





Abstract
REBASE is a comprehensive and fully curated database of information about the components of restriction-modification (RM) systems. It contains fully referenced information about recognition and cleavage sites for both restriction enzymes and methyltransferases as well as commercial availability, methylation sensitivity, crystal and sequence data. All genomes that are completely sequenced are analyzed for RM system components, and with the advent of PacBio sequencing, the recognition sequences of DNA methyltransferases (MTases) are appearing rapidly. Thus, Type I and Type III systems can now be characterized in terms of recognition specificity merely by DNA sequencing. The contents of REBASE may be browsed from the web http://rebase.neb.com and selected compilations can be downloaded by FTP (ftp.neb.com). Monthly updates are also available via email.
OVERVIEW
The previous description of REBASE in the 2010 NAR Database Issue (1) described 3945 biochemically or genetically characterized restriction-modification (RM) systems and included an analysis of just 400 bacterial and archaeal genomes that were present in GenBank (2,3). Since then the number of completely sequenced genomes has risen to more than 5000 and comprehensive descriptions of the RM content of these fully sequenced genomes are available through REBASE in several different formats. In addition, selected genome shotgun sequence sets are also analyzed and the RM system components extracted. The predicted proteins are named according to standard conventions (4) and labeled with the suffix ‘P’ to indicate they are putative. If and when they become biochemically characterized, then the appropriate Roman numeral is used to replace the Open Reading Frame (ORF)-designation initially assigned.
The fastest growing segment of biochemically characterized RM system components are the DNA methyltransferases, currently being found using real-time Single Molecule Real Time (SMRT) sequencing (5–7). In a large number of cases the recognition motifs for DNA methyltransferases arising from such sequencing can be matched with the genes that encode them. For instance, the recognition sequences of Type I systems have a very characteristic bipartite structure and when the genome contains only one such Type I set of genes, then it is clear that the methyltransferase gene and its associated specificity gene are both active and the recognition motif can be assigned. This in turn allows propagation of these specificities both to putative Type I systems and also in many cases allows the identification of active Type I systems in genomes where more than one Type I system is present. These matches are made via REBASE and documented. The same is true for Type III RM systems and again, is made possible because the recognition specificity for the system is encoded in the methyltransferase gene. The enormous growth in these systems is shown in Figure 1, which documents the rate of discovery of Type I and Type III RM system specificities since the very first one was discovered and characterized in 1968 (8).
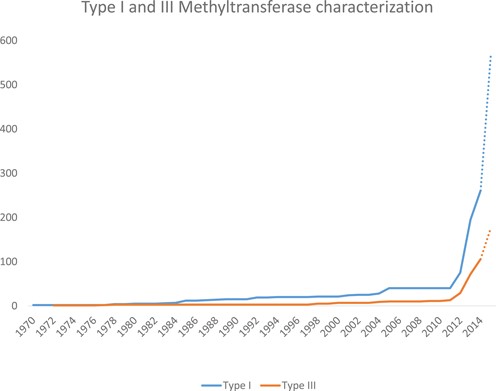
Dotted lines signify methylated motifs awaiting gene assignment. The sharp increase starting in 2012 is the result of the introduction of SMRT sequencing.
Given the unique ability of SMRT sequencing to detect methylated bases it is almost always extremely useful to run the ‘Modification and Motif Analysis’ protocol on such data, to generate the motif_summary.csv file that summarizes the methylation patterns that are present. We encourage everyone sequencing bacterial genomes to generate these summaries and submit them to GenBank as part of their sequence submission. We also welcome their direct submission to REBASE and have an interface specifically for this purpose (http://rebase.neb.com). Such data may be submitted prior to publication and a matching analysis will be performed upon receipt. The results can be kept private until the submitter is ready to publish. For individuals mainly interested in the genome sequence, this further analysis can add much value to the sequence by indicating which RM systems are present and might cause problems during transformation.
Another new feature of REBASE is the identification of a Gold Standard Set of RM system components where each component having a known sequence has been experimentally characterized as having a defined restriction or modification activity. This Gold Standard Set then allows accurate and traceable propagation of annotation into putative genes present in newly sequenced genomes. In future, all RM system specificity predictions will contain traceable annotation back to this reference Gold Standard Set.
From the REBASE website users have a variety of resources available that facilitate the analysis of sequence information, including tools for analyzing sequences (REBASE TOOLS), that allow restriction enzyme recognition sites to be found in submitted sequences (NEBCUTTER) and an implementation of BLAST to allow searching against all sequences in REBASE. Specialty lists of sequence data (REBASE LISTS) such as all Type I specificity subunits or the Gold Standard Set are available for download. In addition, an advanced search feature enables REBASE to be queried by users for specific combinations of information about RM system components, including searching by date of entry. Additional features are added regularly and the site should be consulted for a brief description of any new features as they appear.
Special thanks to many individuals who have so kindly contributed their unpublished results for inclusion in this compilation and to the REBASE users who continue to guide our efforts with their helpful comments. Over the last 2 years many individuals and sequencing centers have contributed their unpublished PacBio data and we are extremely grateful for their willingness to share their data with the wider biological community. We are especially grateful to Karen Otto for administrative help.
FUNDING
REBASE is funded completely by New England Biolabs. Funding for open access charge: New England Biolabs Inc.
Conflict of interest statement. None declared.

{kind=link}
Comments