





Abstract
Members of the serpin (serine proteinase inhibitor) superfamily have been identified in higher multicellular eukaryotes (plants and animals) and viruses but not in bacteria, archaea, or fungi. Thus, the ancestral serpin and the origin of the serpin inhibitory mechanism remain obscure. In this study we characterize 12 serpin-like sequences in the genomes of prokaryotic organisms, extending this protein family to all major branches of life. Notably, these organisms live in dramatically different environments and some are evolutionarily distantly related. A sequence-based analysis suggests that all 12 serpins are inhibitory. Despite considerable sequence divergence between the proteins, in four of the 12 sequences the region of the serpin that determines proteinase specificity is highly conserved, indicating that these inhibitors are likely to share a common target. Inhibitory serpins are typically prone to polymerization upon heating; thus, the existence of serpins in the moderate thermophilic bacterium Thermobifida fusca, the thermophilic bacterium Thermoanaerobacter tengcongensis, and the hyperthermophilic archaeon Pyrobaculum aerophilum is of particular interest. Using molecular modeling, we predict the means by which heat stability in the latter protein may be achieved without compromising inhibitory activity.
Introduction
Serpins are members of a large family of proteinase inhibitors (about 500 identified to date; Irving et al. 2000 ) that inactivate target proteinases using a unique “inhibition by distortion” mechanism (Huntington, Read, and Carrell 2000 ). Whereas the majority of serpins target serine proteinases, several members have been discovered that are capable of inhibiting other classes of proteinases (e.g., the viral serpin crmA with caspase-1 [Ray et al. 1992] and SCCA-1 and MENT with papain-like cysteine proteinases [Schick et al. 1998 ; Irving et al. 2002] ). In addition, several serpins have been identified that do not perform an inhibitory role—for example, the 47-kDa heat shock protein acts as a molecular chaperone (Nakai et al. 1992 ). Serpins have been characterized in many cellular and extracellular contexts, participating in processes such as blood clotting, intracellular signaling, prevention of tissue injury, and hormone binding (Silverman et al. 2001 ).
In contrast to small “rigid” proteinase inhibitors such as those of the Kazal or Kunitz family, inhibitory serpins rely on a complex conformational change to inhibit the target proteinase (Huntington, Read, and Carrell 2000 ). This mechanism requires precise timing to capture the target proteinase: the serpin fold has evolved to function as a finely tuned molecular machine. Some mutations that alter the conformational mobility of the serpin scaffold can result in the spontaneous transition to inactive forms, such as polymers or the latent conformation (see Stein and Carrell 1995 ). Other mutations interfere with the process of conformational change and impair serpin function by promoting substrate-like rather than inhibitory behavior (Hopkins, Carrell, and Stone 1993 ; Stein and Carrell 1995 ).
A fundamental, unanswered question in the field is the evolutionary origin of the serpin fold, which is closely related to the origin of the serpin mechanism itself. In a previous study, we performed an extensive phylogenetic investigation of the serpin superfamily (Irving et al. 2000 ). Despite using sensitive database searching methods such as PSI-BLAST and Hidden Markov Models, we were able to identify serpins only in viruses and higher (multicellular) eukaryotes and were unable to identify any putative prokaryotic or fungal serpin. This observation was somewhat surprising because the presence of serpins in both the animal and plant kingdoms suggests that they should also be found in a common ancestor (e.g., fungi or prokaryotes). Information bearing on the question of whether the ancestral serpin was a proteinase inhibitor would provide insight into the origin of the inhibitory mechanism: did this mechanism develop with the serpin fold or did the first serpin fulfill a noninhibitory function? Current biochemical evidence does not provide a simple answer. The most common serpin targets, trypsin-like serine proteinases, have bacterial homologs (HtrA; Lipinska, Zylicz, and Georgopoulos 1990 ), and serpins have shown activity against bacterial subtilisin (Komiyama et al. 1996 ; Dahlen, Foster, and Kisiel 1997 ) and the caspase-like gingipain K enzyme from Porphyromonas gingivalis (Snipas et al. 2001 ). These enzymes are present in organisms that lack recognizable serpin sequences.
In this study we have identified, from recently released genomic data, the first examples of prokaryotic serpins. Two closely related cyanobacteria, Nostoc punctiforme and Anabaena sp. PCC 7120, the firmicutes Thermobifida fusca, Desulfitobacterium hafniense, Ruminococcus albus, and Thermoanaerobacter tengcongensis, the green nonsulfur bacteria Dehalococcoides ethenogenes, the crenarchaeote Pyrobaculum aerophilum, and the euryarchaeotes Methanosarcina acetivorans and Methanosarcina mazei all contain putative serpin sequences. Sequence motifs and patterns of conservation suggest that they adopt a serpin-like fold and are capable of proteinase inhibition. The distribution of prokaryotic serpin sequences is sporadic. Although the serpin-bearing prokaryotes occur in markedly different environments, it remains unclear as to whether these genes have arisen through lateral gene transfer from eukaryotes or through inheritance of an ancient, prokaryotic serpin ancestor. The existence of a serpin in a hyperthermophilic organism is of particular interest. Using molecular modeling with reference to experimental evidence, we suggest a means by which resistance to polymerization can be obtained without compromising inhibitory activity.
Materials and Methods
Database Searching
A BLAST search was performed, using the protein sequence of human α1-antitrypsin and the tblastn program, against the finished and unfinished microbial genomes at the NCBI (http://www.ncbi.nlm.nih.gov/blast). Seven annotated nucleotide sequences from Anabaena sp. PCC 7120 (Kaneko et al. 2001 ), P. aerophilum (Fitz-Gibbon et al. 2002 ), M. acetivorans (Galagan et al. 2002 ), M. mazei (Deppenmeier et al. 2002 ), T. tengcongensis (Bao et al. 2002 ), and five unannotated draft sequences were identified with an expect score less than 1 × 10−3. The preliminary genome data were obtained from the DOE Joint Genome Institute (JGI; http://www.jgi.doe.gov) and The Institute for Genomic Research (TIGR; http://www.tigr.org). Nucleotide sequence of high quality was obtained for N. punctiforme and T. fusca serpins (phrap score of 90, or 10−7 errors per 100 base pairs) and of reasonable quality for D. hafniense (phrap score of 37–90). Information on sequence quality was not available for D. ethenogenes or R. albus. For each putative serpin, the amino acid translation was used to probe the database a second time but failed to identify further homologous sequences.
Sequence Analysis and Phylogenetic Relationships
The putative serpins were aligned and phylogenetic trees constructed as described previously (Irving et al. 2000 ). Briefly, a structure-based alignment was generated of human α1-antitrypsin (pdb accession 1qlp), human antithrombin (1azx), Manduca sexta serpin 1K (1sek), and chicken ovalbumin (1ova). The bacterial serpins were aligned to this “seed” alignment with reference to secondary structure-specific gap penalties using CLUSTALW (Higgins, Thompson, and Gibson 1996 ). Sites in the alignment were compared with sites strictly conserved in >70% of the serpin superfamily (identified in Irving et al. 2000 ).
A profile alignment was used to incorporate these sequences into an alignment of 219 sequences from the superfamily as a whole, which in turn was used to derive bootstrapped neighbor-joining distance-based trees (500 replicates, JTT substitution model, using MOLPHY; Adachi and Hasegawa 1996 ). Clusters of evolutionarily related sequences were obtained after analysis of the bootstrapped trees using the majority consensus tree approach (Felsenstein 1985 ), the comparison consensus method, and the tree division method (Irving et al. 2000 ). For the tree division method, the “clade-defining” proteins were from (1) vertebrates: human α1-antitrypsin (GenPept accession P01009), heat shock protein 47 (P29043), neuroserpin (Q99574), heparin cofactor II (AAC16324), alpha-2-antiplasmin (Q95121), angiotensinogen (P11859), and ovalbumin (P01012); (2) arthropods: Limulus intracellular coagulation inhibitor-1 (BAA06909) and M. sexta serpin 1K (AAB58491); (3) nematodes: Caenorhabditis elegans (AAB37049); (4) plants: barley protein Z (CAA66232), Triticum aestivum (CAA72274) and Arabidopsis thaliana (AAC27146).
Local Sequence Clusters
Predicted genes at genome positions 892,000–912,000 (Anabaena sp. PCC 7120), 15,000–35,000 (P. aerophilum), 2,750,757–2,773,734; 3,235,948–3,255,573; and 4,189,820–4,190,826 (M. acetivorans), 1,508,868–1,529,055 (T. tengcongensis), and sections 295–297 (accessions AE013513–AE013515, M. mazei) were obtained from the GenBank database. Nucleotide sequence from the region surrounding each putative serpin was obtained for T. fusca (within 10 kb upstream and downstream, on contig 64), N. punctiforme (contig 423), and D. hafniense (contig 3204). Each region was then compared with the nonredundant database using the standalone blastx program (genetic code 11; maximum expect 1 × 10−3; otherwise default parameters) and potential genes were thereby inferred.
Search for Proteinases Correlating with the Presence of Serpins
Sequence alignments representative of all proteinase families in the MEROPS database (http://www.merops.co.uk; Rawlings, O'Brien, and Barrett 2002 ) were obtained. A consensus sequence was derived from each alignment, and in conjunction with tblastn it was used to identify homologs in the prokaryotic genomes.
Molecular Modeling
The serpin from P. aerophilum was modeled using the MODELLER program (Sali and Blundell 1993 ) within the QUANTA package (Accelrys, San Diego, Calif.) using the structure of antithrombin (PDB accession, 1azx) as a template. The alignment between the two proteins was obtained using PSI-BLAST and manually adjusted to take account of elements of secondary structure. Where necessary, manual side-chain refinement was performed using CHARMm. A Ramachandran plot revealed that all residues in the model were in allowed conformations.
Results
A BLAST search of the microbial BLAST database using the protein sequence of α1-antitrypsin identified seven bacterial serpin-like sequences and five archaeal sequences, with significant expect scores (table 1 ). These putative proteins were aligned with human serpins α1-antitrypsin and antithrombin, the tobacco hornworm serpin 1K, and chicken ovalbumin (fig. 1 ). All 12 contain between 23% and 29% sequence similarity with antitrypsin. Although the lower of these values lie within the “twilight zone” of protein sequence comparison, all exhibit the highly conserved motifs characteristic of the serpin superfamily. In particular, the bacterial serpins and most of the archaeal serpins possess 38–47 of the 51 residues strictly conserved in >70% of serpin family members (table 1 and fig. 1 ). This falls well within the observed range for other serpin family members (on average, 43.5 ± 5.9 residues) and indicates that the sequences encode functional serpins. The serpin from P. aerophilum has 32 residues consistent with conserved serpin positions and thus represents the most “diverged” prokaryotic sequence. This is likely to reflect the extreme environment that the organism occupies: deep thermal vents with temperatures that can rise above 100°C (Volkl et al. 1993 ).
We sought to determine whether the presence of serpins in the genomes of these organisms correlates with an endogenous enzyme target. Some inhibition by serpins has been shown for A1 (pepsin), C1 (papain), C14 (caspase), C25 (gingipain), S1 (trypsin), and S8 (subtilisin) proteinase families. A BLAST search revealed that eight of the prokaryotic genomes (with the exception of D. ethenogenes and R. albus) contained subtilisin-like sequences, and all save Methanosarcina spp. contained homologs of the trypsin-like enzyme HtrA. Caspase-like enzymes, a fold that includes the gingipains, have been found in Anabaena, N. punctiforme, and D. ethenogenes species (Aravind and Koonin 2002 ; Koonin and Aravind 2002 ). None of the other proteinase families considered were found. It is clear, however, that the presence of these enzymes does not correlate exclusively with presence of serpins. Of the prokaryotic genomes lacking serpins, subtilisin-like enzymes were present in ∼20%, trypsin-like enzymes in ∼60%, and caspase-like enzymes are also found elsewhere (Koonin and Aravind 2002 ). A similar search using all proteinase classes in the MEROPS database (as on December 17, 2001) failed to identify other types present exclusively in serpin-bearing genomes.
It has been noted that coexpressed genes are frequently situated as contiguous clusters within an operon. But an analysis of regions adjacent to the serpin sequences did not reveal, from one organism to the next, a consistent pattern of neighboring genes that might indicate the presence of a conserved operon. Five of the sequences were taken from unassembled contig data; thus whether they are present in transposable elements or plasmids is not yet clear. The genes from Anabaena sp. PCC 7120, P. aerophilum, T. tengcongensis, M. mazei, and M. acetivorans are localized to the single chromosome of each organism.
Recognizable serpin sequences are not ubiquitous or even widely distributed in the prokaryotic kingdom. Serpins appear to be absent from all other sequenced archaeal organisms (13 finished and two unfinished), from proteobacteria (26 finished and 43 unfinished), and from several other branches less well represented in genome sequencing efforts. Furthermore, one finished and four unfinished cyanobacterial genomes (including N. punctiforme) and 21 complete firmicute genomes (a family which includes T. fusca) also lack serpins.
Discussion
Database searching identified putative serpins in seven species of bacteria and three archaeal species. It is likely that all 12 are functional as proteinase inhibitors because all possess the sequence of small residues within the hinge region, key secondary structural elements, and many of the conserved residues from the family as a whole.
The actinomycete T. fusca (formerly Thermomonospora fusca) is a thermophilic soil bacterium with an optimal growth temperature of 55°C that plays an important role in the degradation of plant detritus, for example in compost heaps (Crawford 1975 ). A related organism, the firmicute T. tengcongensis, grows at 75°C and is found in hot springs (Xue et al. 2001 ). The crenarchaeon P. aerophilum, a member of which has been obtained from a boiling marine water hole, is a hyperthermophile with an optimal growth temperature of 100°C (Fitz-Gibbon et al. 2002 ). The existence of serpins in thermophilic organisms is of particular interest because serpins are metastable in their native state and susceptible to heat-induced polymerization (Lomas et al. 1992 ). Thermophilic organisms' euryarchaeote neighbors, M. acetivorans and M. mazei, are methane-producing organisms that grow at moderate temperatures (35–40°C) and were first isolated from marine sediments (Sowers, Baron, and Ferry 1984 ). Desulfitobacterium hafniense is a gram-positive, endospore-forming, strictly anaerobic bacterium initially isolated from municipal sludge that is capable of dechlorinating both aromatic and alkyl chlorinated compounds (for a review see El-Fantroussi, Naveau, and Agathos 1998 ). The filamentous nitrogen-fixing cyanobacterium N. punctiforme is able to form symbiotic relationships with a variety of terrestrial plants. Furthermore this organism is able to form a partnership with an obligate symbiotic fungus (Zygomycotina) to form the organism Geosiphon pyriforme, the only known example of endocytobiosis (intracellular association of two cells) between a fungus and cyanobacteria (Gehrig, Schussler, and Kluge 1996 ). Dehalococcoides ethenogenes is a eubacterium capable of reducing tetrachloroethene to ethane (Maymo-Gatell et al. 1997 ). Dehalococcoides ethenogenes, along with strain CBDB1 and several uncultivated bacteria, forms part of a clade phylogenetically removed from other bacterial families (Adrian et al. 2000 ). Finally, R. albus is a gram-positive, cellulolytic anaerobe that inhabits the gut of herbivores (Leatherwood 1965 ). Thus serpins appear to be present in a wide variety of prokaryotes that live in diverse environments.
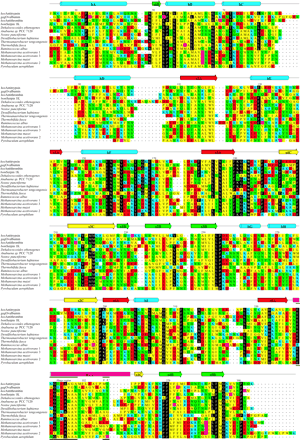
Bootstrapped neighbor-joining trees were constructed on the basis of the alignment of the prokaryotic serpins with other members of the serpin superfamily (identified in Irving et al. 2000 ). The majority consensus tree revealed three strictly conserved associations: the two cyanobacteria, N. punctiforme and Anabaena sp. PCC 7120, clustered together in 100% of the time; the firmicutes D. hafniense, R. albans and T. tengcongensis coincided in 100% of trees; and the euryarchaeote serpins from Methanosarcina spp. also formed a clade with 100% support (fig. 2 ). These associations are not unexpected: they reflect the close evolutionary origin of these prokaryotic species. The comparison consensus method, which identifies underlying relationships that may be obscured by other poorly resolved species, revealed a significant (100%) association between the cyanobacterial and firmicute serpins. It was not possible from these analyses to identify a well-supported relationship between these five bacterial serpins and those from T. fusca, D. ethenogenes, P. aerophilum, the Methanosarcina clade or any other subfamily. These five sequences therefore represent a novel bacterial clade in the serpin superfamily, the four Methanosarcina proteins form an archaeal clade, and the other three serpins are, at present, “orphans” (fig. 2 ).
Using the tree division method, which quantifies the association between members of the bootstrapped phylogenetic trees and certain prenominated sequences, the following question was asked: do the bacterial sequences associate more closely with (1) vertebrate, (2) arthropod, (3) nematode, or (4) plant serpins? The most highly supported association, between the T. fusca serpin and plant sequences, was found in 82.7% of bootstrap trees, suggestive but below the 95% significance threshold of the method. This relationship, although interesting in light of the environmental context of the bacterium, is therefore by no means conclusive. The serpins from cyanobacteria-firmicutes associated more closely with nematode sequences (74.4%) and then with plant sequences (15.4%). Because these associations are fairly strongly biased toward two different lineages, they suggest the possibility of lateral gene transfer.
Of 77 finished genomes, 67 did not contain any identifiable serpin homolog. Three scenarios seem feasible:
Serpins have an ancient origin deep in the prokaryotic tree, but most, because of a tendency to rapidly shed unnecessary genes from their genomes (Makarova et al. 1999 ; Makarova, Ponomarev, and Koonin 2001 ), have lost serpin homologs. This hypothesis is supported in part by the markedly different environments in which some of these organisms exist, which would make lateral transfer of the serpin genes less likely.
Serpins have a relatively recent origin within a subset of the prokaryotic kingdom and have been passed through lateral transfer to other prokaryotes and eukaryotes as well.
Serpins have an origin within eukaryotes and have been passed by lateral transfer to prokaryotes. The phylogenetic data shows some support for this hypothesis.
The latter two scenarios seem more favorable in light of the sporadic distribution of serpin genes in prokaryotic genomes; however, there is no evidence that strongly favors one over the other. With the elucidation of further sequences of prokaryotic organisms living at the boundaries of life, it should be possible to elucidate the evolutionary origin of serpins with greater certainty.
Sequence alignment of the 12 serpins with serpins of known structure reveals that the bacterial sequences, and all but one of the archaeal sequences, are predicted to contain the full complement of elements of secondary structure (fig. 1 ). This is in contrast to viral serpins, several of which contain deletion of entire secondary structure elements (Renatus et al. 2000 ; Simonovic, Gettins, and Volz 2000 ; Guerin et al. 2001 ). As with the viral serpins, the protein from the archaeon P. aerophilum appears to have lost its D-helix; however, we predict that P. aerophilum retains all other structural elements. All 12 prokaryotic serpins contain the pattern of small amino acids in the hinge region of the reactive center loop (RCL) (see fig. 1 ), characteristic of serpins that function as proteinase inhibitors; hence we suggest that the most likely function of these proteins is the inhibition of proteinases.
One striking feature of four of the bacterial serpins (the two Nostoc species, D. ethenogenes and D. hafniense) is that the P1 and P1′ residues (refer to abbreviations for Pn notation) are identical (T-S). The RCL has been noted to be variable in duplicated serpin genes undergoing functional diversification (Inglis et al. 1991 ; Kaiserman et al. 2002 ), and this is indicative of negative (purifying) selection. Two observations further support this hypothesis. First, these serpins are substantially different from one another overall, with 34%–69% identity. Second, three of the sequences share a strong evolutionary relationship (Nostoc spp. and D. hafniense; see fig. 2 ), but R. albus and T. tengcongensis, which are also predicted to be part of this clade, do not have a T-S at the P1-P1′. Furthermore, D. ethenogenes (which does not share a significant evolutionary relationship with Nostoc spp. or D. hafniense) does have a T-S at this site. It therefore appears likely that the four predicted inhibitors from Nostoc spp., D. hafniense, and D. ethenogenes are under selective pressure to target proteinases of similar or identical specificity. Similarly, T. fusca and T. tengcongensis both have the residues A-G; R. albus and M. acetivorans 1 share a P1E; and P. aerophilum and M. acetivorans share P1V.
The sequences A-G and T-S do not fit the substrate profile of any known serine proteinase. But examination of serpins that are known to inhibit papain-like cysteine proteinases reveal that these inhibitors often contain small residues such as Thr at P1 (Schick et al. 1998 ; Irving et al. 2002 ). Furthermore, the P2 position (usually of greater importance in targeting a serpin to a cysteine proteinase) is usually occupied by a large hydrophobic residue that interacts with the S2 specificity pocket. The inhibitors from T. fusca and D. ethenogenes both have a P2 leucine, and we hypothesize that these serpins may be able to inhibit papain-like cysteine proteinases.
The only serpins known to have an acidic P1 residue are the granzyme B inhibitor PI-9 (Sun et al. 2001 ) and the caspase inhibitor crmA (Ray et al. 1992 ). Nevertheless, it seems unlikely that the R. albus serpin (P1-P1′ sequence E-A) would interact with granzyme B or caspase-1 in a physiological context such as the milieu of the rumen. We note that several bacteria have been shown to express enzymes that can cleave substrates with acidic P1 residues (Barbosa, Saldanha, and Garratt 1996 ), including the V8 serine proteinase of the bacterium Staphylococcus aureus, which has a homolog in R. albus.
Methanosarcina acetivorans is unique among the prokaryotes in that it possesses three serpin genes, each of which has a unique P1-P1′ sequence, suggesting that each targets a different enzyme. Methanosarcina mazei appears to have inherited one of these inhibitors. The close predicted evolutionary relationship between M. acetivorans serpin 3 and the M. mazei serpin and the similarity in their RCL sequence (P1-P1′ of G-V and G-M) suggests the two may interact with the same class of proteinase.
We were unable to find a direct correlation between any of the current proteinase family classifications and serpins in the prokaryotic genomes. Therefore, it remains to be determined whether the serpins have evolved to inhibit endogenous proteinases or to target proteinases in the local environment.
The presence of a serpin in P. aerophilum is particularly intriguing because its optimum growth temperature (100°C) is well in excess of the temperature that a typical serpin would be expected to remain in the active inhibitory conformation. Numerous studies have demonstrated that serpins are susceptible to heat-induced polymerization, as a consequence of the metastability of the native state (Lomas et al. 1992 ). This metastability is crucial for serpin inhibitory function: conformational change is required to trap the target proteinase in a distorted, inactive state (Huntington, Read, and Carrell 2000 ). Certain members of the serpin family do demonstrate enhanced stability—for example, the noninhibitory serpin ovalbumin denatures at 73°C (Dong et al. 2000 ). Therefore, while it is conceivable that the serpin from T. fusca serpin may remain active at 55°C through a dramatic increase in stability of the native form, we suggest that the serpin from P. aerophilum must demonstrate some special feature in order for it to use conformational change for inhibition of target proteinases at 100°C. Although it is possible that the P. aerophilum protein no longer undergoes conformational change to inhibit target proteinases, the presence of a typical inhibitory “hinge” in this serpin suggests that this is not the case. To investigate the P. aerophilum serpin further, we built a molecular model using the structure of antithrombin as a template. These data reveal that the predicted P. aerophilum serpin is able to adopt the serpin fold and that all essential elements of the serpin “core” are present (fig. 3 ). We predict that the P. aerophilum serpin lacks the D-helix, however, this is not unprecedented—the X-ray crystal structure of the viral serpin crmA reveals that the D-helix can be “lost” without disrupting the serpin fold (Renatus et al. 2000 ; Simonovic, Gettins, and Volz 2000 ). In comparison with typical serpins, the P. aerophilum serpin is also predicted to contain a significant insertion at the base of the E-helix. Most strikingly, however, we note that the RCL of the P. aerophilum serpin contains a cysteine residue at the P1′ position that we predict would be able to form a disulfide bond with a second cysteine on strand s3C of the C β-sheet. Stabilizing disulfides have been noted in the crystal structure of an intracellular P. aerophilum enzyme, adenylosuccinate lyase (Toth et al. 2000 ). Such an interaction would be predicted to “tie down” the RCL and prevent inappropriate conformational change (such as polymerization). In a study based on antitrypsin, in which a disulfide bond was introduced between the RCL and strand s1C of the C β-sheet, the presence of a covalent linkage prevented the serpin from polymerizing (Chang et al. 1997 ). It was hypothesized that the introduction of this disulfide bond prevented polymerization by restricting conformational change in the RCL and the first strand of the C β-sheet. We predict that a similar situation exists in the P. aerophilum serpin (fig. 4A ). A related study showed that the presence of the introduced disulfide bond in antitrypsin did not affect inhibitory activity (Hopkins et al. 1997 ). Similarly, the predicted disulfide bond in P. aerophilum serpin would not be expected to affect the inhibitory mechanism because the RCL would still be able to rapidly insert into the A β-sheet after cleavage at the P1-P1′ (fig. 4B ). Thus we predict that nature may have used disulfide bonds as a method of stabilizing serpins in the most primitive and extreme environments.
The discovery of serpins in both prokaryotic kingdoms has important implications for the evolution of the serpin superfamily, which was previously believed to be restricted to higher eukaryotic organisms and their viruses. It is not yet clear whether the presence in prokaryotes is the product of gene inheritance or lateral transfer. In time, as sequencing efforts progress, this question may be answered. Nevertheless, these proteins should present useful information on adaptation of the serpin scaffold to the extreme environments in which many prokaryotic organisms live.
Peer Bork, Reviewing Editor
Abbreviations: Pn notation, according to the convention of Schecter and Berger (1967), residues of a peptide substrate N-terminal to the scissile bond are denoted Pn, …, P2, P1 and those C-terminal to the scissile bond are denoted P1′, P2′, …, Pm′ RCL, reactive center loop.
Keywords: serpin prokaryote comparative genomics proteinase inhibitor phylogeny proteinase
Address for correspondence and reprints: James C. Whisstock, Department of Biochemistry and Molecular Biology, Monash University, Clayton Campus, Melbourne, Victoria 3800, Australia. james.whisstock@med.monash.edu.au
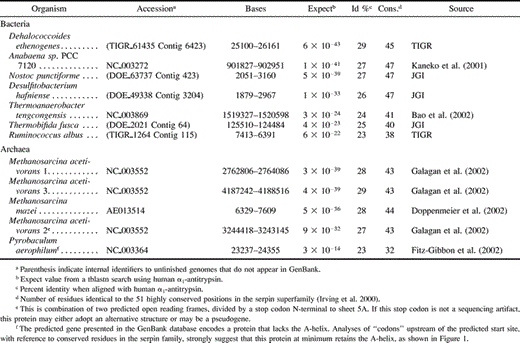
>Fig. 1.—Alignment between the prokaryotic serpins and selected members of the serpin superfamily. Each bacterial serpin contains a 3–71 residue N-terminal sequence not shown in the alignment because it is outside the serpin core. These N-terminal extensions share no significant sequence similarity with any known domain. Residues are colored according to type: polar uncharged (green), acidic (red), basic (blue), and nonpolar (yellow). Those residues which are identical to residues conserved in >70% of the superfamily (Irving et al. 2000 ) appear in white-on-black. Elements of consensus secondary structure are shown at the top. The “hinge region” and P1-P1′ residues are indicated by thick lines. Numbering is according to human α1-antitrypsin
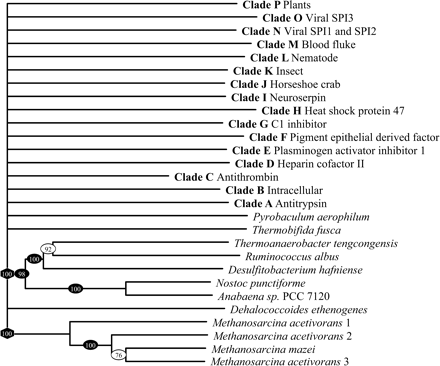
Fig. 2.—Tree illustrating the deduced evolutionary relationships between the bacterial serpins. Conventional bootstrap values are indicated by ovals; the rectangular symbol at the root of the tree indicates the association was determined using the comparison consensus method (Irving et al. 2000 ); and the hexagon indicates the relationship was strictly conserved in all bootstrap trees. The polytomy at the base of the tree indicates that some of the relationships could not be deduced with confidence. The 219 eukaryotic serpins included in the analysis are summarized by their respective clade designations (Irving et al. 2000 ). Branch lengths were determined using CLUSTALW (Higgins, Thompson, and Gibson 1996 )
Fig. 3.—Molecular model of the serpin from P. aerophilum, indicating the position of the predicted disulfide bond linking the RCL to the C β-sheet. The RCL appears at the top of the molecule, highlighted in black; the cysteine residues are shown in ball-and-stick; and the insertion near the E-helix appears in black near the base of the molecule. Selected elements of secondary structure are labeled. The site in the RCL expected to be cleaved by a target proteinase is indicated by a triangle
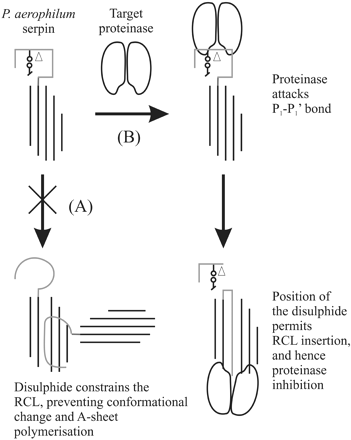
Fig. 4.—A schematic representation of the proposed influence of the RCL–C β-sheet disulfide bond on polymerization, and the serpin inhibitory mechanism. The RCL is shown as a gray line, with the cysteine residues represented by a line-and-circle and the site of cleavage by an empty triangle. The main A β-sheet of the serpin is illustrated by black lines, and the two lobes represent the target proteinase molecule. (A) According to the current model of loop-A sheet polymerization (or “domain-swapping”), the RCL undergoes conformational change and is integrated as a strand into the A β-sheet of another molecule. Consistent with experimental evidence (Chang et al. 1997 ), the presence of a disulfide would be expected to prevent this interaction by constraining the RCL of P. aerophilum serpin. (B) The disulfide bond is C-terminal to the scissile bond cleaved by a target proteinase. Consequently, upon cleavage, the RCL is still able to incorporate into the A β-sheet, as seen with the standard serpin mechanism of inhibition (Huntington, Read, and Carrell 2000 )
We thank the Australian Research Council and the National Health and Medical Research Council of Australia for support. J.C.W. is a NHMRC Senior Research Fellow and Monash University Logan Fellow. A.M.L. is supported by the Wellcome Trust. Preliminary data were obtained from the DOE Joint Genome Institute (JGI) at http://www.jgi.doe.gov and The Institute for Genomic Research (TIGR) at http://www.tigr.org, from projects funded by the DOE (with the exception of R. albus, funded by the United States Department of Agriculture). The sequence data from the JGI have been provided freely by the US DOE Joint Genome Institute for use in this publication only.
References
Adachi J., M. Hasegawa,
Adrian L., U. Szewzyk, J. Wecke, H. Gorisch,
Aravind L., E. V. Koonin,
Bao Q., Y. Tian, W. Li, et al. (21 co-authors)
Barbosa J. A., J. W. Saldanha, R. C. Garratt,
Chang W. S., J. C. Whisstock, P. C. Hopkins, A. M. Lesk, R. W. Carrell, M. R. Wardell,
Crawford D. L.,
Dahlen J. R., D. C. Foster, W. Kisiel,
Deppenmeier U., A. Johann, T. Hartsch, et al. (22 co-authors)
Dong A., J. D. Meyer, J. L. Brown, M. C. Manning, J. F. Carpenter,
El-Fantroussi S., H. Naveau, S. N. Agathos,
Felsenstein J.,
Fitz-Gibbon S. T., H. Ladner, U. J. Kim, K. O. Stetter, M. I. Simon, J. H. Miller,
Galagan J. E., C. Nusbaum, A. Roy, et al. (55 co-authors)
Gehrig H., A. Schussler, M. Kluge,
Guerin J. L., J. Gelfi, C. Camus, M. Delverdier, J. C. Whisstock, M. F. Amardeihl, R. Py, S. Bertagnoli, F. Messud-Petit,
Higgins D. G., J. D. Thompson, T. J. Gibson,
Hopkins P. C., R. W. Carrell, S. R. Stone,
Hopkins P. C., W. S. Chang, M. R. Wardell, S. R. Stone,
Huntington J. A., R. J. Read, R. W. Carrell,
Inglis J. D., M. Lee, D. R. Davidson, R. E. Hill,
Irving J. A., R. N. Pike, A. M. Lesk, J. C. Whisstock,
Irving J. A., S. S. Shushanov, R. N. Pike, E. Y. Popova, D. Bromme, T. H. Coetzer, S. P. Bottomley, I. A. Boulynko, S. A. Grigoryev, J. C. Whisstock,
Kaiserman D., S. Knaggs, K. L. Scarff, et al. (13 co-authors)
Kaneko T. Y. N., C. P. Wolk, et al. (22 co-authors)
Komiyama T., H. Gron, P. A. Pemberton, G. S. Salvesen,
Koonin E. V., L. Aravind,
Leatherwood J. M.,
Lipinska B., M. Zylicz, C. Georgopoulos,
Lomas D. A., D. L. Evans, J. T. Finch, R. W. Carrell,
Makarova K. S., L. Aravind, M. Y. Galperin, N. V. Grishin, R. L. Tatusov, Y. I. Wolf, E. V. Koonin,
Makarova K. S., V. A. Ponomarev, E. V. Koonin,
Maymo-Gatell X., Y. Chien, J. M. Gossett, S. H. Zinder,
Nakai A., M. Satoh, K. Hirayoshi, K. Nagata,
Rawlings N. D., E. O'Brien, A. J. Barrett,
Ray C. A., R. A. Black, S. R. Kronheim, T. A. Greenstreet, P. R. Sleath, G. S. Salvesen, D. J. Pickup,
Renatus M., Q. Zhou, H. R. Stennicke, S. J. Snipas, D. Turk, L. A. Bankston, R. C. Liddington, G. S. Salvesen,
Sali A., T. L. Blundell,
Schecter I., A. Berger,
Schick C., P. A. Pemberton, G. P. Shi, Y. Kamachi, S. Cataltepe, A. J. Bartuski, E. R. Gornstein, D. Bromme, H. A. Chapman, G. A. Silverman,
Silverman G. A., P. I. Bird, R. W. Carrell, et al. (15 co-authors)
Simonovic M., P. G. W. Gettins, K. Volz,
Snipas S. J., H. R. Stennicke, S. Riedl, J. Potempa, J. Travis, A. J. Barrett, G. S. Salvesen,
Sowers K. R., S. F. Baron, J. G. Ferry,
Stein P. E., R. W. Carrell,
Sun J., J. C. Whisstock, P. Harriott, B. Walker, A. Novak, P. E. Thompson, A. I. Smith, P. I. Bird,
Toth E. A., C. Worby, J. E. Dixon, E. R. Goedken, S. Marqusee, T. O. Yeates,
Volkl P., R. Huber, E. Drobner, R. Rachel, S. Burggraf, A. Trincone, K. O. Stetter,

{kind=link}
{kind=link}
{kind=link}
{kind=link}