




Abstract
Throughout the past nearly a decade, the application of high-throughput sequencing to RNA molecules in the form of RNA sequencing (RNA-seq) and its many variations has revolutionized transcriptomic studies by enabling researchers to take a simultaneously deep and truly global look into the transcriptome. However, there is still considerable scope for improvement on RNA-seq data in its current form, primarily because of the short-read nature of the dominant sequencing technologies, which prevents the completely reliable reconstruction and quantification of full-length transcripts, and the sequencing library building protocols used, which introduce various distortions in the final data sets. The ideal approach toward resolving these remaining issues would involve the direct amplification-free sequencing of full-length RNA molecules. This has recently become practical with the advent of nanopore sequencing, which raises the possibility of yet another revolution in transcriptomics. I discuss the design considerations to be taken into account, the technical challenges that need to be addressed and the biological questions these advances can be expected to resolve.
Introduction
The comprehensive and accurate characterization of the transcriptome is critically necessary if we are to achieve a complete understanding of the mechanisms of transcriptional and posttranscriptional gene regulation and of cellular responses to external and internal stimuli requires. The methods for genome-wide measurements of gene expression affect almost all aspects of modern biology, and advances in the field have repeatedly stimulated waves of new discoveries previously out of reach because of technological limitations.
The ability to take a global look at the transcriptome was first provided by microarray technology in the mid-1990s [1, 2], which remained the sole such tool for more than a decade. However, microarrays had numerous issues that made them far from an ideal tool for measuring transcriptomes: separate arrays had to be designed and manufactured for each species (a costly process out of reach for most laboratories), the results from different platforms were difficult to integrate and compare directly [3], much variability was reported between individual laboratories [4], noise levels were high, base-pair resolution was not provided and others.
The advent of high-throughput sequencing platforms in the mid-2000s enabled the development of RNA sequencing (RNA-seq), with the first protocols appearing in 2008 [5–9]. RNA-seq promised to eliminate most of the imperfections of microarrays by measuring transcriptomes at base-pair resolution, using essentially the same protocol for any species, with a high dynamic range and minimal noise levels. These promises have been largely delivered. RNA-seq has enabled researchers to obtain more accurate than ever before measurements of gene expression, to more easily and reliably annotate newly sequenced genomes and to improve the annotation of existing assemblies, to directly quantify alternative splicing events at base-pair resolution and to precisely measure allele-specific expression and RNA editing events. The noncoding portions of the transcriptome became accessible at base-pair resolution, and whole subfields are today dedicated to the study of long intergenic RNAs (lincRNAs) and circular RNAs, whose diversity and abundance in eukaryote genome became apparent thanks to RNA-seq [10, 11]. RNA-seq also made it possible to analyze environmental samples at the RNA level using metatranscriptomic methods, and to study the proteomes of species without sequenced genomes by generating reference-free de novo transcriptome assemblies, enormously empowering evolutionary genomics and phylogenetics. Finally, single-cell RNA-seq (scRNA-seq) has become a burgeoning field [12], providing researchers with the means to characterize gene expression genome-wide at the level of large numbers of individual cells in a wide variety of systems.
The list of accomplishments is impressive, but the success has nevertheless not yet been complete, as a number of challenges remain unresolved. While existing RNA-seq protocols and sequencing platforms provide more accurate quantifications on a genome-wide level, some distortions in the data are still introduced by existing library generation and sequencing protocols [13]. The problems of transcript assembly and transcript quantification at the level of individual alternatively spliced isoforms have not been fully solved, especially in large and complex genomes (such as those of mammals), where alternative transcript processing is most prevalent and of highest relevance [14, 15]; this is because sequencing reads are much shorter than the length of mature messenger RNAs (mRNAs) and often simply do not provide enough information for accurate inference of the structure and relative abundances of the complete transcripts. Finally, while scRNA-seq studies have provided invaluable insights into the transcriptomes of individual cells, nobody has yet fully measured the transcriptome of an individual cell. To do that, each and every mRNA molecule in a cell has to be captured, successfully converted into complementary DNA (cDNA) and sequenced. However, the numerous steps in scRNA-seq protocols result in the loss of the majority of the transcripts present in each cell, leading to high levels of technical noise and stochasticity [14, 16].
It has become clear that radically different sequencing technologies will be needed for the resolution of these issues, methods capable of sequencing full-length transcripts and without amplification. Nanopore sequencing [17] has held such a promise for a long time, and has finally become a practical reality in the past few years. More recently, initial direct RNA sequencing experiments were reported using the Oxford Nanopore Technologies (ONT) platform [18], bringing us closer to the desired full-length amplification-free RNA-seq. The achievement of that goal depends on whether nanopore platforms will be able to meet certain requirements regarding throughput and the efficiency of capture and sequencing of individual RNA molecules, which are discussed in detail below.
Short-read RNA-seq and its limitations
RNA-seq, as established, currently takes two broad approaches to converting RNA molecules into sequencing libraries. By far, the most common, as implemented in most commercially available kits, begins with the random fragmentation of RNA; cDNA is then generated and amplified to produce the final library. While the earliest protocols provided reads randomly originating from either genomic strand, various methods were later developed for preserving information about the strandedness of the original molecule [19]. Alternatively, full-length cDNAs can be generated (with or without subsequent amplification), and then fragmented and amplified into a final library [20]. In all variations of the assay, size selection is applied at some steps to ensure a tight and short distribution of fragment sizes in the library.
There are several limitations inherent to existing protocols. One is that the process of reverse transcription is far from unbiased. The not-so-random nature of random priming [21] and the negative influence of certain secondary structures on the processivity of reverse transcription [22] are well-known phenomena resulting in uneven coverage along the length of transcripts and the under- and overrepresentation in libraries of certain sequences.
Another is that, as with all polymerase chain reaction (PCR)-based methods, sequencing biases exist. This is probably not a dramatic issue in most cases when working with organisms with fairly ‘normal’ GC content (such as humans and most other common model organisms), but it is a major problem for certain individual transcripts and especially when working with genomes with extreme GC compositions and with environmental samples (consisting of mixtures of organisms, whose genomes’ GC contents vary wildly). Sequences with, for example, high AT% do not amplify as well as more GC-normal ones, distorting the original relative abundances of fragments in the final library. Solutions to issues with PCR biases exist in the form of linear amplification protocols for library generation [23], but they have not been widely adopted.
An important problem without an optimal solution is transcript assembly and the accurate quantification of gene expression at the transcript level for genes with a complex array of alternatively spliced isoforms. These tasks can be carried out in genome space (only possible when a sequenced genome is available) or in reference-free transcriptome space. A wide variety of software packages dealing with each of these problems have been developed, aiming at reconstructing transcripts from short reads and estimating their relative abundances. While the level of sophistication of these algorithms is high, and they work well for relatively simple cases, they do not produce fully reliable results, as demonstrated by a number of single-locus real-life examples and simulation studies [14, 15]: transcripts are often misassembled, and relative isoform abundances are incorrectly estimated in a significant portion of cases, the more so, the more complex (and often, biologically interesting because of that) the loci in question are.
It is unlikely that a completely satisfactory computational solution to these problems will be developed, as the underlying reason for the failures of existing algorithms is the absence of sufficient information in the data sets. The average eukaryote mRNA is ∼2–2.5 kb long, with transcripts much longer than that being widespread, while the average fragment size of RNA-seq libraries is no longer than 250–300 bp. Long-range connectivity information is thus simply not present in the data, and there is a limit to what probabilistic methods can achieve without it.
It is important to note that increasing fragment sizes is not an option for RNA-seq. Illumina sequencing has advanced greatly from the early days of single-end 25 bp reads, and today, it is common to sequence ≥500 bp from the same sequencing fragment in paired-end mode. However, transcriptomes are complex and heterogeneous, with both short and long transcripts being produced by the cell. While it helps with assembly, increasing library fragment sizes introduces a bias against short transcripts (e.g. a hypothetical 500 bp long RNA molecule can be fragmented into two pieces with a uniform probability anywhere along its length, and if the average fragment size is ∼450±50 bp, ∼60% of the time, both pieces will be excluded by size selection) and exacerbates reverse transcription and PCR biases, thus leading to inaccurate quantifications. For the purposes of quantification at the gene level, it is in fact optimal to sequence short fragments.
The short reads of current RNA-seq also make it difficult to accurately identify the 5′ and 3′ ends of transcripts, necessitating the integration of RNA-seq with other techniques, specifically designed to capture transcript ends, such as Rapid Amplification of cDNA Ends (RACE) [24], CAGE [25], RAMPAGE [26] and poly(A)-site profiling, for the purposes of transcript reconstruction. This is an especially acute problem for transcripts of low abundance such as most lincRNAs [24, 27].
These limitations of short-read methods leave incompletely resolved on a global, whole-genome level a number of major biological questions. These concern the prevalence and functional importance of alternative splicing, the precise identity and impact on the proteome of alternatively spliced isoforms, the accurate delineation of rare noncoding transcripts and numerous others. For individual loci, detailed experimental validation of hypotheses generated based on short-read RNA-seq has been necessary, slowing down the pace of scientific progress.
Single-cell RNA-seq
Issues and considerations specific to single-cell transcriptomics (scRNA-seq) need to be examined separately. The minute amounts of RNA present in a single cell necessitate the application of library generation protocols tailored to the task, of which a number have been developed [12]. Of note, multiple scRNA-seq protocols only generate sequences from the 3′ or 5′ ends of transcripts, thus measuring the expression of genes, but largely preventing the analysis of alternative splicing or allele-specific expression at the single-cell level.
Single-cell transcriptomics differs fundamentally from bulk RNA-seq in that the starting material, an individual cell, contains a limited number of original RNA molecules, and ideally each and every one of them should be accounted for in the final data sets. However, the multiple biochemical steps between original RNA molecules and final sequencing libraries are far from completely efficient, meaning that many original RNA molecules are not captured and sequenced. These inefficiencies can be measured in total by the single-molecule capture probability (psmc) [16], which refers to the probability that any individual RNA molecule will be successfully converted into the final library and sequenced. The values of psmc vary between different protocols and probably vary between individual transcripts within the same reaction in at present poorly understood ways; but on average, and in the cases in which they have been reported, they have not exceeded psmc = 0.5, and in fact seem to be mostly in the 0.1–0.2 range [16, 20, 28], meaning that often only 1 in ∼5–10 RNA molecules is captured. Capture inefficiency is less of a problem for physically larger cells with higher RNA content, but unfortunately, many human cell types contain on average in the low hundreds of thousands of transcripts [16, 29], and some important functional classes of genes, e.g. transcription factors, are often primarily expressed in the range where stochastic effects are dominant [16]. The scaling of RNA amount per cell as a function of cell volume [30] is such that this is likely to be a major issue for the majority of organisms.
Two general approaches toward dealing with technical stochasticity in scRNA-seq can be applied. Computationally, noise can be accounted for through statistical modeling [31]. Stochasticity can also be defeated through the force of numbers, which has been greatly facilitated by the development of automated microfluidic platforms for the massively parallel gene expression profiling of large numbers of cells [28, 32, 33]. These have been successful in extracting valuable biological insights from single-cell transcriptomic data sets, but it remains the case that alternative methods eliminating technical stochasticity directly are highly desirable.
Epitranscriptomics
Epitranscriptomics, the study of posttranscriptional RNA modifications using functional genomic tools, is an exciting emerging area of research. RNA molecules, in particular noncoding ones, are subject to a huge array of such modifications [34], and for several of them, such as m1A, m6A, m5C, and 2’-O-methylation, sequencing methods for their genome-wide profiling have been developed in the past few years [35–41]. However, not all of these have base-pair resolution, as some rely on immunoprecipitation with antibodies specific to the modification, and all of them are specialized protocols that are not as readily available to everyone as general RNA-seq. More directly and widely applicable methods for mapping RNA modifications would greatly speed up research in the area.
Existing alternative approaches
The challenges outlined so far have been addressed to a varying extent using existing technologies and protocols, but no fully satisfactory solutions have been devised yet. A brief review of these efforts follows below.
Direct and nearly direct RNA sequencing with short reads
The issues associated with the use of PCR in library generation have been long recognized, and two methods for amplification-free RNA-seq have been developed in the past.
The first such method used the unique properties of the Helicos single-molecule sequencing instrument, and used the 3′ ends of transcripts as direct templates for Helicos sequencing [42–44], thus avoiding amplification altogether. However, this approach only generated short 3′ tags without much information regarding the rest of the transcript. In addition, the Helicos platform is no longer in production, and even when it was on the market, it had a limited distribution in laboratories worldwide.
The FRT-seq assay [45] is based on the Illumina platform, carrying out cDNA synthesis on the surface of the sequencing flow cell (thus, it is probably better referred to as ‘nearly-direct’ RNA-seq). FRT-seq requires nonstandard manipulation of flow cells and the sequencing instrument, which is probably why it has not seen a widespread adoption within the genomics community yet.
Both of these direct RNA-seq methods generate only short reads, thus leaving the challenging problems of transcript reconstruction and isoform-level quantification without a solution.
Long-read transcriptome sequencing and transcript reconstruction
Sequencing platforms generating long reads are now available, making it possible to sequence complete or nearly complete cDNAs. Mammalian transcriptomes were first surveyed in depth using 454 sequencing [46], which generated reads of ≤ 1 kb length. More recent such surveys have relied on the much longer read lengths provided by Pacific Biosciences’ (PacBio) SMRT sequencing method [47, 48] or using synthetic long reads [49] (generated by isolating founder molecules, fragmenting them and tagging the fragments with unique barcodes allowing the identification and reconstruction of the parent molecules). Finally, nanopore sequencing using the ONT platform has been applied to the problem, although with a somewhat more limited scope [50].
In addition, the structure of rare transcripts has been resolved in detail using targeted capture and enrichment, for example using the CaptureSeq method [27], or by applying RACE coupled with high-throughput sequencing [24]. These approaches have been successful in the context of targeted studies, but they require major front-end investments in probe and primer design, necessarily limiting their wide applicability.
A practical limitation with the application of all long-read platforms to transcriptomics has been their low throughput (a subject discussed in more depth below). They provide invaluable information for transcript structure, but the low number of reads makes transcript quantification difficult.
Not only that but quantification will probably not be made possible by potential future drastic increases in read numbers or decreases in cost per read because of the need to build sequencing libraries in the form of DNA, which distorts transcript representation.
The deepest problem with long-read RNA sequencing is the structure of the transcriptome. The cell produces a wide diversity of transcripts, some short, some long and everything in between. It is at present not possible to generate and then sequence libraries without major distortions in relative transcript abundances being introduced by the differential success in making it through all the steps of the protocol of transcripts with significantly different length, differential resistance to reverse transcription or differing sequence compositions. Indeed, PacBio transcriptome sequencing protocols usually physically separate transcripts in different length classes, and these are then sequenced separately. This is a limitation that appears to be fundamental to all methods of library generation that involve reverse transcription and amplification, and one that can be only overcome using a radically different approach to the problem.
The ideal sequencing platform
Based on these and other considerations, we can identify a set of requirements that the ideal sequencing platform that will allow the unbiased comprehensive, fully informed measurement of the transcriptome should conform to:
It should generate long reads that capture the full length of transcripts.
It should be able to sequence RNA directly, without reverse transcription and amplification, eliminating the biases associated with them.
It should exhibit high efficiency in transcript capture, making it applicable to low-input measurements.
It should be able to detect and recognize modified nucleotides in RNA directly.
It should preferably have a low error rate, facilitating all the types of studies of sequence variation that the reliably low error rates of Illumina sequencing enables.
At present, such an ideal sequencing platform does not exist, and it may in fact be the case that it never will because of the inherent difficulty of achieving high sequencing accuracy with single-molecule sequencing technologies, but a major advance in several of these directions might be made possible by the advent of nanopore sequencing.
Nanopore-based direct long-read rna-seq
The idea behind nanopore sequencing—to translocate a nucleic acid molecule through a pore, through which electrical current passes, and read off individual bases based on the characteristic changes in the current that each base induces—is simple, beautiful and has been around for more than two decades [17, 51]. But implementing it into a practical sequencing instrument has not been easy, and has only become a practical reality in the past few years with the appearance of ONT [52].
The advantages of nanopores are numerous: the reads are long, amplification is not necessary, they do not appear to have a significant sequence bias [53] and they can potentially read not only the classic four nucleotides but also distinguish epigenetically modified nucleotides from their non-modified versions [18, 54]. The downsides are a high error rate and a somewhat limited until recently throughput (though the latter limitation is not in principle too difficult to overcome).
In principle, the same approach can be used to sequence not only DNA but also RNA (and potentially even proteins), and indeed, work on direct nanopore-based detection and sequencing of RNA has been active [55–57]. Most recently, the first application of ONT’s technology for direct transcriptome sequencing was presented [18], providing amplification-free full-length mapping of transcripts as well as the direct detection of . Quantification on the whole-genome level was not explored, but overall, the prospects are tantalizing. Nanopores will likely always have a higher error rate because of the single-molecule nature of the technology, somewhat limiting their potential in applications requiring high-precision sequencing, but they can in principle simultaneously fully resolve transcript structure and quantify alternatively spliced isoforms with high accuracy and without bias while also greatly facilitating epitranscriptomics.
In addition, they might achieve an improvement on existing single-cell transcriptomic techniques, both by the sequencing of full transcripts (not featured in a number of current protocols) and by the improvement of single-molecule capture efficiency. Stochastic error in scRNA-seq is because of the numerous enzymatic steps through which an RNA molecule has to be taken to be sequenced; therefore, the best way to eliminate stochastic error is to sequence RNA directly. Microfluidics has been the main driver of advances in the scRNA-seq field, and one can imagine that a combination of a microfluidic device for the separation and isolation of individual cells and nanopore sequencing might achieve significant improvements in performance over currently available options.
What milestones do nanopores need to achieve to meet these expectations and transform transcriptomics?
The direct RNA sequencing process
Direct RNA sequencing differs in major ways from PCR-based methods in what is measured and how. When carrying out conventional RNA-seq, fragmentation and PCR generate what can be effectively treated as an approximately infinite population of fragments from which sequencing reads are sampled.
In contrast, in direct RNA-seq, the starting population of molecules is finite. Given that the goal is to estimate its composition as accurately as possible, stochastic sampling effects become important.
Where c refers to individual cells within a cell population C, G is the set of all genes and is the number of transcripts of gene g in cell c (note that the same reasoning applies for individual transcripts/splice isoforms, i.e. ).
These transcripts have to be then converted into a sequenceable form. Even with nanopores, direct RNA-seq is not completely ‘direct’. In the existing protocol, the poly(A) tails of mRNAs are ligated to sequencing adapters, and a cDNA molecule is synthesized (even though what is read is the RNA part of the resulting RNA-DNA hybrid). Improvements in methods are undoubtedly coming in the future, but some inefficiency in conversion is expected to always remain. In addition, RNA might have to be purified, which is also associated with losses. We can think of the combined effect of RNA handling as plib, or the probability that an individual molecule originally present in the sample is successfully converted into a sequenceable form. The expected number of such molecules for a given gene is therefore:
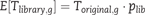
Where Rg is the number of sequenced reads for gene g.
Direct RNA-seq accuracy
The parameters most relevant to the quantification of direct RNA-seq are therefore:
How many reads need to be sequenced for accurate quantification?
What is the minimum number of cells/amount of RNA needed for accurate quantification?
What is the value of the psmc parameter?
The psmc value is especially relevant to the application of the method to low-input RNA-seq and scRNA-seq.
To understand the relative importance of these parameters, we can carry out simulations over a range of values (in this case: 1, 10, 102, 103, 104, 105, 106 and 107 cells; psmc = 0.01, 0.1, 0.5 and 0.9) using relative transcript abundances estimated from short-read data (in this case, from the human K562 cell line; see the Supplementary Methods section for more details) and compare results with traditional RNA-seq (Figures 1 and 2; Supplementary Figures S1–S8). Note that for organisms with much smaller or larger cell, genome and transcriptome sizes, somewhat different result may be obtained.
![Consistency of short-read RNA-seq quantification as a function of sequencing depth. RNA-seq data for the K562 cell line, generated by the Encyclopedia of DNA Elements (ENCODE) Consortium [58], were downloaded from the Consortium’s data portal. Mapped fragments were randomly subsampled to the indicated sequencing depths (see the Supplementary Methods section for more details). Gene expression values were calculated for each subsample. Genes were then split in abundance classes as shown, and the fraction of genes whose quantification was within 10% of their final (i.e. derived from the full set of reads) FPKM value was calculated for each class. Note that the final value is not necessarily the correct one, as any biases of the library generation protocol and the sequencing process will affect all subsamples equally, but the shape of the curves (whether they asymptote toward the final value within the examined range of sequencing depths or not) is helpful for determining what the optimal depth of sequencing is. (A) Gene-level. (B) Transcript-level.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bfg/16/6/10.1093_bfgp_elw043/1/m_elw043f1.jpeg?Expires=1716407097&Signature=2-bGc5cRdWEGe8-Ivmm7LcvB03XQRhjJzfL8ClkP2bhpxhOczl9xc-ltseztdQxq~~C95pXoWpSmoRlgTVskyZThihbWg6F8h8O6yD9KEAUH-7BvWDBrM0I6tivN2Fj0wwQeKogG-YzyE5rur9OuWWNTSPrC7hDjKUoyxw4bV0qAAzV4mHRtCFBAqN38GBpSzot-TMxMXJdcX4gsQVHTsxX71sB1jlvF4XslIFoPPLQnMGLAcVf2W3WLxlwyFmX7eWX~hj~n9wJh38O91wOXjU4SkeG94vBckhwA39Kt7qFJGOGxvsaNwA8tIadS9FdvQo6dT4DOtLjqfskNHWjmkA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Consistency of short-read RNA-seq quantification as a function of sequencing depth. RNA-seq data for the K562 cell line, generated by the Encyclopedia of DNA Elements (ENCODE) Consortium [58], were downloaded from the Consortium’s data portal. Mapped fragments were randomly subsampled to the indicated sequencing depths (see the Supplementary Methods section for more details). Gene expression values were calculated for each subsample. Genes were then split in abundance classes as shown, and the fraction of genes whose quantification was within 10% of their final (i.e. derived from the full set of reads) FPKM value was calculated for each class. Note that the final value is not necessarily the correct one, as any biases of the library generation protocol and the sequencing process will affect all subsamples equally, but the shape of the curves (whether they asymptote toward the final value within the examined range of sequencing depths or not) is helpful for determining what the optimal depth of sequencing is. (A) Gene-level. (B) Transcript-level.
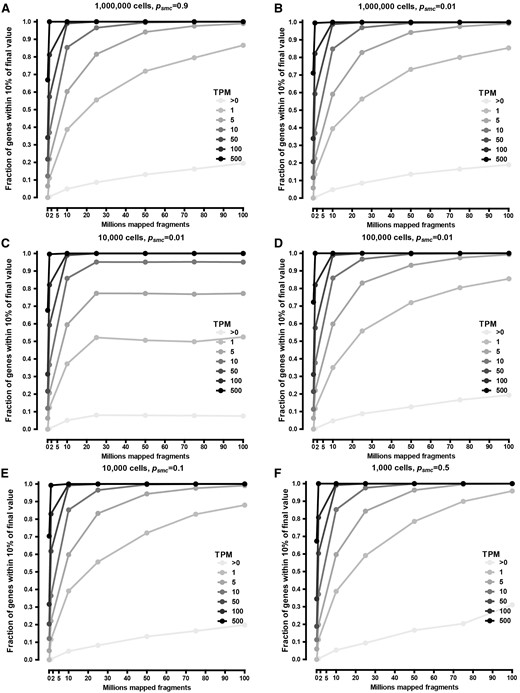
Accuracy of direct RNA-seq quantification at the gene level as a function of input cell number, psmc, and sequencing depth. Gene expression in human K562 cells was simulated using the GENCODE V16 annotation as described in the Methods section. (A) 106 cells, psmc=0.01. (B) 106 cells, psmc=0.9. (C) 104 cells, psmc=0.01. (D) 105 cells, psmc=0.01. (E) 104 cells, psmc=0.1. (F) 103 cells, psmc=0.5. See Supplementary Figures S1–8 for the full set of simulation results.
Figure 1 shows the stability of quantification as a function of sequencing depth for short-read RNA-seq (it is important to note that the ground truth is not known here, and what is measured is not how accurate it is in absolute terms, but how quickly quantification asymptotes toward the final values, which might not be entirely accurate because of various biases in the protocol). Quantification at the level of individual isoforms (Figure 1B) is significantly more unstable than it is for genes (Figure 1A), underscoring the need for long-read direct RNA-seq. Most highly expressed genes already approach their final quantification values even with just a few million mapped reads/fragments, while for genes expressed at 1 or only a few copies per cell [1–5 FPKMs (fragments per kilobase of transcript per million mapped reads)], the same consistency is achieved at ∼40×106 mapped fragments.
The comparative performance of direct RNA-seq depends on the capture efficiency and the input amount of RNA. At a near-perfect psmc = 0.9 and using 106 human cells as input, each containing on average 200 000 transcripts per cell, the expected performance of direct RNA-seq compares favorably with the short-read version. High level of quantification accuracy can be achieved, and most genes expressed at as low as ∼1 transcript copy per cell (∼5 TPM) can be accurately quantified with ∼25 × 106 mapped reads, more highly expressed genes with as few as 5–10 × 106 (Figure 2A).
As long as the input number of cells is high, the efficiency of transcript capture seems to be of little concern, as essentially identical results can be expected with psmc = 0.01 (Figure 2B) (and with even lower values; not shown).
However, some of the most exciting applications of direct RNA-seq would target low-input samples, such as isolated rare cell populations. What are the limits on cell number? With a psmc approaching 1, single cells can be accurately profiled, but with poorer efficiencies, larger cell populations are needed. At a low psmc = 0.01, 105 cells appear to be sufficient, while 104 cells might not provide a sufficiently accurate representation of the transcriptome (Figure 2C and D). But at psmc = 0.1, 104 cells should be sufficient (Figure 2E), and at psmc = 0.5, 103 and even fewer cells can be profiled (Figure 2F).
The real-life values of these parameters are not known at present (other than perhaps the contribution to pseq of poor-quality reads), and it would be important to extensively characterize them in the future. The inclusion of synthetic spike-in controls [59, 60] in carefully predetermined amounts could allow the measurement of capture efficiency within each individual experiment.
The numbers of reads generated by ONT’s instruments that have been reported so far in the literature have been significantly below the millions and tens of millions of reads identified here as necessary for comprehensive transcriptome profiling [52, 53], but the throughput of the platform has been continuously increasing, and further improvements promise to eventually bring it to the desired level [61]. Of note, because of the nature of direct RNA-seq, the reads should ideally be generated from a single flow cell, as there is no DNA library to go back to, especially for minute input samples.
Error rates
The error rates of nanopore sequencing, especially when a sequence is run through the pore only once (1D-reads) have been relatively high (up to ∼15%). The initially reported error rates for direct RNA-seq are even higher, up to 20% [18], and the option for generating consensus ‘2D-reads’ is not immediately available (although it has been suggested that this might be possible by covalently linking the RNA strand to the cDNA strand and sequencing both, thus improving accuracy significantly).
These error rates are much higher than what traditional RNA-seq methods have been delivering, and will make certain types of analyses that require high sequencing precision difficult. To what extent they will affect the application of direct RNA-seq to the problems it is best suited to address (accurate quantification, resolution of alternative splicing isoforms) remains to be seen. Accurate alignment when a fully sequenced and well-annotated genome is available is possible even with high error rates, but while most transcripts differ from each other over sufficiently long stretches of sequence, there are also numerous examples of alternatively spliced isoforms in mammalian genomes that only differ by a few nucleotides [14]. Recent paralogs and pseudogenes are also likely to pose a challenge for data sets with high error rates.
These issues will be investigated in depth and addressed computationally once sufficiently many data sets from diverse sources become available.
‘Dark matter’ transcriptomics
The functional genomics era has brought to light the existence of a bewildering array of noncoding RNA species, but it has also generated significant amounts of controversy regarding how much of the genome is transcribed, what the precise nature of all these transcripts is, which ones have an important biological function and what it is [62–64]. While the latter two questions cannot be fully answered without direct genetic analysis, much could be learned from the precise delineation of the coordinates of ncRNAs. Because they are often expressed at low levels, it has in many cases been difficult to do that precisely on a genome-wide scale, without the application of targeted enrichment strategies.
The ability to directly sequence full-length transcripts is of obvious attraction in this context. How useful in practice that approach will turn out to be will depend on the development of specific protocols for the capture of total RNA into nanopores. The initial direct RNA-seq protocol relies on the existence of a poly(A) tail to convert a transcript into a sequenceable form, and a different approach will be needed if total RNA is to be sequenced. Such protocols will likely decrease the capture efficiency, especially given that they will also have to involve the depletion of ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs), but such studies are usually carried out on large numbers of cells from cell lines, making this less of a concern.
Given sufficiently many cells as input, the probability of capture of a rare transcript is estimated to be , where R is the number of reads, Cc is the average number of copies per cell for the transcript of interest and Tall is the average total number of transcripts per cell (coding and noncoding, excluding the fraction of rRNAs and tRNAs that is removed before sequencing). There are no good estimates of the typical values of Tall in mammalian cells, but if we assume , with reads (the range around which direct RNA-seq becomes highly quantitative) even transcripts present in only 1 in 5 cells should be detectable with a probability close to 1 (Supplementary Figure S9). The use of direct RNA-seq for the comprehensive annotation of the noncoding transcriptome therefore holds great promise.
Single-cell transcriptomics
As discussed above, the biggest challenge in scRNA-seq studies has been the technical stochasticity associated with the efficiency of transcript capture, and the best way to satisfactorily resolve that issue is to eliminate as much of library preparation as possible. Direct RNA-seq through nanopores offers the fewest library preparation steps imaginable at present and is thus the most promising candidate for a technological solution to the problem of technical noise.
Whether these promises will hold out in practice remains to be seen in the future when microfluidic devices combining single-cell isolation and lysis with nanopore sequencing are developed. Even direct RNA-seq with nanopores is not entirely direct, as some manipulation of RNA molecules is necessary to bring them to the pore; this process certainly proceeds with less than complete efficiency (plib). The sequencing process is not entirely efficient either, as a molecule has to successfully engage with the pore and the resulting read has to be of sufficient quality to be mapped back to the genome (pseq). If the best achievable values of these parameters turn out to be sufficiently high that exceeds ∼0.5, nanopore-based scRNA-seq will be a clear winner, but even if the total capture efficiency psmc is around the level of existing methods (around 0.1), the advantage of providing full-length reads (rather than 3′ or 5′ tags) that nanopores have will be attractive in a number of biological contexts.
Epitranscriptomics
Another area of great potential for direct RNA-seq is the direct detection of modified nucleotides in RNA. Such nucleotides induce changes in the current through nanopores that differ from those of unmodified bases, allowing for their detection without the use of specific methods for each modification. So far, the detection of has been reported, but the principle is general and should apply to all such modifications.
The expected difficulty is that the known array of RNA modifications is vast [34]. Base calling for nanopore sequencing is challenging as it is (thus the high error rate of the platform). Whether adding a large number of additional components to the models of base-calling algorithms is going to be feasible in practice, and whether the necessary precision will be achieved remains to be seen.
Nevertheless, nanopores promise to open up the field of epitranscriptomics to everyone equipped with a nanopore sequencer, and to greatly accelerate research in the area.
Conclusions
In summary, while it may still fall short of the dreamed-of perfection in transcriptomic measurements, nanopore-based direct RNA-seq promises to either resolve or at the least advance over the current state of art with respect to a number of existing issues in the field, including the accurate gene- and transcript-level quantification of gene expression, transcript reconstruction, the profiling of low-input RNA samples and the measurement of modified nucleotides. These prospects make it the most exciting development in transcriptomic research, as the revolutionary impact of the initial RNA-seq methods developed around 2008, and turning them into practical reality will constitute a major area of active research over the coming years.
Key Points
Existing RNA-seq methods are powerful, but they still have serious limitations in several important areas.
The development of direct reverse transcription and PCR-free RNA-seq using nanopores promises to resolve a number of these issues.
The major areas to which direct RNA-seq is most relevant are gene- and transcript-level quantification, scRNA-seq and epitranscriptomics.
The key performance milestones and parameters that need to be met and their relevance to biological questions are discussed.
Supplementary Data
Supplementary data are available online at https://academic.oup.com/bfg.
Funding
This material is based on work supported by the National Science Foundation under grant number CNS-0521433.
Georgi K. Marinov received his PhD from the California Institute of Technology in 2014. His research uses functional genomic tools to understand the mechanisms and evolution of gene regulation in eukaryotes.

{kind=link}
{kind=link}