal generalizations of the last element of the pattern as parents (removing the last element if it corresponds to the highest level in the DAG).
In order to perform pruning correctly, we have to make sure that all considered parents have been tested before a pattern is tested, i.e. the spanning tree of the amino acids and their properties has to be constructed in a way that, in depth-first traversal, all DAG parents of a node are visited before the node itself. The tree shown in Figure 1 fulfills this constraint.
2.3.5 Implementation
We provide a Perl implementation of the proposed algorithm, called MERCI (Motif—EmeRging and with Classes—Identification), at http://dtai.cs.kuleuven.be/ml/systems/merci. Pseudo code is given in Supplementary Table S1. Counting frequencies is done by Perl's pattern matching operator, using regular expressions to represent classes. The implementation provides the following additional features:
The user is not restricted to use one of the classifications discussed here, but can define his own classification scheme of amino acid properties.
There is an option to find gapped motifs. The algorithm is easily extended to include gaps by splitting the extension operator into two basic operations: (i) add a top-level candidate to the end of the pattern, and (ii) add a gap followed by a top-level candidate to the end of the pattern. The program supports gaps of variable length, i.e. the user provides a maximal gap length L, and a gap symbol then denotes any number of amino acids between 0 and L. The maximal number of gap symbols is also set by the user.
The program includes a searching tool, which can be used to locate the discovered motifs' occurrences in any set of sequences.
2.4 Meloidogyne incognita dataset
We first describe how the positive and negative protein sets were constructed. Statistics about the resulting sets are given in Table 3. Then we explain how the resulting motifs are evaluated.
Meloidogyne incognita protein set statistics
| Positive set | Negative set | |
|---|---|---|
| Number of sequences | 100 | 459 |
| Shortest/longest sequence length (residues) | 43/902 | 57/2106 |
| Average sequence length (residues) | 270.3 | 438.8 |
| Sequences with signal peptide | 57 | 43 |
| Positive set | Negative set | |
|---|---|---|
| Number of sequences | 100 | 459 |
| Shortest/longest sequence length (residues) | 43/902 | 57/2106 |
| Average sequence length (residues) | 270.3 | 438.8 |
| Sequences with signal peptide | 57 | 43 |
Meloidogyne incognita protein set statistics
| Positive set | Negative set | |
|---|---|---|
| Number of sequences | 100 | 459 |
| Shortest/longest sequence length (residues) | 43/902 | 57/2106 |
| Average sequence length (residues) | 270.3 | 438.8 |
| Sequences with signal peptide | 57 | 43 |
| Positive set | Negative set | |
|---|---|---|
| Number of sequences | 100 | 459 |
| Shortest/longest sequence length (residues) | 43/902 | 57/2106 |
| Average sequence length (residues) | 270.3 | 438.8 |
| Sequences with signal peptide | 57 | 43 |
2.4.1 Positive set
We constructed a positive set with proteins that are known to be secreted or likely to be secreted by the nematode into plant root tissue (Bellafiore et al., 2008; Ding et al., 2000; Dubreuil et al., 2007; Huang et al., 2003; Wang et al., 2007). The data consists of 59 proteins whose expression in subventral and/or dorsal secretory glands has been shown, 38 proteins that have been identified in the secretome of root-knot nematodes, and 3 translated EST contigs identified by mass spectrometry in nematode secretions. The resulting 100 sequences were scanned with SignalP 3.0 (Emanuelsson et al., 2007) for the presence of a potential signal peptide. The criterion used was detection of a signal peptide with either one of the two methods (artificial neural networks or hidden Markov models) integrated in SignalP. In total, 57 sequences have a predicted signal peptide.
2.4.2 Negative set
As negative set, we used a series of proteins encoded by single-copy genes widely conserved throughout evolution. Such proteins are very unlikely to be secreted by plant-parasitic nematodes in plants as they are highly conserved in non-parasitic species. To identify these proteins, we ran an all versus all comparison of seven proteomes (M.incognita, M.hapla, Brugia malayi, Pristionchus pacificus, Caenorhabditis elegans, Caenorhabditis briggsae, and Drosophila melanogaster) using OrthoMCL (Li et al., 2003). We identified 459 groups of conserved proteins present as a single copy in all of the seven different proteomes. We retrieved the corresponding proteins in M.incognita and checked for the presence of a signal peptide using SignalP. We found that 43 out of these 459 proteins bear a predicted signal peptide. Presence of proteins with a signal peptide in the negative set avoids biasing for motifs indicating the presence of a signal peptide.
2.4.3 Evaluating motifs
The identified motifs were scanned against the proteome of M.incognita (Abad et al., 2008), consisting of 20 359 proteins. Moreover, the genome of M.incognita is known to encode a repertoire of plant cell wall-degrading proteins. Among these proteins, cellulases (Béra-Maillet et al., 2000; Ledger et al., 2006; Rosso et al., 1999), xylanases (Mitreva-Dautova et al., 2006), polygalacturonases (Jaubert et al., 2002) and pectate lyases (Huang et al., 2005) have been shown to be expressed in M.incognita secretory glands. A total of 16 full-length proteins bearing a signal peptide and corresponding to these cell wall-degrading enzymes were identified from the genome annotation and from the literature. Half of them were initially included in the positive set, the rest can be used as a positive control.
3 DISCUSSION
3.1 Finding proteins secreted into plant tissues by M.incognita
Using the datasets described in Section 2.4, we have identified motifs that are specific to the positive proteins, i.e. we searched for motifs that are absent from the negative set by setting FN to zero.3
In a first set of experiments, we searched for the top five motifs, without considering physico-chemical properties. Disabling the use of gaps resulted in the motifs shown in Table 4 (top). We can make two observations from this result. First, the result contains more than five motifs, since there are multiple motifs with the cut-off frequency of 5 (see Section 2.3.2). Secondly, we notice several motifs that are very similar. More precisely, motif <TLLIIS> is present together with its two parent motifs <TLLII> and <LLIIS>, the latter being the most frequent motif in the result. Many pattern discovery algorithms restrict their output to a set of closed patterns, i.e. patterns that do not have any specializations with the same frequency, and would thus discard <TLLII>. Instead, in this work we output the complete set of top K motifs. The reason is that our motifs can be used as discriminators to identify unknown positive sequences. Depending on the application, one might be more interested in maximizing precision (the proportion of positive predictions that are correct), in which case one would prefer to use the most specific motifs, or in maximizing recall (also called sensitivity, the proportion of positive sequences that are correctly predicted), in which case one would use the most general motifs.
Motifs found in the secreted proteins
| Classific. | Motif | freq(Motif,P) |
|---|---|---|
| None | <L L I I S> | 8 |
| (no gaps) | <E G A G> | 6 |
| <A S K Y> | 5 | |
| <A E G D> | 5 | |
| <T L L I I> | 5 | |
| <T L L I I S> | 5 | |
| None | <F x(0,5) L I I S> | 8 |
| (1 gap) | <F x(0,5) L L I I S> | 8 |
| <L L I I S> | 8 | |
| <L L I I S x(0,5) I> | 8 | |
| + 5 motifs | 7 | |
| Koolman | <L L aliphatic I S aliphatic aliphatic> | 9 |
| and Rohm | <L L aliphatic I neutral aliphatic aliphatic> | 9 |
| (no gaps) | <L I I S aliphatic aliphatic> | 8 |
| <L L I I S> | 8 | |
| <L aliphatic I I S> | 8 |
| Classific. | Motif | freq(Motif,P) |
|---|---|---|
| None | <L L I I S> | 8 |
| (no gaps) | <E G A G> | 6 |
| <A S K Y> | 5 | |
| <A E G D> | 5 | |
| <T L L I I> | 5 | |
| <T L L I I S> | 5 | |
| None | <F x(0,5) L I I S> | 8 |
| (1 gap) | <F x(0,5) L L I I S> | 8 |
| <L L I I S> | 8 | |
| <L L I I S x(0,5) I> | 8 | |
| + 5 motifs | 7 | |
| Koolman | <L L aliphatic I S aliphatic aliphatic> | 9 |
| and Rohm | <L L aliphatic I neutral aliphatic aliphatic> | 9 |
| (no gaps) | <L I I S aliphatic aliphatic> | 8 |
| <L L I I S> | 8 | |
| <L aliphatic I I S> | 8 |
The symbol x(M, N) denotes a gap of minimal length M and maximal length N. The last column denotes the frequency of the motifs in the positive proteins.
Motifs found in the secreted proteins
| Classific. | Motif | freq(Motif,P) |
|---|---|---|
| None | <L L I I S> | 8 |
| (no gaps) | <E G A G> | 6 |
| <A S K Y> | 5 | |
| <A E G D> | 5 | |
| <T L L I I> | 5 | |
| <T L L I I S> | 5 | |
| None | <F x(0,5) L I I S> | 8 |
| (1 gap) | <F x(0,5) L L I I S> | 8 |
| <L L I I S> | 8 | |
| <L L I I S x(0,5) I> | 8 | |
| + 5 motifs | 7 | |
| Koolman | <L L aliphatic I S aliphatic aliphatic> | 9 |
| and Rohm | <L L aliphatic I neutral aliphatic aliphatic> | 9 |
| (no gaps) | <L I I S aliphatic aliphatic> | 8 |
| <L L I I S> | 8 | |
| <L aliphatic I I S> | 8 |
| Classific. | Motif | freq(Motif,P) |
|---|---|---|
| None | <L L I I S> | 8 |
| (no gaps) | <E G A G> | 6 |
| <A S K Y> | 5 | |
| <A E G D> | 5 | |
| <T L L I I> | 5 | |
| <T L L I I S> | 5 | |
| None | <F x(0,5) L I I S> | 8 |
| (1 gap) | <F x(0,5) L L I I S> | 8 |
| <L L I I S> | 8 | |
| <L L I I S x(0,5) I> | 8 | |
| + 5 motifs | 7 | |
| Koolman | <L L aliphatic I S aliphatic aliphatic> | 9 |
| and Rohm | <L L aliphatic I neutral aliphatic aliphatic> | 9 |
| (no gaps) | <L I I S aliphatic aliphatic> | 8 |
| <L L I I S> | 8 | |
| <L aliphatic I I S> | 8 |
The symbol x(M, N) denotes a gap of minimal length M and maximal length N. The last column denotes the frequency of the motifs in the positive proteins.
When enabling the use of a gap position (see Table 4, middle), with a maximal gap length of 5, we see that the <LLIIS> motif, which was the most frequent pattern in the previous experiment, can be extended to the left and to the right without decreasing the frequency. Note that the FP threshold has changed from 5 to 7.
In a second set of experiments, we allowed physico-chemical properties in the motifs, starting with the simple Koolman and Rohm (1996) classification. Table 4 (lower part) shows the results, only the results without gaps are shown. Again, we see the <LLIIS> motif, this time together with a number of degenerate variants.
Closer inspection of the <LLIIS> motif and its variants showed that they always occur near the start of the protein sequences. This is consistent with most reported cases in the literature, which state that the signals that control compartment of destination of proteins are often positioned at their N-terminal region. Therefore, in the next experiment, we searched for motifs specifically in this region. We only considered the 30 first positions in this analysis, both for positive and negative sequences. As motifs controlling protein localization are usually short, we set a maximal motif length of 15, and disabled gaps. Without classification, the motif with the maximal frequency is <LIIS> (note the slight difference with the previous <LLIIS>), which occurs in 10 positive sequences. When using the more complex RasMol (Sayle and Milner-White, 1995) classification, the maximal motif frequency obtained is 38 (see Table 5 for an example). When reporting the top 100 motifs, 97 motifs have a frequency of at least 35, with a total coverage of 68 positive proteins. Taking a closer look at the coverage, we observed several things. First, some sequences are covered by almost all motifs, meaning that there is a lot of overlap between the motifs. Secondly, we see that the identified motifs are preferentially found in positive proteins bearing a signal peptide (SP). Since SPs are present in the negative set, and none of the negatives is covered, these motifs do not indicate the SP itself, but a pattern within the SP that is probably related to secretion in plants. We, therefore, focused our analysis on the subset of motifs that cover as many as possible of the 57 SP-bearing positive proteins and none of the non-SP-bearing positives. This subset still contains 66 motifs, covering all but one of the SP-bearing positives. To reduce this number, we applied a heuristic set covering algorithm4 to find a small subset of motifs with the same coverage as the 66 motifs. This procedure resulted in a subset of four motifs, shown in Table 5.
Motifs at N-terminal using RasMol's classification. The last column denotes the frequency of the motifs in the positive proteins
| Motif | freq(Motif,P) |
|---|---|
| <neutral buried neutral large buried neutral neutral neutral hydrophobic hydrophobic neutral acyclic acyclic acyclic buried> | 38 |
| <large hydrophobic neutral buried neutral neutral buried buried neutral acyclic acyclic hydrophobic neutral acyclic acyclic> | 35 |
| <hydrophobic neutral buried acyclic neutral neutral neutral buried neutral large neutral acyclic neutral polar acyclic> | 35 |
| <neutral neutral L buried hydrophobic buried neutral hydrophobic neutral neutral acyclic neutral> | 35 |
| Motif | freq(Motif,P) |
|---|---|
| <neutral buried neutral large buried neutral neutral neutral hydrophobic hydrophobic neutral acyclic acyclic acyclic buried> | 38 |
| <large hydrophobic neutral buried neutral neutral buried buried neutral acyclic acyclic hydrophobic neutral acyclic acyclic> | 35 |
| <hydrophobic neutral buried acyclic neutral neutral neutral buried neutral large neutral acyclic neutral polar acyclic> | 35 |
| <neutral neutral L buried hydrophobic buried neutral hydrophobic neutral neutral acyclic neutral> | 35 |
Motifs at N-terminal using RasMol's classification. The last column denotes the frequency of the motifs in the positive proteins
| Motif | freq(Motif,P) |
|---|---|
| <neutral buried neutral large buried neutral neutral neutral hydrophobic hydrophobic neutral acyclic acyclic acyclic buried> | 38 |
| <large hydrophobic neutral buried neutral neutral buried buried neutral acyclic acyclic hydrophobic neutral acyclic acyclic> | 35 |
| <hydrophobic neutral buried acyclic neutral neutral neutral buried neutral large neutral acyclic neutral polar acyclic> | 35 |
| <neutral neutral L buried hydrophobic buried neutral hydrophobic neutral neutral acyclic neutral> | 35 |
| Motif | freq(Motif,P) |
|---|---|
| <neutral buried neutral large buried neutral neutral neutral hydrophobic hydrophobic neutral acyclic acyclic acyclic buried> | 38 |
| <large hydrophobic neutral buried neutral neutral buried buried neutral acyclic acyclic hydrophobic neutral acyclic acyclic> | 35 |
| <hydrophobic neutral buried acyclic neutral neutral neutral buried neutral large neutral acyclic neutral polar acyclic> | 35 |
| <neutral neutral L buried hydrophobic buried neutral hydrophobic neutral neutral acyclic neutral> | 35 |
Scanning the complete proteome of M.incognita, we found that the 4 motifs cover 2579 proteins (12% of the genome). A total of 2073 of these proteins (80.3%) are predicted to have an SP, while only 17% (3487 out of 20 359) of the M.incognita proteins have a predicted SP. Interestingly, if we only consider the proteins that are covered by at least 2 motifs, then 1106 out of 1162 (95%) have an SP. The 4 motifs cover 7 out of the 8 cell wall-degrading proteins from the evaluation set. Additionally, using the OrthoMCL analysis performed to construct the set of negatives (see Section 2.4.2), we noticed that 1817 of the 2579 proteins are parasite specific, i.e. they do not have orthologs in C.elegans, C.briggsae, D.melanogaster and P.pacificus. Given that the complete proteome contains 12 234 such proteins, the identification of this subset forms an important contribution to the pipeline of experiments necessary to identify the whole set of candidate effectors. It also introduces the open question of how these motifs in the signal peptide regulate secretion in plants.
3.2 Related work
A large body of literature in the area of motif discovery exists. Here, we focus on systems that learn discriminative and/or degenerate motifs among biological or other sequences.
A lot of research has been carried out in frequent or discriminative substring mining. Most string mining approaches make use of efficient data structures to represent the string dataset (Fischer et al., 2006; Weese and Schulz, 2008) and avoid the candidate generation approach. However, these techniques are limited to finding motifs defined over the same alphabet as the sequences. Some form of degenerate motifs can be obtained by searching for so-called approximate frequent motifs (Ji and Bailey, 2007; Zhu et al., 2007), meaning that some mismatches are allowed when counting the frequency of a motif.
In the area of mining frequent sequential patterns, often used in marketing applications where patterns are searched in ordered lists of transactions, an algorithm is proposed that can integrate user-defined taxonomies in the patterns. GSP (Srikant and Agrawal, 1996) is a candidate generation algorithm, where candidates are generated by joining frequent sequences of the previous level, pruning away sequences that have a non-frequent subsequence. In order to incorporate taxonomies, each data sequence is replaced by an extended sequence, by adding all ancestor items to each transaction. It does not exploit the generalization structure inherent to the search space and does not find discriminative patterns.
Two other pattern discovery approaches allow the user to provide sets of amino acids, which are considered equivalent. However, they do not search for discriminative motifs. Teiresias (Rigoutsos and Floratos, 1998) uses a convolution technique to generate new motifs from two smaller motifs. It returns the set of closed patterns that are frequent in a (positive) set of sequences. The amino acid sets are introduced without any modification to the algorithm, i.e. no generalization relation is exploited. The Pratt algorithm (Jonassen, 1997) uses the concept of a pattern graph to guide the search, and uses a mix of specialization and generalization operators to generate candidates. In a first stage, it searches motifs consisting of specific amino acids, and in an optional refinement stage, these motifs are made more general by replacing amino acids by the amino acid sets. However, it can operate in an exhaustive manner as well.
Another set of related systems is based on probabilistic models. These systems return the discovered motifs as position-specific weight matrices, which specify a score for each residue/position pair. This results in degenerate motifs by enumerating possible alternative residues for each position, in contrast to describing possible alternatives, as in our approach. The enumeration does not take into account any classification. Example systems that find discriminative motifs are DEME (Redhead and Bailey, 2007), which uses a combination of global and local search to find a single best motif, and the widely used MEME software (Bailey and Elkan, 1994), which uses an expectation maximization algorithm, and was recently extended to incorporate negative sequences as input (Bailey et al., 2010). However, these algorithms find motifs that are overrepresented in the positive and underrepresented in the negative set; it is not possible to require total absence from the negatives.
Finally, we mention two logic-based methods. Warmr (King et al., 2001) is an inductive logic programming system searching frequent patterns. It can take background knowledge as input, that could be used to represent the DAG. However, the system does not find discriminative patterns and, since it was not specifically designed for sequences, requires complex data formatting and language bias descriptions from the user. MineSeqlog (Lee and De Raedt, 2004) is a system for mining discriminative logical sequences. It finds motifs by applying a frequent subsequence mining algorithm twice: once to find the set of most specific patterns that are frequent in the positive sequences, and once to find the most specific patterns that are frequent in the negative sequences. The resulting patterns are those that are more general than the former set and more specific than the latter set. The double application of the frequent pattern miner results in a less efficient approach.
3.3 Comparison
We have compared the output of MERCI to four methods from the related work section, that also output degenerate motifs. We applied each method to identify motifs corresponding as much as possible to a common setup: we used the amino acid properties based on the Koolman and Rohm (1996) classification, allowed at the most 1 gap of maximal length 2, requiring a minimal occurrence in the positives of 10% and absence from the negative set. We looked for the 10 best motifs with these constraints. Parameters that are not discussed were left to their default value. The systems MERCI, Pratt and DEME were installed and run locally, thus we can also compare their running times (run on an Intel Q9400 2.66 GHz processor).
3.3.1 MERCI
Using MERCI with the above settings, we found three motifs, see Table 6. These are the only motifs (with a single gap) that occur in at least 10 positive sequences, and do not occur in any negatives, and hence can be used as a reference. The running time of MERCI was 119 s.
Motifs found using MERCI
| Motif | freq(Motif,P) |
|---|---|
| <L L aliph I S x(0,2) aliph aliph> | 10 |
| <L L aliph aliph neutral aliph x(0,2) A> | 10 |
| <L aliph aliph aliph neutral L x(0,2) aliph aliph> | 10 |
| Motif | freq(Motif,P) |
|---|---|
| <L L aliph I S x(0,2) aliph aliph> | 10 |
| <L L aliph aliph neutral aliph x(0,2) A> | 10 |
| <L aliph aliph aliph neutral L x(0,2) aliph aliph> | 10 |
Motifs found using MERCI
| Motif | freq(Motif,P) |
|---|---|
| <L L aliph I S x(0,2) aliph aliph> | 10 |
| <L L aliph aliph neutral aliph x(0,2) A> | 10 |
| <L aliph aliph aliph neutral L x(0,2) aliph aliph> | 10 |
| Motif | freq(Motif,P) |
|---|---|
| <L L aliph I S x(0,2) aliph aliph> | 10 |
| <L L aliph aliph neutral aliph x(0,2) A> | 10 |
| <L aliph aliph aliph neutral L x(0,2) aliph aliph> | 10 |
3.3.2 Teiresias
As Teiresias cannot take a negative set as input, we only used the positive sequences. The maximum gap length of 2 was simulated by setting parameters L = 2 and W = 4. We used the seq version, and applied equivalence-based pattern discovery, with the equivalence sets based on the (Koolman and Rohm, 1996) classification. Even though Teiresias only reports closed patterns, the result set contains 328 135 motifs. Many of the reported motifs are overly general, e.g. <aliphatic x(1) x(1) aliphatic> (where x(1) denotes a fixed gap of one position), which not only occurs in all positive sequences, but also in all negative sequences. Calculating a significance score could not be performed within the execution time limit that comes with the web version of Teiresias. Surprisingly, the three motifs found by MERCI are not in Teiresias's output list. The reason is that MERCI uses variable length gaps, while Teiresias does not. For instance, the second MERCI motif occurs three times with a gap length of 0, five times with a gap length of 1 and two times with a gap length of 2. Thus, Teiresias would need three motifs to represent this motif, and these motifs would have a frequency of 3, 5 and 2.
3.3.3 Pratt
Pratt also has a single input set of proteins. We set the maximal gap length to 2 and allowed a single variable length gap. We required Pratt to perform exhaustive search, in order to maximize the output similarity to MERCI, which also performs an exhaustive search. Pratt scores patterns based on the information content. We considered the 10 best patterns, and counted their frequency in the negative sequences. Only half of the patterns are absent from the negative sequences, they are shown in Table 7, and are very similar to the patterns found by MERCI. The reason why there are more patterns and they have a larger frequency is that Pratt also introduces fixed length gaps, e.g. x(2), and their number cannot be restricted by the user. The running time required by Pratt was 2566 s, which is more than 20 times slower than MERCI.
Motifs found using Pratt
| Motif | freq(Motif,P) |
|---|---|
| <L L aliph aliph neutr aliph aliph x(0,2) aliph x(2) E> | 12 |
| <L L aliph aliph neutr aliph x(0,2) aliph aliph x(2) E> | 12 |
| <L L x(1) I S x(1) aliph x(0,2) aliph x(2) E> | 10 |
| <L L aliph x(1) neutr aliph aliph x(0,2) aliph x(1) neutr E> | 10 |
| <L aliph aliph aliph x(1) aliph I x(0,2) aliph x(1) neutr E> | 10 |
| Motif | freq(Motif,P) |
|---|---|
| <L L aliph aliph neutr aliph aliph x(0,2) aliph x(2) E> | 12 |
| <L L aliph aliph neutr aliph x(0,2) aliph aliph x(2) E> | 12 |
| <L L x(1) I S x(1) aliph x(0,2) aliph x(2) E> | 10 |
| <L L aliph x(1) neutr aliph aliph x(0,2) aliph x(1) neutr E> | 10 |
| <L aliph aliph aliph x(1) aliph I x(0,2) aliph x(1) neutr E> | 10 |
Motifs found using Pratt
| Motif | freq(Motif,P) |
|---|---|
| <L L aliph aliph neutr aliph aliph x(0,2) aliph x(2) E> | 12 |
| <L L aliph aliph neutr aliph x(0,2) aliph aliph x(2) E> | 12 |
| <L L x(1) I S x(1) aliph x(0,2) aliph x(2) E> | 10 |
| <L L aliph x(1) neutr aliph aliph x(0,2) aliph x(1) neutr E> | 10 |
| <L aliph aliph aliph x(1) aliph I x(0,2) aliph x(1) neutr E> | 10 |
| Motif | freq(Motif,P) |
|---|---|
| <L L aliph aliph neutr aliph aliph x(0,2) aliph x(2) E> | 12 |
| <L L aliph aliph neutr aliph x(0,2) aliph aliph x(2) E> | 12 |
| <L L x(1) I S x(1) aliph x(0,2) aliph x(2) E> | 10 |
| <L L aliph x(1) neutr aliph aliph x(0,2) aliph x(1) neutr E> | 10 |
| <L aliph aliph aliph x(1) aliph I x(0,2) aliph x(1) neutr E> | 10 |
3.3.4 MEME
The web version of the MEME software does allow to give a negative sequence set as input. However, in contrast to the previous systems, MEME neither does allow the use of a specific classification scheme nor does it use gaps. We used the zoops (zero or one per sequence) motif distribution, set the motif width between 2 and 50 and let MEME search for 10 motifs. In the result, three motifs have an E-value less than 1. The top scoring motif is very similar to our LLIIS-type motifs. The corresponding motif logo is shown in Figure 3. This motif was input to the corresponding motif occurrence locator program FIMO to search for occurrences in both positive and negative sequence sets. Using the default significance threshold, the motif was found in 32 positive sequences and also in 36 negative sequences. We conclude that, while MEME allows negative sequences, it should not be used to search for discriminating motifs. In fact, MEME and MERCI have different target applications. While MERCI searches for motifs that can be used directly as a discriminator when classifying new sequences, MEME searches for motifs that describe a set of sequences. For instance, if the application is to find motifs shared by orthologous proteins, then it can help to include a negative set to guide the search toward significant motifs, while it is allowed that some negative sequences also contain the motif.

Top motif found by MEME.
3.3.5 DEME
DEME reports a single motif, with a width given by the user. Again, gaps are not supported, and no classification scheme can be specified. We have searched for a motif of length 6, 8 and 10, respectively. The consensus sequences, together with their frequencies [as calculated by applying a threshold of 0.5 on the resulting probabilities, see Redhead and Bailey (2007)], are given in Table 8. The running times required to obtain these motifs were 972, 1032 and 1069 s, respectively, resulting in a total running time that is 25 times as high as MERCI's running time. DEME finds motifs that are highly frequent in the positive set, and infrequent in the negative set, and therefore gives more useful results for our task than MEME. However, the motifs are still present in the negative sequences, and we believe the fixed motif width is an important drawback.
Motifs found using DEME
| Motif (consensus) | freq(Motif,P) | freq(Motif,N) |
|---|---|---|
| <K G E G D A> | 38 | 3 |
| <L F I I S L I G> | 54 | 2 |
| <L L H I S L I A P N> | 59 | 2 |
| Motif (consensus) | freq(Motif,P) | freq(Motif,N) |
|---|---|---|
| <K G E G D A> | 38 | 3 |
| <L F I I S L I G> | 54 | 2 |
| <L L H I S L I A P N> | 59 | 2 |
Motifs found using DEME
| Motif (consensus) | freq(Motif,P) | freq(Motif,N) |
|---|---|---|
| <K G E G D A> | 38 | 3 |
| <L F I I S L I G> | 54 | 2 |
| <L L H I S L I A P N> | 59 | 2 |
| Motif (consensus) | freq(Motif,P) | freq(Motif,N) |
|---|---|---|
| <K G E G D A> | 38 | 3 |
| <L F I I S L I G> | 54 | 2 |
| <L L H I S L I A P N> | 59 | 2 |
4 CONCLUSION
We propose an algorithm for the de novo identification of protein motifs specific to a set of proteins. The motifs are not restricted to a sequence of specific amino acids, but can involve physico-chemical amino acid properties. The algorithm combines a variety of existing and new algorithmic contributions into a practical tool, that is freely available, and is able to include user-defined amino acid properties. We provide additional software to scan sequence databases for the occurrence of the identified motifs. To our knowledge, no method is currently available to identify discriminative motifs that are degenerated according to a classification scheme.
Our tool was used to discover motifs specific to root-knot nematode proteins that are secreted into plant tissues. We showed that by allowing properties in the motifs, we are able to find motifs with a higher frequency in a positive set of proteins, while still being absent from a negative set. Using a set of four identified motifs as discriminators, we detected a total of 2579 proteins in the proteome of M.incognita that can be considered as new putative effectors.
We have compared the motifs discovered by our tool to the result of four other tools that find degenerate motifs. We conclude that our tool is the only one that finds a set of motifs using predefined amino acid classes, that is completely absent from the negative set.
While we have focused on finding protein motifs, the tool can be used on any kind of sequence dataset, with any kind of research question for which a positive and negative set can be defined.
1We have discarded the classes positive and negative, since they are equivalent to acidic and basic. We also changed the classification of H: we only classify it as basic, instead of both as basic and neutral.
2The number of motifs that are frequent in the positive set can be very large, especially when using amino acid properties. Therefore, memory requirements can be an issue in breadth-first search.
3In this discussion, we focus on the motifs found by MERCI. For more information about running times, refer to Supplementary Section S2.
4Initially none of the sequences is covered, and we iteratively add a motif with maximal score, until the maximal coverage is obtained. The score of a motif is defined as the sum of the sequence scores of the not-yet-covered sequences it covers, where each sequence is scored by the inverse of the number of motifs present in the sequence. This ensures that sequences that are covered by few motifs have a high score. In case of multiple motifs with the highest score, the one with the shortest length is taken.
ACKNOWLEDGEMENTS
The authors would like to thank Hendrik Blockeel and Siegfried Nijssen for making valuable suggestions with respect to the algorithm.
Funding: This work was supported by the Research Foundation - Flanders (FWO) through a postdoctoral grant (to C.V.).
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: John Quackenbush


{kind=link}
{kind=link}
{kind=link}