




Abstract
Microlissencephaly is a rare brain malformation characterized by congenital microcephaly and lissencephaly. Microlissencephaly is suspected to result from abnormalities in the proliferation or survival of neural progenitors. Despite the recent identification of six genes involved in microlissencephaly, the pathophysiological basis of this condition remains poorly understood. We performed trio-based whole exome sequencing in seven subjects from five non-consanguineous families who presented with either microcephaly or microlissencephaly. This led to the identification of compound heterozygous mutations in WDR81, a gene previously associated with cerebellar ataxia, intellectual disability and quadrupedal locomotion. Patient phenotypes ranged from severe microcephaly with extremely reduced gyration with pontocerebellar hypoplasia to moderate microcephaly with cerebellar atrophy. In patient fibroblast cells, WDR81 mutations were associated with increased mitotic index and delayed prometaphase/metaphase transition. Similarly, in vivo, we showed that knockdown of the WDR81 orthologue in Drosophila led to increased mitotic index of neural stem cells with delayed mitotic progression. In summary, we highlight the broad phenotypic spectrum of WDR81-related brain malformations, which include microcephaly with moderate to extremely reduced gyration and cerebellar anomalies. Our results suggest that WDR81 might have a role in mitosis that is conserved between Drosophila and humans.
Introduction
The development of the human cerebral cortex is an orchestrated process involving the birth of neural progenitors in the periventricular germinal zones, cell proliferation characterized by both symmetric and asymmetric mitoses and the subsequent migration of post-mitotic neurons to their final destinations in six highly ordered, functionally-specialized layers (Bystron et al., 2008; Rakic, 2009). Understanding of the molecular mechanisms guiding these intricate processes is in its infancy, and substantially driven by the discovery of rare mutations that cause malformations of cortical development (MCD) (Barkovich et al., 2012; Guerrini and Dobyns, 2014; Jamuar and Walsh, 2015). These mutations are a major cause of intellectual disability, epilepsy, and neurological deficits, often resulting in life-long support and treatments. MCD are classified based on the stage of development at which the developmental process was likely disturbed: neural progenitor proliferation or apoptosis, neuronal migration and cortical organization (Manzini and Walsh, 2011; Barkovich et al., 2012).
Abnormal proliferation of neural progenitors and neuronal migration can result in a wide range of disease phenotypes such as microcephaly (MIC, MIM 251200) and lissencephaly (LIS, MIM 607432) (Gilmore and Walsh, 2013). Congenital microcephaly is classified as a disorder of neurogenesis suggesting either a decrease in the number of proliferative divisions or an increase in apoptotic cell death of neural progenitors. Underlying cellular mechanisms include abnormal mitotic spindle structure, structurally abnormal or extra centrosomes, altered cilia function, impaired DNA repair, DNA damage response signalling and DNA replication, along with attenuated cell cycle checkpoint proficiency (Thornton and Woods, 2009; Bilgüvar et al., 2010; Kaindl et al., 2010; Bettencourt-Dias et al., 2011). In contrast to microcephaly, human lissencephaly manifests as a lack of normal cortical folding. The lack of folding reflects abnormal histological organization of the cortical layers and is most often due to disruption of neuronal migration (Bystron et al., 2008). Microlissencephaly (MLIS, MIM 616212) is an extremely rare entity characterized by the combination of extreme primary microcephaly with disordered cortical lamination. Most cases of microlissencephaly are described in consanguineous families suggesting an autosomal recessive inheritance with only a few disease-causing genes identified thus far. These include biallelic mutations in NDE1 (Alkuraya et al., 2011), KATNB1 (Hu et al., 2014; Mishra-Gorur et al., 2014), and WDR62 (Bilgüvar et al., 2010). NDE1 encodes a partner of dynein that localizes to the centrosome and mitotic spindle poles (Alkuraya et al., 2011). KATNB1 encodes a microtubule-severing enzyme that localizes to microtubules and centrosomes (Hu et al., 2014; Mishra-Gorur et al., 2014) and WDR62 encodes another centrosomal protein (Chen et al., 2014). Dominant mutations in tubulin genes, TUBA1A, TUBB2B and TUBB3 (Bahi-Buisson et al., 2014; Fallet-Bianco et al., 2014) have also been identified in patients with microlissencephaly, underlining the critical role of the microtubule cytoskeleton in this condition. Remarkably, most of the genes involved in microlissencephaly can also cause microcephaly, suggesting both conditions belong to the same disease spectrum. Despite significant progress in understanding the genetic basis of these conditions, the causative genes and pathophysiological mechanisms underlying most cases of microcephaly/microlissencephaly remain unknown. This makes it challenging to provide families with a genetic diagnosis, prenatal testing and genetic counselling (Bahi-Buisson et al., 2014).
In the present study, we report on seven novel compound heterozygous mutations in WDR81 leading to microcephaly with moderate to extremely reduced gyration with or without cerebellar anomalies ranging to cerebellar hypoplasia to cerebellar atrophy. In addition, we show that in patient fibroblasts and in Drosophila, WDR81 disruption is associated with an increased mitotic index and delayed prometaphase/metaphase transition.
Overall, our results expand the phenotypic spectrum of WDR81 mutations and suggest a highly conserved role of this gene in mitotic progression.
Patients and methods
As part of our ongoing research program on cortical malformations, we performed whole exome sequencing in individuals in whom clinically available molecular genetic tests had failed to detect any mutation, and for whom the phenotype shows specific features that could allow the identification of additional cases for a replicative cohort if a novel gene is identified.
In all cases, metabolic work up and array comparative genomic hybridization were normal. Next-generation sequencing on a panel of genes involved in MCD, including NDE1, WDR62 and tubulin genes did not demonstrate any mutations in 50 genes of MCD. The genetic analysis performed was subject to informed consent procedures and approved by the Institutional Review Boards at Necker Enfants Malades Hospital and Paris Descartes University.
Whole-exome sequencing
Whole exome sequencing of peripheral blood DNA from proband and both parents was performed using the Agilent SureSelect Human All Exon Kits v5, and sequence was generated on a HISeq2500 machine (Illumina). Sequences were aligned to hg19 by using BWA v.0.6.1, and single nucleotide variants (SNVs) and indels were called by using GATK v.1.3. Annotation of variants was performed with GATK Unified Genotyper. All calls with read coverage of ≤2 ×or a Phred-scaled SNP quality score of ≤20 were removed from consideration. The annotation process was based on the latest release of the Ensembl database. Variants were annotated and analysed using the Polyweb software interface designed by the Bioinformatics platform of University Paris Descartes and Imagine Institute.
Filters used for variant screening were as following: (i) all variants previously observed (in dbSNP138 and/or in in-house projects) were excluded; (ii) only variants leading to abnormal protein sequence (splicing, non-synonymous, frameshift, stop) were retained; (iii) we considered the PolyPhen-2 and SIFT prediction status as informative but not restrictive; and (iv) because most MCD are autosomal recessive, we searched for autosomal recessive events. Because all patients were sporadic cases from unrelated parents, the following models of inheritance in the variant screening were considered, by order of priority: autosomal recessive (in particular compound heterozygous but without excluding homozygous variants), X-linked and de novo SNVs.
Validation was achieved by standard bidirectional Sanger methods (primer sequences available on request).
Cell culture
Available fibroblast cells from Patients IM-MCD_606 and CerID-22 and from age-matched controls were grown in Opti-MEM® supplemented with 1% foetal bovine serum, 1% penicillin-streptomicin and 1% Fungizone at 37°C in humid air containing 5% CO2. For cell proliferation studies, patient and control cells were plated at the same concentration and synchronized by serum starvation for 24 h after which serum was reintroduced for 24 h. For ciliogenesis studies, confluent fibroblasts were serum starved for 48 h.
Real-time reverse-transcription PCR
RNA was extracted from patient and control fibroblasts and Drosophila brain tissues using Qiagen RNA kit, including on-column DNase digestion. RNA was quantified using the spectrophotometer NanoDrop 2000 (Thermo Fisher Scientific). cDNA synthesis from total RNA was conducted using the GeneAmp RNA PCR Core Kit (Applied Biosystems) or Verso cDNA kit with random hexamers. ACTB was used as internal control gene. cDNA Sanger sequencing was performed as described above for genomic DNA. Real time RT-PCR was performed using the GoTaq® qPCR Master Mix (Promega) and the WDR81 Forward/Reverse primer set. HPRT and GUSB genes were used as internal control for normalization in fibroblasts, and Gapdh1 for normalization in Drosophila tissues. (Sequences of primers are available upon request).
Immunocytochemistry
Fibroblast cells were fixed in 4% paraformaldehyde and treated in 50 mM NH4Cl, 0.5% Triton™ X-100 followed by 1 h in phosphate-buffered saline-bovine serum albumin (PBS-BSA) 3% Tween 0.1% blocking solution or treated in 0.5 M glycine pH 7.5, 0.2% saponin followed by 1 h in PBS-saponin 0.2% BSA 0.2% for endolysosomal membrane integrity and structural preservation. Cells were then incubated with mouse monoclonal acetylated α-tubulin (1:1000, Sigma clone 6-11B-1), rabbit polyclonal pericentrin (1:1000, Abcam ab4448), rabbit monoclonal Ki67 (1:200, Abcam ab16667), mouse monoclonal pH3 (1:200, Cell Signaling #9706), monoclonal Anti-phospho-Histone3 H2A.X (Ser139), clone JBW301 (1:200, Millipore 05-636), rabbit monoclonal EEA1 (1:1000, Cell Signaling #C45B10), mouse monoclonal anti-Lamp2 (1:500, Abcam ab25631) antibodies for 1 h at room temperature. After three washes, cells were incubated with Alexa Fluor® fluorescent dye coupled secondary antibodies: Alexa Fluor® 488 goat anti-rabbit IgG (1:200, Invitrogen), Alexa Fluor® 555 donkey anti-mouse IgG (1:200, Invitrogen). Images were taken using a Leica SP8 confocal microscope. At least 150 control and patient cells were analysed per experiment.
Drosophila stocks and genetics
Flies were raised on cornmeal medium at 25°C. The following stocks were used: wild-type W1118, w;worniu-GAL4, w[1118]; y[1] w[*]; P{Act5C-GAL4-w}E1/CyO (Bloomington stock centre), cg6734 RNAi lines used were P{KK102429}VIE-260B (obtained from VDRC, stock number v106697), y1 v1; P{TRiP.HMJ22542}attP40/CyO (Bloomington stock center) and hsflp; tub-FRT-Gal80-FRT-Gal4, UAS–mCD8–mRFP (gift from F. Bosveld). The two independent cg6734 RNAi lines showed similar results. Crosses were maintained at 25°C and laid eggs were then transferred to 29°C to achieve maximum efficacy of the UAS-Gal4 system. Neural stem cells clones were induced in second instar larvae by heat shock (1 h 37 °C) and mid third instar larval brains were analysed.
Immunohistochemistry and tissue imaging
Immunohistochemistry was performed as described (Rujano et al., 2013, 2015). Briefly, staged brains from third instar larvae were dissected in PBS, fixed for 20 min in 4% paraformaldehyde in PBS with 0.1% Triton™ X-100; washed once in PBS-T (PBS, 0.3% Triton™ X-100) and incubated overnight at 4°C with primary antibodies diluted in PBS-T. After washing in PBS-T, brains were incubated overnight at 4°C with secondary antibodies, Hoechst (0.5 µg/ml) to stain the nuclei and phalloidin to stain actin cortex (Rhodamine or 488, Molecular Probes) followed by three 10-min washes with PBS and mounted in mounting media (1.25% n-propyl gallate, 75% glycerol, 25% H2O). Primary antibodies used: guinea pig anti-Deadpan (Dpn) (1:100, gift from R. Basto), mouse anti-α-tubulin (T6199) (1:1000, Sigma-Aldrich), mouse anti-phospho-Histone H3 (Ser10) (6G3) (1:200, Ab9706, Cell Signalling), rabbit anti-Rab7 (1:2000, gift from A. Nakamura), rat anti-RFP (1:1000, Chromotek 5f8-100). Secondary antibodies used: fluorescent conjugated Alexa Fluor® 488, Alexa Fluor® 555 and Alexa Fluor® 647 (Molecular probes). All images were acquired on a Leica Sp8 confocal microscope with a 60× 1.4 NA objectives using NIS Element software. Images were processed with Fiji 63 and Adobe Photoshop.
Quantification of mitotic neuroblasts
Quantifications were performed manually using the cell counter tool in the Fiji software. Total number of neuroblasts and mitotic neuroblasts were always quantified on the ventral side of the brains at the plane were the maximum number of neuroblasts are present. Sixteen and 20 lobes from individual larvae were quantified for wild-type and dWDR81 knockdown, respectively.
Statistical analysis
Data were analysed with Prism (GraphPad Software) or Excel (Microsoft Office) to generate bar graphics. Error bars represent ± standard error of the mean (SEM) as indicated in figure legends. The two-tailed unpaired t-test was used for statistical analysis of two groups of samples. Two-way ANOVA with a Bonferroni post-test was used to evaluate statistical significance of multiple groups of samples. P < 0.05 was considered significant.
Results
Identification of WDR81 variations by whole exome sequencing
Trio based exome sequencing was performed in all cases with a mean depth of coverage for each sample of at least 136-fold and with at least 99% of the exome covered 15-fold or greater. De novo, autosomal-recessive, X-linked nonsynonymous SNVs and frameshift indels were identified with the publicly available SOLVE-Brain package (https://github.com/Paciorkowski-Lab/SOLVE-Brain.git). This was then used to annotate candidate genes with brain expression. Exome read-depth metrics are summarized in Supplementary Table 1, as well as other non-pathogenic variations identified in other genes. Exome sequencing revealed that all of these patients harbour rare predicted damaging nonsense or missense mutations in the WDR81 gene (Gene Bank Reference Sequence: NM_001163809.01) (Fig. 1B and Supplementary Table 1). Each mutation was confirmed by Sanger sequencing and segregated in the expected autosomal-recessive pattern in all available family members. None of the mutations were observed in the public databases including dbSNP, 1000 Genomes, the NHLBI Exome Variant Server (EVS) and the Exome Aggregation Consortium (ExAC) browser.
![WDR81 mutations identified in five affected families. (A) Pedigrees (P = proband); (B) schematic representation of WDR81 gene and (C) predicted protein domain structure with the positions of the variations identified [red: Patient Im-MCD_606 (Family 1), blue: Patient Im-MCD_752 (Family 2), pink: Patient CerID-22 (Family 3), green: Patient Rdb-MIC_233 (Family 4), yellow: Patient Nan-MCD-001 (Family 5); RefSeq NM_001163809.01]. BEACH = Beige and Chediak-Higashi domain; C = cytosol; EC = extracellular; MFS = major facilitator superfamily domain; WD40 = WD40 beta-propeller domain.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/140/10/10.1093_brain_awx218/3/m_awx218f1.jpeg?Expires=1716359171&Signature=jNW7qEIYq4lxhLcP6yS9jchzvgmUlkGEp919FgbjNqJawCXyIgyabZGrmz64Lvh7oKkrUcd~-ztQ-7ZE2gsIz-mCipADrFJ14tjOR2ZwH1GaP95KIOw5NozykwYAaO6hNhii388xa615UBDksf~Jsn2gkCO~J2sY52M4Cr4hPmsZ-pggnkUMCG8uFgXm6mVu3dXYN0GOx5fckBh5lwmKd~HR8tW2F-7izHEtqfNV2VpBOTkSBpjOsHU5hce646dryU0RH94kqB5Vto1n3aRwFrV60ZS5rJJ7q1iUhyIpCi2WJ0ebObkBK7q8YSSx9BspQZ1IkvrInyF1SJWH8Cg1AA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
WDR81 mutations identified in five affected families. (A) Pedigrees (P = proband); (B) schematic representation of WDR81 gene and (C) predicted protein domain structure with the positions of the variations identified [red: Patient Im-MCD_606 (Family 1), blue: Patient Im-MCD_752 (Family 2), pink: Patient CerID-22 (Family 3), green: Patient Rdb-MIC_233 (Family 4), yellow: Patient Nan-MCD-001 (Family 5); RefSeq NM_001163809.01]. BEACH = Beige and Chediak-Higashi domain; C = cytosol; EC = extracellular; MFS = major facilitator superfamily domain; WD40 = WD40 beta-propeller domain.
Further direct Sanger Sequencing of the 10 exons of WDR81 in a replicative cohort of 17 patients and foetuses with unexplained sporadic microcephaly/microlissencephaly did not lead to the identification of additional mutations (Supplementary Table 2).
Phenotype of patients with WDR81 mutations
WDR81 mutations were identified in seven individuals from non-consanguineous healthy parents including four sporadic cases (i.e. three unrelated children and one foetal case) and one family with one living boy and two twin sisters diagnosed prenatally with subsequent termination of pregnancy (Fig. 1).
The living Patients Im-MCD_606 (Family 1), CerID-22 (Family 3), Rdb-MIC_233 (Family 4) and Nan-MCD-001 (Family 5), carrying WDR81 mutations were 3, 22, 13 and 17 years of age at their last evaluation and all presented with extreme microcephaly. All were born at term, after uneventful pregnancies and normal prenatal ultrasound at 30 weeks of gestation. All presented initially with a minor or mild microcephaly from −1 to −3 standard deviations (SD) without growth deficiency. In all cases, microcephaly proceeded to worsen with age reaching −5 to −10 SD at last evaluation. Neurological development was extremely impaired in all cases; none were able to hold their head, develop any voluntary hand use, visual contact or acquire any babbling. They developed early onset dyskinetic movement disorder and spastic tetraplegia from 2 months of age. Two developed drug resistant epilepsy (Patients Rdb-MIC_233 and Nan-MCD-001). Apart from microcephaly, they grew normally, were not dysmorphic and did not have any other congenital anomalies. Ophthalmological and auditory investigations were normal in all cases. There was no evidence of neurological decline or immune deficiency.
Brain MRI revealed extreme gyral simplification with abnormal gyral pattern comprising foci of extremely reduced sulcation and agyria, with increased subarachnoid spaces and deformed and enlarged ventricles, with mild cerebellar atrophy (Fig. 2). Detailed clinical data are provided in Table 1.
Detailed clinical data
| Family | Family 1 | Family 2 | Family 3 | Family 4 | Family 5 | |||
|---|---|---|---|---|---|---|---|---|
| Patient | Im-MCD_606 | Im-MCD_752 | CerID-22 | Rdb-MIC_233 | Rdb-MIC_234 | Rdb-MIC_235 | Nan-MCD-001 | Published case |
| Gender | Male | Foetus female (25 GW) | Female | Male | Foetus female (30 GW) | Foetus female (30 GW) | Female | |
| Gene (RefSeq) WDR81 (NM_001163809.1) | ||||||||
| Nucleotide variation 1 | c.1882 C>T | c.2834_2837delTGTT | c.1582 C>T | c.1735G>A | c.1735G>A | c.1735G>A | c.3820_3835del | c.2567C>T |
| Nucleotide variation 2 | c.3713 C>G | c.5464 C>T | c.4036_4041dup | c.1358 dup | c.1358 dup | c.1358 dup | c.5453G>T | |
| Protein variation 1 | p.Gln628* | p.Phe946Serfs*17 | p.His528Tyr | p.Gly579Arg | p.Gly579Arg | p.Gly579Arg | p.Pro1274Thrfs*56 | p.Pro856Leu |
| Protein variation 2 | p.Pro1238Arg | p.Arg1822* | p.Val1346_Thr1347dup | p.Tyr453* | p.Tyr453* | p.Tyr453* | p.Gly1818Val | |
| Sporadic case | Sporadic case | Sporadic case | Familial case | Familial case | Familial case | Sporadic case | Familial cases (n = 5) | |
| Ethnicity | European | European | European | European | European | European | European | Turkish |
| Transmission | AR | AR | AR | AR | AR | AR | AR | AR |
| References | This series | This series | This series | This series | This series | This series | This series | Gulsuner, 2011 |
| Clinic | ||||||||
| Head circumference at birth, cm | 30.5 (−2.7 SD) | NA | 32 (−1.8 SD) | 30 (−3 SD) | N/A | N/A | 33 (−1 SD) | N/K |
| Birth weight, g | 3620 (+0.5 SD) | NA | 3180 (−0.4 SD) | 3320 (−0.1 SD) | N/A | N/A | 3700 (+1 SD) | N/K |
| Birth length, cm | 48 (−1.4 SD) | 50.5 (−0.3 SD) | 49 (−1 SD) | 51.5 (+1.5 SD) | N/K | |||
| Age at last evaluation | 27 months | 25 GW | 22 years | 13 years | 33 GW | 33 GW | 17 years | 28 years |
| Head circumference, cm | 37.5 (−9 SD) | 23 (−1 SD) | 44 (−7 SD) | 41 (<−8 SD) | 25.7 (<−8 SD) | 27.4 (−4 SD) | 42 (−10 SD) | Disproportionate short stature (150 cm) |
| Growth parameter (weight) | 9300 g (−1 SD) | 970 g (+1 SD) | 30 kg (−5 SD) | 32 kg (−1.5 SD) | 1561 g (−2 SD) | 2056 g (+0.1 SD) | 50 kg (−1 SD) | Normal |
| Growth parameter (height), cm | 71 | 34.5 (+1 SD) | N/K | 121 (−5 SD) | 41.5 (−1.5 SD) | 42.2 (−1 SD) | 158 (−1 SD) | N/K |
| Level of neurological development | Virtually no development | N/A | Virtually no development | Virtually no development | N/A | N/A | Virtually no development | Intellectual disability |
| Neurological examination | Spastic tetraplegia | N/A | Spastic tetraplegia | Spastic tetraplegia | N/A | N/A | Spastic tetraplegia | No spasticity |
| Epilepsy | Absent | N/A | Absent | Infantile spasms (2.5 years) | N/A | N/A | Drug resistant > 1 seizures/day | Absent |
| Movement disorder | Generalized dyskinesia | N/A | Generalized dyskinesia | Dystonia | N/A | N/A | Absent | Cerebellar ataxia |
| Other signs | Nystagmus (neonatal period) | N/A | Nystagmus | Nystagmus (neonatal period) | N/A | N/A | Scoliosis, precocious puberty | Bilateral external ophthalmoplegia |
| MRI | ||||||||
| Age at MRI | 22 months | 25 GW (US) | 14.5 years | 4 years | 30 GW (foetal MRI) | 30 GW (foetal MRI) | 6 years | |
| Cortex | LIS/extremely reduced gyration | Delayed primary gyration | Gyral simplification | LIS/extremely reduced gyration | Delayed primary gyration | Delayed primary gyration | Cortical atrophy | Normal/brain atrophy |
| Basal ganglia | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| Corpus callosum | Thin | Corpus callosum agenesis | Thin | Thin | Thin | Thin | Thin | Thin |
| Brainstem | Normal | Severe hypoplasia | Normal | Normal | Normal | Normal | Normal | Normal |
| Cerebellum | Normal | Severe hypoplasia | Cerebellar atrophy | Normal | Normal | Normal | Cerebellar atrophy | Cerebellar atrophy + vermis midline cleft |
| Myelination | Immature/dysmyelination Enlarged ventricles and subarachnoid space | N/A | Gliosis (periventricular) | Immature/dysmyelination Enlarged ventricles and subarachnoid space | N/A | N/A | Immature/dysmyelination | N/A |
| Family | Family 1 | Family 2 | Family 3 | Family 4 | Family 5 | |||
|---|---|---|---|---|---|---|---|---|
| Patient | Im-MCD_606 | Im-MCD_752 | CerID-22 | Rdb-MIC_233 | Rdb-MIC_234 | Rdb-MIC_235 | Nan-MCD-001 | Published case |
| Gender | Male | Foetus female (25 GW) | Female | Male | Foetus female (30 GW) | Foetus female (30 GW) | Female | |
| Gene (RefSeq) WDR81 (NM_001163809.1) | ||||||||
| Nucleotide variation 1 | c.1882 C>T | c.2834_2837delTGTT | c.1582 C>T | c.1735G>A | c.1735G>A | c.1735G>A | c.3820_3835del | c.2567C>T |
| Nucleotide variation 2 | c.3713 C>G | c.5464 C>T | c.4036_4041dup | c.1358 dup | c.1358 dup | c.1358 dup | c.5453G>T | |
| Protein variation 1 | p.Gln628* | p.Phe946Serfs*17 | p.His528Tyr | p.Gly579Arg | p.Gly579Arg | p.Gly579Arg | p.Pro1274Thrfs*56 | p.Pro856Leu |
| Protein variation 2 | p.Pro1238Arg | p.Arg1822* | p.Val1346_Thr1347dup | p.Tyr453* | p.Tyr453* | p.Tyr453* | p.Gly1818Val | |
| Sporadic case | Sporadic case | Sporadic case | Familial case | Familial case | Familial case | Sporadic case | Familial cases (n = 5) | |
| Ethnicity | European | European | European | European | European | European | European | Turkish |
| Transmission | AR | AR | AR | AR | AR | AR | AR | AR |
| References | This series | This series | This series | This series | This series | This series | This series | Gulsuner, 2011 |
| Clinic | ||||||||
| Head circumference at birth, cm | 30.5 (−2.7 SD) | NA | 32 (−1.8 SD) | 30 (−3 SD) | N/A | N/A | 33 (−1 SD) | N/K |
| Birth weight, g | 3620 (+0.5 SD) | NA | 3180 (−0.4 SD) | 3320 (−0.1 SD) | N/A | N/A | 3700 (+1 SD) | N/K |
| Birth length, cm | 48 (−1.4 SD) | 50.5 (−0.3 SD) | 49 (−1 SD) | 51.5 (+1.5 SD) | N/K | |||
| Age at last evaluation | 27 months | 25 GW | 22 years | 13 years | 33 GW | 33 GW | 17 years | 28 years |
| Head circumference, cm | 37.5 (−9 SD) | 23 (−1 SD) | 44 (−7 SD) | 41 (<−8 SD) | 25.7 (<−8 SD) | 27.4 (−4 SD) | 42 (−10 SD) | Disproportionate short stature (150 cm) |
| Growth parameter (weight) | 9300 g (−1 SD) | 970 g (+1 SD) | 30 kg (−5 SD) | 32 kg (−1.5 SD) | 1561 g (−2 SD) | 2056 g (+0.1 SD) | 50 kg (−1 SD) | Normal |
| Growth parameter (height), cm | 71 | 34.5 (+1 SD) | N/K | 121 (−5 SD) | 41.5 (−1.5 SD) | 42.2 (−1 SD) | 158 (−1 SD) | N/K |
| Level of neurological development | Virtually no development | N/A | Virtually no development | Virtually no development | N/A | N/A | Virtually no development | Intellectual disability |
| Neurological examination | Spastic tetraplegia | N/A | Spastic tetraplegia | Spastic tetraplegia | N/A | N/A | Spastic tetraplegia | No spasticity |
| Epilepsy | Absent | N/A | Absent | Infantile spasms (2.5 years) | N/A | N/A | Drug resistant > 1 seizures/day | Absent |
| Movement disorder | Generalized dyskinesia | N/A | Generalized dyskinesia | Dystonia | N/A | N/A | Absent | Cerebellar ataxia |
| Other signs | Nystagmus (neonatal period) | N/A | Nystagmus | Nystagmus (neonatal period) | N/A | N/A | Scoliosis, precocious puberty | Bilateral external ophthalmoplegia |
| MRI | ||||||||
| Age at MRI | 22 months | 25 GW (US) | 14.5 years | 4 years | 30 GW (foetal MRI) | 30 GW (foetal MRI) | 6 years | |
| Cortex | LIS/extremely reduced gyration | Delayed primary gyration | Gyral simplification | LIS/extremely reduced gyration | Delayed primary gyration | Delayed primary gyration | Cortical atrophy | Normal/brain atrophy |
| Basal ganglia | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| Corpus callosum | Thin | Corpus callosum agenesis | Thin | Thin | Thin | Thin | Thin | Thin |
| Brainstem | Normal | Severe hypoplasia | Normal | Normal | Normal | Normal | Normal | Normal |
| Cerebellum | Normal | Severe hypoplasia | Cerebellar atrophy | Normal | Normal | Normal | Cerebellar atrophy | Cerebellar atrophy + vermis midline cleft |
| Myelination | Immature/dysmyelination Enlarged ventricles and subarachnoid space | N/A | Gliosis (periventricular) | Immature/dysmyelination Enlarged ventricles and subarachnoid space | N/A | N/A | Immature/dysmyelination | N/A |
AR = autosomal recessive; GW = weeks of gestation; LIS = lissencephaly; N/A = non-applicable; NK = not known; US = ultrasound.
Detailed clinical data
| Family | Family 1 | Family 2 | Family 3 | Family 4 | Family 5 | |||
|---|---|---|---|---|---|---|---|---|
| Patient | Im-MCD_606 | Im-MCD_752 | CerID-22 | Rdb-MIC_233 | Rdb-MIC_234 | Rdb-MIC_235 | Nan-MCD-001 | Published case |
| Gender | Male | Foetus female (25 GW) | Female | Male | Foetus female (30 GW) | Foetus female (30 GW) | Female | |
| Gene (RefSeq) WDR81 (NM_001163809.1) | ||||||||
| Nucleotide variation 1 | c.1882 C>T | c.2834_2837delTGTT | c.1582 C>T | c.1735G>A | c.1735G>A | c.1735G>A | c.3820_3835del | c.2567C>T |
| Nucleotide variation 2 | c.3713 C>G | c.5464 C>T | c.4036_4041dup | c.1358 dup | c.1358 dup | c.1358 dup | c.5453G>T | |
| Protein variation 1 | p.Gln628* | p.Phe946Serfs*17 | p.His528Tyr | p.Gly579Arg | p.Gly579Arg | p.Gly579Arg | p.Pro1274Thrfs*56 | p.Pro856Leu |
| Protein variation 2 | p.Pro1238Arg | p.Arg1822* | p.Val1346_Thr1347dup | p.Tyr453* | p.Tyr453* | p.Tyr453* | p.Gly1818Val | |
| Sporadic case | Sporadic case | Sporadic case | Familial case | Familial case | Familial case | Sporadic case | Familial cases (n = 5) | |
| Ethnicity | European | European | European | European | European | European | European | Turkish |
| Transmission | AR | AR | AR | AR | AR | AR | AR | AR |
| References | This series | This series | This series | This series | This series | This series | This series | Gulsuner, 2011 |
| Clinic | ||||||||
| Head circumference at birth, cm | 30.5 (−2.7 SD) | NA | 32 (−1.8 SD) | 30 (−3 SD) | N/A | N/A | 33 (−1 SD) | N/K |
| Birth weight, g | 3620 (+0.5 SD) | NA | 3180 (−0.4 SD) | 3320 (−0.1 SD) | N/A | N/A | 3700 (+1 SD) | N/K |
| Birth length, cm | 48 (−1.4 SD) | 50.5 (−0.3 SD) | 49 (−1 SD) | 51.5 (+1.5 SD) | N/K | |||
| Age at last evaluation | 27 months | 25 GW | 22 years | 13 years | 33 GW | 33 GW | 17 years | 28 years |
| Head circumference, cm | 37.5 (−9 SD) | 23 (−1 SD) | 44 (−7 SD) | 41 (<−8 SD) | 25.7 (<−8 SD) | 27.4 (−4 SD) | 42 (−10 SD) | Disproportionate short stature (150 cm) |
| Growth parameter (weight) | 9300 g (−1 SD) | 970 g (+1 SD) | 30 kg (−5 SD) | 32 kg (−1.5 SD) | 1561 g (−2 SD) | 2056 g (+0.1 SD) | 50 kg (−1 SD) | Normal |
| Growth parameter (height), cm | 71 | 34.5 (+1 SD) | N/K | 121 (−5 SD) | 41.5 (−1.5 SD) | 42.2 (−1 SD) | 158 (−1 SD) | N/K |
| Level of neurological development | Virtually no development | N/A | Virtually no development | Virtually no development | N/A | N/A | Virtually no development | Intellectual disability |
| Neurological examination | Spastic tetraplegia | N/A | Spastic tetraplegia | Spastic tetraplegia | N/A | N/A | Spastic tetraplegia | No spasticity |
| Epilepsy | Absent | N/A | Absent | Infantile spasms (2.5 years) | N/A | N/A | Drug resistant > 1 seizures/day | Absent |
| Movement disorder | Generalized dyskinesia | N/A | Generalized dyskinesia | Dystonia | N/A | N/A | Absent | Cerebellar ataxia |
| Other signs | Nystagmus (neonatal period) | N/A | Nystagmus | Nystagmus (neonatal period) | N/A | N/A | Scoliosis, precocious puberty | Bilateral external ophthalmoplegia |
| MRI | ||||||||
| Age at MRI | 22 months | 25 GW (US) | 14.5 years | 4 years | 30 GW (foetal MRI) | 30 GW (foetal MRI) | 6 years | |
| Cortex | LIS/extremely reduced gyration | Delayed primary gyration | Gyral simplification | LIS/extremely reduced gyration | Delayed primary gyration | Delayed primary gyration | Cortical atrophy | Normal/brain atrophy |
| Basal ganglia | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| Corpus callosum | Thin | Corpus callosum agenesis | Thin | Thin | Thin | Thin | Thin | Thin |
| Brainstem | Normal | Severe hypoplasia | Normal | Normal | Normal | Normal | Normal | Normal |
| Cerebellum | Normal | Severe hypoplasia | Cerebellar atrophy | Normal | Normal | Normal | Cerebellar atrophy | Cerebellar atrophy + vermis midline cleft |
| Myelination | Immature/dysmyelination Enlarged ventricles and subarachnoid space | N/A | Gliosis (periventricular) | Immature/dysmyelination Enlarged ventricles and subarachnoid space | N/A | N/A | Immature/dysmyelination | N/A |
| Family | Family 1 | Family 2 | Family 3 | Family 4 | Family 5 | |||
|---|---|---|---|---|---|---|---|---|
| Patient | Im-MCD_606 | Im-MCD_752 | CerID-22 | Rdb-MIC_233 | Rdb-MIC_234 | Rdb-MIC_235 | Nan-MCD-001 | Published case |
| Gender | Male | Foetus female (25 GW) | Female | Male | Foetus female (30 GW) | Foetus female (30 GW) | Female | |
| Gene (RefSeq) WDR81 (NM_001163809.1) | ||||||||
| Nucleotide variation 1 | c.1882 C>T | c.2834_2837delTGTT | c.1582 C>T | c.1735G>A | c.1735G>A | c.1735G>A | c.3820_3835del | c.2567C>T |
| Nucleotide variation 2 | c.3713 C>G | c.5464 C>T | c.4036_4041dup | c.1358 dup | c.1358 dup | c.1358 dup | c.5453G>T | |
| Protein variation 1 | p.Gln628* | p.Phe946Serfs*17 | p.His528Tyr | p.Gly579Arg | p.Gly579Arg | p.Gly579Arg | p.Pro1274Thrfs*56 | p.Pro856Leu |
| Protein variation 2 | p.Pro1238Arg | p.Arg1822* | p.Val1346_Thr1347dup | p.Tyr453* | p.Tyr453* | p.Tyr453* | p.Gly1818Val | |
| Sporadic case | Sporadic case | Sporadic case | Familial case | Familial case | Familial case | Sporadic case | Familial cases (n = 5) | |
| Ethnicity | European | European | European | European | European | European | European | Turkish |
| Transmission | AR | AR | AR | AR | AR | AR | AR | AR |
| References | This series | This series | This series | This series | This series | This series | This series | Gulsuner, 2011 |
| Clinic | ||||||||
| Head circumference at birth, cm | 30.5 (−2.7 SD) | NA | 32 (−1.8 SD) | 30 (−3 SD) | N/A | N/A | 33 (−1 SD) | N/K |
| Birth weight, g | 3620 (+0.5 SD) | NA | 3180 (−0.4 SD) | 3320 (−0.1 SD) | N/A | N/A | 3700 (+1 SD) | N/K |
| Birth length, cm | 48 (−1.4 SD) | 50.5 (−0.3 SD) | 49 (−1 SD) | 51.5 (+1.5 SD) | N/K | |||
| Age at last evaluation | 27 months | 25 GW | 22 years | 13 years | 33 GW | 33 GW | 17 years | 28 years |
| Head circumference, cm | 37.5 (−9 SD) | 23 (−1 SD) | 44 (−7 SD) | 41 (<−8 SD) | 25.7 (<−8 SD) | 27.4 (−4 SD) | 42 (−10 SD) | Disproportionate short stature (150 cm) |
| Growth parameter (weight) | 9300 g (−1 SD) | 970 g (+1 SD) | 30 kg (−5 SD) | 32 kg (−1.5 SD) | 1561 g (−2 SD) | 2056 g (+0.1 SD) | 50 kg (−1 SD) | Normal |
| Growth parameter (height), cm | 71 | 34.5 (+1 SD) | N/K | 121 (−5 SD) | 41.5 (−1.5 SD) | 42.2 (−1 SD) | 158 (−1 SD) | N/K |
| Level of neurological development | Virtually no development | N/A | Virtually no development | Virtually no development | N/A | N/A | Virtually no development | Intellectual disability |
| Neurological examination | Spastic tetraplegia | N/A | Spastic tetraplegia | Spastic tetraplegia | N/A | N/A | Spastic tetraplegia | No spasticity |
| Epilepsy | Absent | N/A | Absent | Infantile spasms (2.5 years) | N/A | N/A | Drug resistant > 1 seizures/day | Absent |
| Movement disorder | Generalized dyskinesia | N/A | Generalized dyskinesia | Dystonia | N/A | N/A | Absent | Cerebellar ataxia |
| Other signs | Nystagmus (neonatal period) | N/A | Nystagmus | Nystagmus (neonatal period) | N/A | N/A | Scoliosis, precocious puberty | Bilateral external ophthalmoplegia |
| MRI | ||||||||
| Age at MRI | 22 months | 25 GW (US) | 14.5 years | 4 years | 30 GW (foetal MRI) | 30 GW (foetal MRI) | 6 years | |
| Cortex | LIS/extremely reduced gyration | Delayed primary gyration | Gyral simplification | LIS/extremely reduced gyration | Delayed primary gyration | Delayed primary gyration | Cortical atrophy | Normal/brain atrophy |
| Basal ganglia | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| Corpus callosum | Thin | Corpus callosum agenesis | Thin | Thin | Thin | Thin | Thin | Thin |
| Brainstem | Normal | Severe hypoplasia | Normal | Normal | Normal | Normal | Normal | Normal |
| Cerebellum | Normal | Severe hypoplasia | Cerebellar atrophy | Normal | Normal | Normal | Cerebellar atrophy | Cerebellar atrophy + vermis midline cleft |
| Myelination | Immature/dysmyelination Enlarged ventricles and subarachnoid space | N/A | Gliosis (periventricular) | Immature/dysmyelination Enlarged ventricles and subarachnoid space | N/A | N/A | Immature/dysmyelination | N/A |
AR = autosomal recessive; GW = weeks of gestation; LIS = lissencephaly; N/A = non-applicable; NK = not known; US = ultrasound.
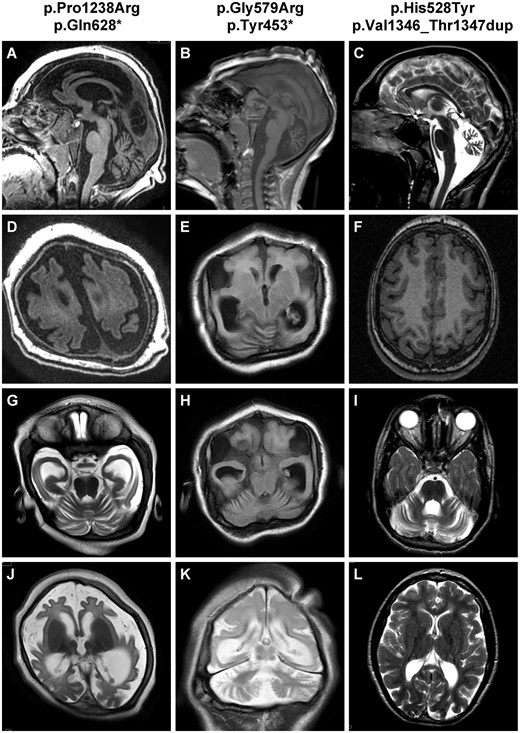
Representative brain MRI of patients with WDR81 mutations. Patients Im-MCD_606 (p.Pro1238Arg; p.Gln628*), Rdb-MIC_233 (p.Gly579Arg; p.Tyr453*) and CerID-22 (p.His528Tyr; p.Val1346_Thr1347dup) respectively aged 22 months (A, D, G and J), 4 years (B, E, H and K), and 14.5 years (C, F, I and L) at the time of MRI. Sagittal sections T1-weighted (A and B) or T2-weighted (C) show moderate (A) to severe cerebellar atrophy (C), and thin corpus callosum in all cases. Axial weighted sections show extreme gyral simplification (D and E) or milder (F), with area of absence of gyri (agyria) without thickening of cortex. At the level of the cerebellar hemispheres, cerebellar atrophy with excess of visibility of hemisphere foliation (G, H and I); T2-weighted sections also demonstrate delayed myelination at 22 months (J), hypomyelination at 9 years (K) or periventricular white matter hyperintensities (L).
WDR81 mutations were also identified in three foetal cases (Families 2 and 4). The first (Patient Im-MCD_752) was a sporadic female case diagnosed by foetal ultrasound at 25 weeks of gestation with corpus callosum agenesis and pontocerebellar hypoplasia. The two remaining cases (Patients Rdb-MIC_234 and 235) were dizygotic twins, sisters of Patient Rdb-MIC_233 in whom foetal MRI at 30 weeks of gestation showed deceleration of head growth and delayed gyration. In accordance with the French law, these brain malformations lead to medical termination of pregnancy. Macroscopic examinations showed extreme microcephaly with absent to extremely delayed gyration pattern in all cases, corpus callosum agenesis (1/3) and pontocerebellar hypoplasia (2/3). The neuropathological classification would be within microcephaly with extremely reduced gyration with or without pontocerebellar hypoplasia and with or without corpus callosum agenesis and normal growth.
Expression of WDR81 in patient fibroblasts
To study the functional consequences of the WDR81 mutations at the cellular level, we used dermal fibroblast cultures established from skin biopsies of two patients (Patients Im-MCD_606 and CerID-22; Table 1) and age-matched controls. Expression of WDR81 in Patient Im-MCD_606, harbouring a nonsense mutation (p.Gln628*), was reduced by almost 40%, indicating nonsense-mediated mRNA decay (Supplementary Fig. 1A).
Cellular consequences of WDR81 variations in patient fibroblasts
In view of the cellular phenotypes associated with other microcephaly causing genes including multipolar spindle and supernumerary cilia, we next analysed mitotic spindle and centrosome structures and ciliogenesis in patient fibroblasts. Immunostaining experiments did not reveal any impact on mitotic spindle organization nor any abnormal primary cilia structure and number (Supplementary Fig. 1B and C). Microcephaly has also been associated with altered DNA damage response, yet no spontaneous accumulation of double-strand breaks was observed in patient fibroblasts compared to controls (Supplementary Fig. 2). Disruptions in cell cycle progression and decreased proliferation can also induce microcephaly, but no difference was observed in the percentage of cycling cells between patient and control cells (Fig. 3A). However, using a mitotic DNA specific marker (phospho-histone H3; PH3), we analysed the mitotic index and found that it was significantly increased in Patient Im-MCD_606 fibroblasts (carrying a stop mutation and subsequent nonsense-mediated decay), and also in fibroblasts from Patient CerID-22 (no nonsense-mediated decay) although not significant (Fig. 3A). To gain further insights into the mechanisms leading to this increase, we next analysed the proportion of cells in each mitotic phase and found a significant increase in the number of cells in prometaphase/metaphase in both patient fibroblasts as compared to control cells (Fig. 3B).
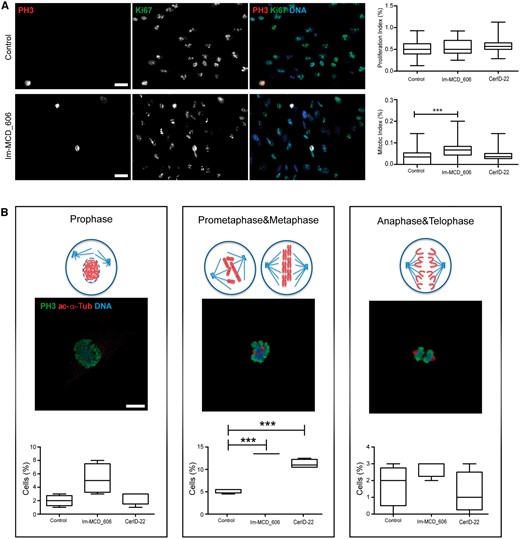
WDR81 mutant cells exhibit an increased mitotic index associated with delayed mitotic progression. (A) Immunostaining with a static marker of proliferative activity (Ki67; AbCam Ab16667) and DAPI revealed no difference in proliferative index in patient cells as compared to control cells (control: 0.5192% ± 0.01974%, Patient Im-MCD_606: 0.5478 ± 0.02149 (not significant) and Patient CerID-22: 0.5825% ± 0.01782% (not significant). Staining with a mitosis specific marker (phospho-histone H3; PH3; Cell Signaling #9706) showed increased mitotic index in patient cells as compared to controls (0.038% ± 0.0038% for the control, 0.069% ± 0.005% for Patient Im-MCD_606 (P < 0.0001) and 0.041% ± 0.0037% for Patient CerID-22 (not significant); n > 400 mutant and control cells in four independent experiments. Scale bar = 10 µm. (B) Percentage of cells at each mitosis stage measured after staining using PH3 and acetylated alpha-tubulin (Sigma clone 6-11B-1; mitotic spindle) antibodies. WDR81 mutant cells showed a significant increase in the number of mutant cells in prometaphase/metaphase as compared to control cells (control = 4.75% ± 0.25%, Patient Im-MCD_606 = 13% ± 0% (P < 0.0001) and CerID-22 = 10.75% ± 0.48% (P < 0.0001); n > 400 cells in three independent experiments) indicating mitotic progression delay in WDR81 mutant cells. Numbers represent the average ± SEM. The P-values were calculated with the two-tailed unpaired t-test (***P < 0.001). Scale bar = 5 µm.
Knockdown of the WRD81 orthologue in Drosophila neuroblasts results in mitotic delay
To investigate the consequences of WDR81 disruption further, we extended our analyses to Drosophila, an in vivo model previously shown to be a valuable tool to assess the underlying mechanisms of MCDs (Rujano et al., 2013; Mishra-Gorur et al., 2014; Yamamoto et al., 2014). The Drosophila larval brain contains proliferating neural stem cells that give rise to all neurons and glial cells present in the adult fly brain. We used an RNAi-mediated approach to downregulate CG6734, the fly orthologue of WDR81 (hereafter referred to as dWDR81). Since no antibody against dWDR81 was available, we used Act5c-Gal4 to drive expression of dWDR81 RNAi ubiquitously and determined the efficiency of the RNAi-mediated knockdown by quantitative PCR (qPCR) in brain RNA extracts. RNA levels were reduced by ∼40% (Fig. 4B) in larval brains resulting in a partial decrease in dWDR81 expression. We subsequently used Worniu-Gal4 to drive expression of dWDR81 RNAi exclusively in neural stem cells (neuroblasts) and analysed mitoses. Neuroblasts were identified by their large size (Fig. 4A) and Deadpan (Dpn, a marker for neuroblasts) expression. Defects in overall brain size or viability were not observed. However, at the cellular level we found that the partial knockdown of dWDR81 resulted in an increased mitotic index in the neuroblasts from the central brain, with an increase in the proportion of mitotic cells in prometaphase/metaphase and a decrease in anaphase/telophase, indicating a delay in mitotic progression (Fig. 4C, E and F). As with the patient fibroblasts, no defects in centrosome number or mitotic spindle organization were observed (Fig. 4D). Taken together, these results indicate that the Drosophila orthologue of WDR81 plays an important function in brain development in the fly and, similarly to other previously described microcephaly-related proteins, dWDR81 likely plays a role in mitotic progression of proliferating neural stem cells (Novorol et al., 2013; Chen et al., 2014; Mishra-Gorur et al., 2014). Furthermore, our observations suggest a conserved mechanism in humans and Drosophila resulting from WDR81 depletion.
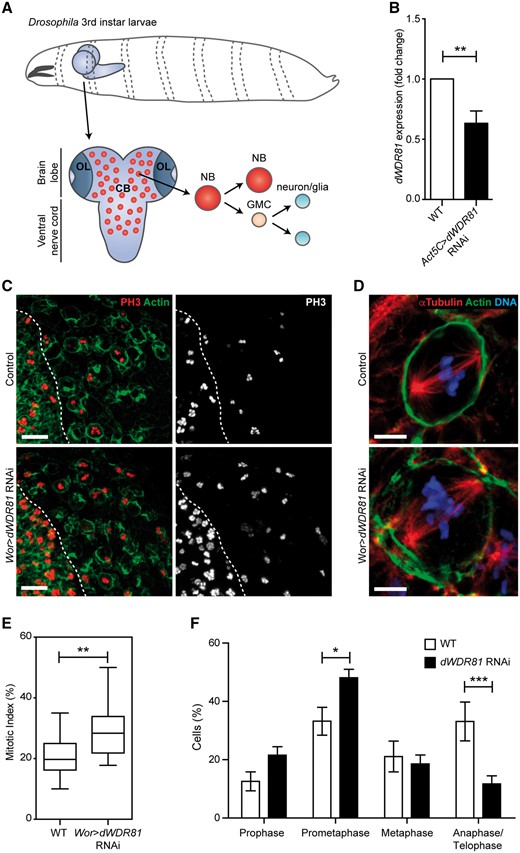
Knockdown of WDR81 orthologue in Drosophila results in increased mitotic index and a delay in mitotic progression. (A) Schematic representation of the Drosophila third instar larval brain depicting the central brain (CB) and optic lobes (OL). Neural stem cells (neuroblasts, NB) in the central brain divide asymmetrically to produce a self-renewed neural stem cell and a ganglion mother cell (GMC) that divides once more to give rise to two neurons or glial cells. (B) dWDR81 mRNA levels measured by qPCR in brain tissue of wild-type larvae and larvae expressing dWDR81 RNAi under Act5C-Gal4. (C) Expression of dWDR81 RNAi in neural stem cells with Worniu-Gal4 (Wor > WDR81 RNAi) results in an increase in the number of mitotic cells in the central brain (right side of the dashed line) as seen with PH3. Actin was used to visualize cell contours. Scale bar 20 = µm. (D) No alteration in mitotic spindle organization (visualized with α-tubulin) was observed in dWDR81 depleted neural stem cells. Scale bar = 5 µm. (E) Quantification of the mitotic index showed a significant increase in the amount of neural stem cells in mitosis when dWDR81 is depleted (29% ± 8.6%) as compared to controls (21% ± 6.8%) (P = 0.0026); n = 694 dWDR81 RNAi cells from three independent experiments and n = 552 wild-type cells from five independent experiments. Numbers represent the average ± SD. Statistical significance was assessed by a two-tailed unpaired t-test. (F) Quantification of the number of mitotic cells in prophase, prometaphase, metaphase and anaphase/telophase in wild-type and dWDR81 depleted neural stem cells, shows an increase in prometaphase state (33% in wild-type versus 48% in dWDR81 RNAi (P < 0.05) and a decrease in the number of cells in anaphase/telophase (33% in wild-type versus 12% in dWDR81 RNAi (P < 0.001), indicative of a delay in mitotic progression. Bars represent the mean and lines SEM. Statistical significance was assessed by a two-way ANOVA followed by a Bonferroni post-test.
Discussion
Our data demonstrate that compound heterozygous mutations in WDR81 are responsible for microcephaly with extremely reduced gyration or agyria with or without cerebellar anomalies ranging from pontocerebellar hypoplasia to cerebellar atrophy expanding the phenotypic spectrum associated with WDR81 mutations. Our results suggest that WDR81 has a role in normal cell proliferation. Patient fibroblasts exhibit increased mitotic index associated with prolonged prometaphase/metaphase suggestive of chromosome congression and/or separation defects (Nasmyth, 2002; Lara-Gonzalez et al., 2012). These mitotic defects may lead to reduced neurogenic cell divisions, alterations of neural cell fates or to a failure to maintain the progenitor cell population. As previously demonstrated, with mutations in other microcephaly/microlissencephaly-causing genes, these defects would lead to a greatly reduced cerebral cortex (Nicholas et al., 2010; Alkuraya et al., 2011; Hu et al., 2014; Mishra-Gorur et al., 2014).
In 2011, the WDR81 homozygous missense mutation p.Pro856Leu was reported in one large Turkish family suffering from cerebellar hypoplasia and quadrupedal locomotion (CAMRQ2, MIM 610185) (Gulsuner et al., 2011), a brain disorder that falls into the category of autosomal recessive cerebellar ataxia (Tan, 2006). A subsequent study performed on an ENU-induced mouse line carrying a distinct missense WDR81 mutation (p.Leu1349Pro) suggested that the WDR81 mutation resulted in the formation of abnormal spheroid-like mitochondria and the progressive death of Purkinje cells and photoreceptor cells (Traka et al., 2013). This finding was consistent with the phenotype of slowly progressing degenerative process of cerebellar atrophy that may represent the less severe end of the spectrum of the disorder described here. Remarkably, in our series, the mildest phenotype was associated with missense and in-frame deletion, while the most severe case carried nonsense and frameshift variants suggesting putative phenotype-genotype correlations. Of note, none of these cases demonstrated retinal anomalies or photoreceptor degeneration as described in the initial report (Gulsuner et al., 2011).
WDR81 encodes WD repeat-containing protein 81, [UniProt Knowledgebase (UniProtKB), see ‘Web resources’ section], a large protein (1941 amino acids) of unclear function. WDR81 is a poorly characterized gene that encodes nine protein isoforms (Traka et al., 2013). The longest human isoform is predicted to be a transmembrane protein, composed of an N-terminal BEACH (Beige and Chediak-Higashi) domain and belonging to the major facilitator superfamily (MFS) consisting of solute carrier proteins (Pao et al., 1998). Additionally, this isoform contains a C-terminal WD40 beta-propeller domain (Stirnimann et al., 2010; Xu and Min, 2011), which is shared with common to other previously identified microcephaly genes, WDR62 (Bilgüvar et al., 2010; Nicholas et al., 2010; Chen et al., 2014), KATNB1 (Hu et al., 2014; Mishra-Gorur et al., 2014) and LIS1 (Reiner et al., 1993; Hattori et al., 1994). Importantly, the mutations are distributed along the entire length of the gene and predicted to affect the different crucial domains of the protein. In Patient Im-MCD_606, harbouring the nonsense mutation p.Gln628*, we demonstrated the existence of nonsense-mediated decay. We would therefore anticipate that all the nonsense mutations identified would also result in nonsense-mediated decay, although this could not be tested as no patient material was available.
WDR81 is stably expressed in neural progenitor cells and post-mitotic neurons in the developing human brain (http://www.mitocheck.org). WDR81 expression remains detectable during infancy and in the adult brain particularly in neocortex, hippocampus and striatum. This expression pattern suggests a continuing role of WDR81 in the proliferation of neural progenitors, neuronal migration and laminar organization of the human cortex (Neumann et al., 2010). To understand the cellular role of WDR81 in the proliferation of neural progenitors, we used Drosophila as a model system. The larval brain of Drosophila has already been used to investigate the function of microcephaly-related genes (Rujano et al., 2013; Mishra-Gorur et al., 2014; Yamamoto et al., 2014). Our studies show that partial loss of WDR81 orthologue in the fly leads to an increased mitotic index of neural stem cells. In human, we found that WDR81 patient fibroblasts also exhibit increased mitotic index associated with delayed prometaphase/metaphase transition. These data are in accordance with a previous high-throughput siRNA screen in HeLa cells, which showed that WDR81 is a cell division gene whose inactivation leads to mitotic delays (Neumann et al., 2010). Increased mitotic index was also shown in WDR62-depleted cells (Nicholas et al., 2010) suggesting common pathophysiological mechanisms. However, in contrast to the cellular phenotypes associated to WDR62 and KATNB1 depletion, we found that centrosomes, mitotic spindle structure, and primary cilia number and structure remain normal in WDR81 patient fibroblasts and Drosophila neuroblasts.
Recently, the orthologue of WDR81 in C. elegans, SORF-1, together with SORF-2 (WDR91 orthologue) was found to have a critical role in facilitating endosomal maturation and delivery of cargo to late endo-lysosomes. In C. elegans as well as mammalian Hela cells, WDR81 acts in a complex with WDR91 and Beclin1 to regulate endosomal phosphatidyl inositol phosphate 3 (PtdIns3) levels by suppressing phosphatidylinositol 3-kinase activity. Loss of function of WDR81 leads to the elevation and prolonged existence of endosomal PtdIns3P, which induces excessive fusion of early endosomes and delays early to late endosome conversion (Liu et al., 2016; Rapiteanu et al., 2016). We have analysed early and late endosomes in our patient’s fibroblasts and Drosophila neural stem cells knockdown for WDR81, and neither model showed defects in the morphology of these compartments. The differences with the previous studies (Liu et al., 2016; Rapiteanu et al., 2016), might be due to residual activity of WDR81 in patient fibroblasts and Drosophila neural stem or due to cell type related differences. Future research using neuronal cell lines may shed light on the link between delayed mitotic progression observed in our patients and models and altered early to late endosome conversion.
Overall, our study reports ten WDR81 mutations in seven cases from five families with microcephaly with extremely reduced gyration including agyria, thus expanding the phenotypic spectrum of WDR81 mutations with potential phenotype–genotype correlations. These findings will contribute to improving the diagnosis of such extremely rare disorders. Taken together, our functional data in both Drosophila and patient cells provide new clues as to the role of WDR81 and sheds light onto its highly conserved function.
Web resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
World Health Organization Align GVGD, http://agvgd.iarc.fr/agvgd_input.php/
dbSNP, http://www.ncbi.nlm.nih.gov.gate2.inist.fr/projects/SNP/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
Uniprot, http://www.uniprot.org/uniprot/
SIFT, http://sift.bii.a-star.edu.sg/
ExAC, http://exac.broadinstitute.org/
MitoChek database: http://www.mitocheck.org/
Acknowledgements
The authors would like to thank the affected individuals and their families for participation in this study as well as the clinicians in charge of these patients who may not be cited. We would also like to thank Brian Popko for providing WDR81 antibodies. We thank Renata Basto for the Dpn antibody and the Vienna Drosophila Resource Center for the RNAi lines. The Imaging platform in the Imagine Institute for help and advice with image acquisition. We thank Mr Michaël Nicouleau for technical assistance.
Funding
Research reported in this publication was supported by the Agence Nationale de la Recherche (ANR-10-IAHU-01) (M.C., M.R., C.M., S.L., S.T., N.B.B.), The Agence Nationale de la Recherche (ANR 16-CE16-0011; R16198KK) (M.C., C.M., N.B.B.), the Fondation Maladies Rares, the Fondation pour la Recherche Médicale (FRM funding within the frame of the programme Equipe FRM; J.C–DEQ20130326477), the Fondation Maladies Rares, Fondation NRJ – Institut de France, Agence Nationale de Recherche (ANR Blanc 1103 01, project R11039KK; ANR E-Rare-012-01, project E10107KP; ANR-13-BSV-0009-01), the EU-FP7 project GENECODYS (grant number 241995) and DESIRE (grant agreement 602531) and Agence Nationale de Recherche (ANR Blanc-SVSE1-2013; ANR-13-BSV1-0027) (ST,TAB), M.S. has been supported by the ATIP-Avenir program, the Fondation Bettencourt-Schueller (Liliane Bettencourt Chair of Developmental Biology) as well as State funding by the Agence Nationale de la Recherche (ANR) under the “Investissements d'avenir” program (ANR-10-IAHU-01) and the NEPHROFLY (ANR-14-ACHN-0013) grant.
Supplementary material
Supplementary material is available at Brain online.
Abbreviation
References
Author notes
Mara Cavallin, Maria A. Rujano, Sophie Thomas and Nadia Bahi-Buisson authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}