

Abstract
The family of 2D materials has expanded quite rapidly, especially with the addition of transition metal carbides and nitrides called MXenes, in the last decade. Since their discovery in 2011, about 30 different MXenes have been synthesized, and the structure and properties of several dozens have been predicted by first-principles approaches. Given the outstanding advances in the MXene field, it is thus appropriate to review the most relevant properties of these MXenes and point out their potential applications. In this article, the structural, transport, magnetic, vibrational, mechanical, and electrochemical properties of MXenes are overviewed. The goal is to illustrate how the chemical versatility in the intrinsic composition and surface terminations combined with the potential addition of a fourth element enable to tune MXenes properties to meet the targeted applications.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
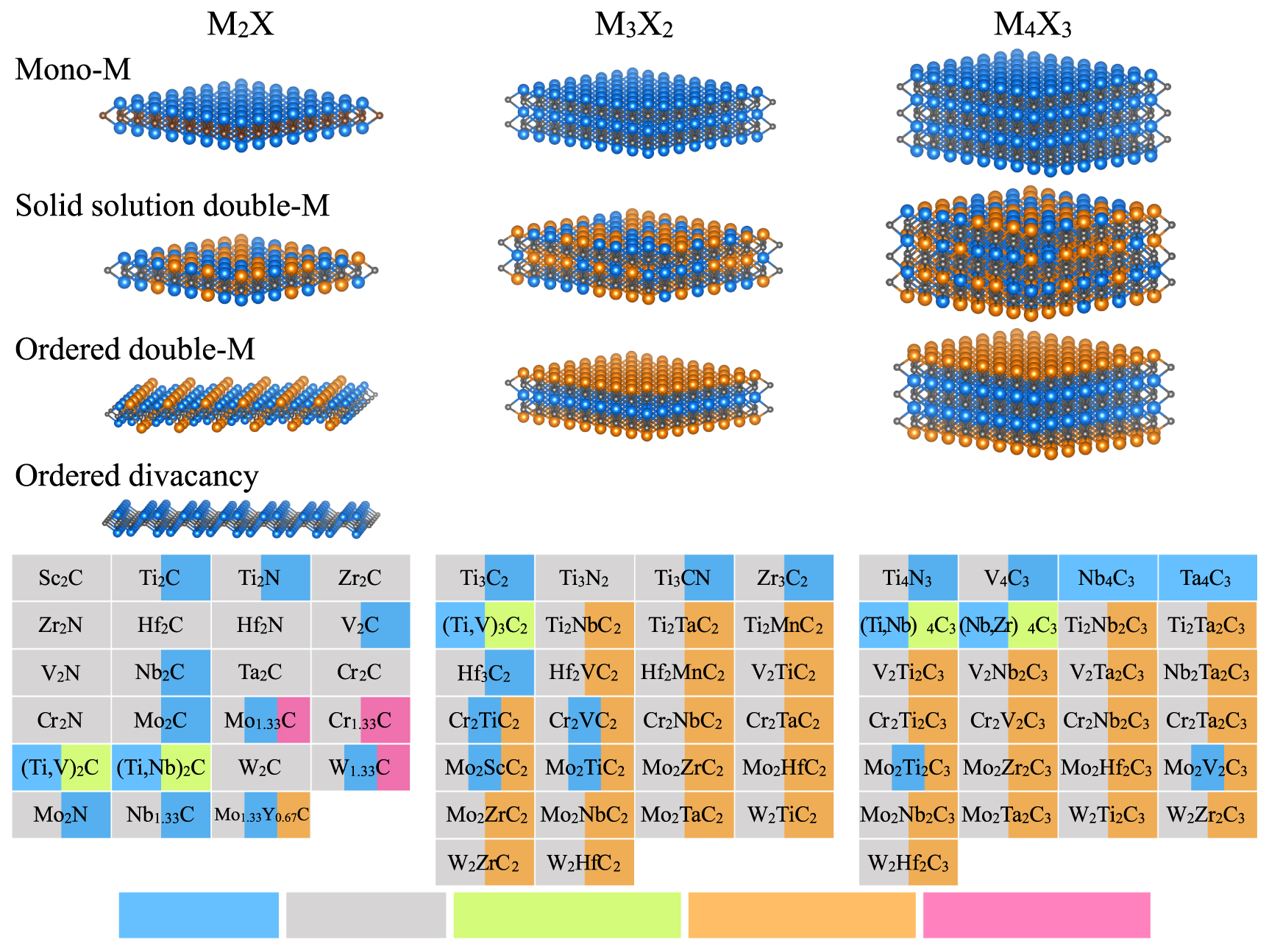
Since the exfoliation of graphene in 2004 and the characterization of its outstanding properties [1, 2], two-dimensional (2D) materials have received considerable attention in the field of materials science and device processing. 2D crystals are a subclass of nanomaterials that exhibit peculiar physical properties due to the quantum confinement of their electrons [3]. Owing to their large surface areas combined with unique electronic, mechanical, and optical properties, 2D materials have been intensively studied for their possible use in electronic devices, composite materials, and energy-related applications [4–7]. Additionally, they can be employed as building blocks to obtain the so-called van der Waals heterostructures, known to exhibit unusual properties and new phenomena [5]. At present, the existence and stability of a few dozens of 2D crystals have been reported, including hexagonal boron nitride (h-BN) [8], transition-metal dichalcogenides (MoS2, MoSe2, WS2, WSe2, NbSe2,...) [9], thin oxide layers (TiO2, MoO3, WO3,...), silicene [10], germanene [11, 12], phosphorene [13], etc. Following the technique initially used to isolate a monolayer graphene from its three-dimensional (3D) counterpart, graphite, most 2D materials were obtained from the mechanical exfoliation of a parent 3D phase. This was feasible thanks to the weak van der Waals (vdW) interlayer interactions intrinsically present in the parent phases. In 2011, Naguib and coworkers showed that 3D nanolaminate MAX phases with strong interlayer bonds could also be exfoliated into 2D crystals, using a combination of chemical etching and sonication techniques [14]. Since then, about 30 transition-metal carbides and nitrides, called MXenes, with different chemical compositions and orders have been reported [15–17], and up to 70 compositions have been predicted theoretically (figure 1) [17, 18]. Depending on the 3D precursor, MXenes materials exhibit the formula Mn + 1Xn Tz (n = 1, 2, 3) or M1.33XTz , where M is an early transition metal (Sc, Y, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, or W), X represents either a carbon or a nitrogen atom, and Tz are termination groups such as −F, −OH, =O, or −Cl, mostly depending on the nature of the chemical environment [16]. The thickness of the MXene monolayer is about 1 nm and varies with the value of the n index and the nature of the terminal groups [17].

Figure 1. Chemical versatility of all existing MXenes systems, including conventional Mn + 1Xn (n = 1, 2, 3), double-M solid solutions, out-of-plane ordered, in-plane ordered double-M, and divacancy ordered MXene systems. [17] (2019) (Springer Nature Switzerland AG 2019). With permission of Springer.
Download figure:
Standard image High-resolution imageMost MXenes (25 out of 30) were obtained by selective etching of the Al planes from the MAX phases [19]. To date, only one non-Al-containing MAX phase, Ti3SiC2, has been successfully etched into MXene [20]. It should also be noted that MXenes can be synthesized from non-MAX phase precursors, i.e. Mo2CTz was obtained by etching Ga layers from Mo2Ga2C [21], Zr3C2Tz by etching Al3C3 layers from Zr3Al3C5 [22], and Hf3C2Tz by etching (Al,Si)4C4 layers from Hf3(Al,Si)4C6 [23]. Although hydrofluoric acid (HF) treatment remains the most common synthesis pathway, it is highly hazardous and often requires a delamination step through the intercalation of metallic cations or organic molecules to separate the MXene sheets. Consequently, various alternatives have been explored, including in situ formation of HF via the mixture of HCl and LiF solutions [24], NH4HF2 [25, 26], or electrochemical etching [27, 28]. More information can be found in two recent reviews by Verger et al [29, 30].
The chemical variety of the MAX phases, with over 150 compositions reported so far [31, 32], explains the resulting chemical versatility in 2D MXenes. The rapid interest in synthesizing new MXenes has caught the attention from the MAX community to discover new MAX phase compositions. In this regard, many isostructural MAX phase solid solutions have been reported [33], together with the more recent reports of chemically ordered quaternaries, where the ordering of two M elements results in out-of-plane ordered (o-MAX) [34] or in-plane ordered (i-MAX) systems [35, 36]. Interestingly, this ordering allowed the addition of non-traditional MAX phase elements, such as Sc, Y, W, and rare-earth elements [37–40]. Since most of the newly discovered MAX phases are Al-based, the family of 2D MXenes is expected to further expand in the near future. To date, a few o-MAX phases have already been exfoliated to form out-of-plane ordered double transition metal MXenes, e.g. Cr2TiC2Tz [41], Mo2TiC2Tz [41], Mo2ScC2TZ [42], and Mo2Ti2C3Tz (figure 1) [41]. More interestingly, depending on the etching conditions, the i-MAX phases can be exfoliated to form two different types of MXenes. In-plane ordered double transition metal MXenes, such as (Mo2/3Y1/3)2CTz [43], are obtained from the selective etching of the Al layers in the parent MAX phase, while ordered divacancy MXenes, such as Mo1.33CTz [35] and W1.33CTz [42] result from the removal of the minority M element (Sc or Y) together with the Al element (figure 1). Additionally, Halim et al [44] reported the exfoliation of the (Nb2/3Sc1/3)2AlC solid solution into Nb1.33CTz with disordered vacancies.
In summary, the chemical versatility in their intrinsic composition (M and X sites) and surface terminations (Tz ), combined with the potential addition of a fourth element through alloying on the M site, give the key advantage to tune MXenes properties to meet the targeted applications. It is now well-established that MXenes exhibit a unique combination of chemical and physical properties that makes them promising building blocks of an impressive number of potential applications [17, 19, 45], including energy storage devices [46], such as hydrogen storage [46–49], Li and multivalent ion batteries [50–54], and electrochemical capacitors [24, 55–59], thermoelectric materials [60–63], electromagnetic interference shielders [64], transparent conductors [25, 65–68], structural composites [69], catalysts [70–72], sensing devices [73–76], and many more. Additional applications have been proposed in theoretical studies, based on the important diversity in MXenes electronic, magnetic, optical, and electrochemical properties [77]. However, most of the theoretical studies to date are based on defect-free crystal structures, considering pristine or homogeneously-terminated surfaces. Experimentally, the lack of control in surface termination prevents the formation of such ideal MXenes. Therefore, more research is needed on both experimental and theoretical sides, respectively, to produce high-quality single-flakes with homogeneous terminations and to simulate more realistic MXenes crystals. In this context, it is of high importance to gain insights into the mechanisms that govern the MAX phase etchability and to understand the role played by the chemistry and the structure of MXenes on their physical properties. With over 2,300 publications since the discovery of the first MXene system, it is timely to update our knowledge on the MXenes properties and related applications.
In this review article, complementarily to the recent works of Khazaei and coworkers [77, 78], we summarize the latest first-principles studies on the structural, electronic, magnetic, vibrational, mechanical, and electrochemical properties of MXenes and derivatives. In section 2, the structural properties of 2D MXenes, o-MXenes, and i-MXenes are investigated, highlighting the important role played by density functional theory (DFT) calculations in the predictive search of new MXenes. A discussion on the energetically-favored position of the terminal groups in functionalized MXenes is also presented. In section 3, we give insights into the exfoliation potential of the 3D MAX phases into 2D MXenes, through the computation of the bonding strengths and exfoliation energies. Section 4 summarizes the latest studies on the transport (4.1), magnetic (4.2), vibrational (4.3), mechanical (4.4), and electrochemical (4.5) properties. For each property, some related potential applications are proposed. A few additional properties and potential applications are also mentioned in section 4.6. Given that the scientific research on MXenes materials is still an emerging field, there is plenty of work to still be achieved. Outlook and perspectives are widely discussed in section 5. Lastly, conclusions are drawn in section 6.
2. Structure and stability of MXenes
The structure of pristine MXenes can be constructed by removing the A element from the parent MAX phases. The structures of conventional pristine MXenes with the general formula Mn + 1Xn (n = 1, 2, 3) are depicted in figure 1. Consistent with the symmetry of the precursor MAX phases, the derived MXenes are hexagonal. The atoms are arranged in a layered structure where the X layers are alternatively sandwiched between the M layers. Similarly, the o-MXenes with the general formula M'2M"X2 or M'2M"2X3 also have an hexagonal symmetry (figure 1). In o-MXenes, the outer-layers are exclusively formed with the M' element, while the inner-layer(s) consist of M" element. The exfoliation of the in-plane ordered MAX phases results either in 2D i-MXenes with the general formula (M'2/3M"1/3)2X, or in 2D M1.33X systems with ordered divacancies. Their pristine structures are illustrated in figure 1. The presence of two different transition metal elements in each layer involves a small out-of-plane shift of the larger M" atoms from their usual positions, which, in turn, results in a change in the system symmetry from hexagonal to monoclinic [79].
In the search for new stable MXene structures, theoretical approaches are of high interest. In 2013, Khazaei et al [18] investigated the formation and stability of various M2C and M2N systems with F, OH, and O surface terminal groups, using first-principles calculations. The dynamical stability is assessed through the absence of imaginary frequencies in the calculated phonon spectra. Anasori et al [41] examined the relative stability of 32 o-MXenes, considering different chemical ordering in both fully-ordered and partially-ordered configurations. Depending on the elemental combination of transition metals, ordered MXenes are, in some cases, more stable than their solid-solution counterparts. In total, about 25 different ordered MXenes have been predicted, among which only 6 have been experimentally synthesized [17]. In recent works [80, 81], the dynamical stability of several MXene 2 H phases has been predicted, with structures similar to those of 2 H TMDCs [82]. Additionally, in the search for new MXene systems, several theoretical works have been conducted to find new MAX precursors. In this context, Dahlqvist et al [36, 79, 83, 84] developed a systematic procedure to predict the phase stability and chemical ordering of several quaternary i-MAX and o-MAX phases. This procedure involves two main steps. First, the computation of the formation enthalpy in order to ascertain the thermodynamic stability. Compounds are thus considered as stable if their formation enthalpy is negative. Second, a linear optimization procedure which considers all known competing phases and their relative stability is used to check the potential decomposition of the studied phase into any of these competing phases [83]. Altogether, Dahlqvist et al [84] reported on the stability of 15 new o-MAX phases, additionally to the 7 existing ones. Moreover, the combination of theoretical and experimental approaches has led to the prediction and synthesis of 30 i-MAX phases, including the very recent rare-earth containing MAX phases [38, 39]. Most of these newly-discovered o-MAX and i-MAX still need to be experimentally converted into 2D MXenes.
As expected from the experimental observations, MXenes become thermodynamically more stable upon surface functionalization, and the stability is further enhanced for fully-terminated MXenes [18]. Based on the unit cell of pristine M2X MXene, functionalized MXene structures are constructed, with the general formula M2Xn T2 with T = F, OH, and O terminations. As depicted in figure 2(a), two types of hollow sites on the surface can be distinguished: A sites correspond to fcc sites for which no X atom is present under the M atoms, while B sites correspond to hcp sites located on the top of a X atom. As proposed by Khazaei et al [18] different functionalization models can be built depending on the relative positions of the terminal groups. The first model (MD1) considers two functional groups of the same type positioned on the top of the two transition-metal atoms (figure 2(b)). In the second model (MD2), the two functional groups are located on the top of hollow sites A (figure 2(c)). The third model (MD3) considers one functional group on the top of hollow site A and a second functional group on the top of hollow site B (figure 2(d)). At last, model 4 (MD4) presents two functional groups positioned on the top of hollow sites B (figure 2(e)). Given that the properties of MXenes can strongly vary with the nature and position of the terminal groups, it is mandatory to systematically investigate all possible configurations and evaluate their respective stability, in order to find the ground-state structure. As a general trend, in functionalized M2X system, MD1 is energetically less stable than the other three models. Depending on the ionic state of the transition-metal element and the nature of the terminal group (−F, −OH, =O), either MD2, MD3, or MD4 is found to be the most stable configuration [18].

Figure 2. Atomic structure of (a) pristine M2X with the two types of hollow sites A and B, and terminated M2XT2 in the four configurations: (b) MD1, (c) MD2, (d) MD3, and (e) MD4. M, X, and T elements are respectively in red, brown, and blue. Reprinted figure with permission from [85], Copyright (2018) by the American Physical Society.
Download figure:
Standard image High-resolution imageUsing the same four models, we investigated the formation of heterogeneously-terminated V2CTz
MXenes (T = F, O, OH) and found that an equivalent of MD2 is the most energetically favorable configuration [85]. The heterogeneity of the terminal groups induces a difference in the V-T bond lengths on both sides of the MXene layer. As the reduced atomic coordinates are modified, the Wyckoff positions are influenced, and the space group is moved from  to P3m1. Alhtough there are much less reports on hetereogeneous terminations, Hu et al [86] investigated the stability of Ti2CTz
, Ti3C2Tz
and Nb4C3Tz
systems with respect to heterogeneous terminations (T = F, OH, and O). The most stable structure of all three systems corresponds to the fully O-terminated configuration, while the fully OH-terminated configuration is found to be the least energetically favorable.
to P3m1. Alhtough there are much less reports on hetereogeneous terminations, Hu et al [86] investigated the stability of Ti2CTz
, Ti3C2Tz
and Nb4C3Tz
systems with respect to heterogeneous terminations (T = F, OH, and O). The most stable structure of all three systems corresponds to the fully O-terminated configuration, while the fully OH-terminated configuration is found to be the least energetically favorable.
3. Synthesis of MXenes
The experimental process used to convert MAX phases into MXenes is quite complex. Moreover, given that the interlayer bonds in multilayered MXenes are 2 to 6 times stronger than the weak long-range vdW interactions as in graphite or bulk MoS2, an intercalation step is often required to weaken the bonds before the delamination into 2D MXene sheets [29]. This strategy explains the difficulty in using first-principles techniques to predict the etchability of the MAX phases. Nevertheless, ab initio molecular dynamics (AIMD) calculations have been used to simulate the etching process of Ti3AlC2 in HF solution [87] and DFT calculations have allowed to gain insight into the exfoliation potential of several MAX phases into 2D MXenes.
The MD simulation explains how the HF solution interacts with the Ti3AlC2 MAX phase (figure 3) [87]. After the spontaneous dissociation of HF molecules, the F ions are attracted by the A and M elements, resulting in a weakening of the M-A bonds. As the HF concentration increases, the M-A bonds are broken and AlF3 molecules are formed. Progressively, with the removal of the AlF3 molecules, an interlayer gap is opened, facilitating further insertion of HF molecules. In contrast to F species, H ions are moderately attracted by the MAX phase and form H2 molecules instead. In the end of the etching process, a fluorinated MXene is formed, together with AlF3 and H2 molecules. Additional researches have concluded that the termination process is much more complex and might additionally include = O and −OH functional groups [14].

Figure 3. Schematic representation of the chemical exfoliation of Ti3AlC2 phase in the presence of HF acid, from AIMD simulation. The number of HF molecules increases from (a) zero, (b) one, (c) two, (d) three, (e) four, (f) five, (g) six, and (h) seven molecules. During the exfoliation process, AlF3 and H2 molecules are produced. Ti, Al, C, F, and H elements are respectively represented by small white, orange, blue, cyan, and pink spheres. Reprinted with permission from ACS Applied Materials & Interfaces 8, 2016 [87]. Reprinted with permission from [87]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution imageTheoretically, the mechanical exfoliation of the MAX phases into 2D MXenes has been investigated [88]. Given that, in some cases, the bonding in the ab-plane is stronger than the one perpendicular to the plane ( ) [89, 90], the theoretical breaking of the M-A bonds upon tensile stress has been revealed, highlighting the possibility to form 2D MXenes through the mechanical exfoliation of 3D MAX phases. This theoretical prediction has been experimentally achieved in a very recent work. Gkountaras and coworkers [91] successfully reported on the mechanical exfoliation of four MAX phase single crystals, using the adhesive tape method. They coined the resulting crystals 'MAXenes', since the presence of the A element in the 2D flakes was confirmed. The thickness of the flakes could be reduced down to a monolayer. Most importantly, crystals that were to date chemically unetchable, such as Cr2AlC, Ti2SnC, and even the ferromagnetic Mo4Ce4Al7C3, have been mechanically exfoliated. This opens the door to the formation of less defective crystals and the in-depth characterization of their electronic, magnetic, and optical properties.
) [89, 90], the theoretical breaking of the M-A bonds upon tensile stress has been revealed, highlighting the possibility to form 2D MXenes through the mechanical exfoliation of 3D MAX phases. This theoretical prediction has been experimentally achieved in a very recent work. Gkountaras and coworkers [91] successfully reported on the mechanical exfoliation of four MAX phase single crystals, using the adhesive tape method. They coined the resulting crystals 'MAXenes', since the presence of the A element in the 2D flakes was confirmed. The thickness of the flakes could be reduced down to a monolayer. Most importantly, crystals that were to date chemically unetchable, such as Cr2AlC, Ti2SnC, and even the ferromagnetic Mo4Ce4Al7C3, have been mechanically exfoliated. This opens the door to the formation of less defective crystals and the in-depth characterization of their electronic, magnetic, and optical properties.
The prediction of potential MAX candidates for the exfoliation into 2D MXenes can be performed by evaluating the bonding strengths and exfoliation energies [92]. A first way to evaluate the bonding strengths consists in the computation of the force constants FCi related to a specific element i. For the MAX phases to be successfully transformed into 2D MXenes, M-A bonds need to be weak, while M-X bonds must be strong enough to ensure the MXene integrity after the exfoliation process. Interestingly Khazaei et al [92] found that M-X bonds are the strongest in MAX phases and the global trend is that shorter bonds are stronger. Based on the computed force constants for the experimentally exfoliated MAX phases, some thresholds are set on the FCA and FCX such that the exfoliation would only occur if FCA ≤21.855 eV/Å2 and FCX ≥40.511 eV/Å2 [92].
Another method to gain insight into the bonding strengths is the crystal orbital Hamilton population (COHP) analysis. The COHP technique allows to partition the electronic band structure into bonding, nonbonding, and antibonding contributions of the localized atomic basis sets [94–96]. By integrating the COHP up to the Fermi energy (ICOHP), indication about the relative bonding strengths and the covalency of the bonds can be obtained. All the COHP calculations are performed using the local orbital basis suite towards electronic structure reconstruction (LOBSTER) code [94–96], with the pbeVaspFit2015 basis set [97]. This technique has confirmed that the M-X bonds are stronger than the M-A bonds in all the studied MAX phases, as depicted in figure 4(a)). Dahlqvist et al [98] used the COHP analysis to investigate the bonding strengths within two W-based i-MAX phases. This technique allowed to understand the removal of both Al and M" elements from (W2/3M"1/3)2AlC (M = Sc, Y) during etching and the resulting formation of divacancy ordered W1.33C MXenes [42]. More recently, we have used the COHP technique to shed light on the bonding strengths into rare-earth (RE) containing i-MAX phases with the general formula (Mo2/3RE1/3)2AlC (RE = Nd, Sm, Gd, Tb, Dy, Ho, Er, Tm, and Lu) [93, 99]. In all systems, the Mo-C and RE-C bonds are stronger than the Mo-Al and RE-Al bonds (figure 4(a)), suggesting their exfoliation is feasible.

Figure 4. (a) Integrated COHP (bond strength) for both M-A and M-X bonds, and (b) computed static exfoliation energy as a function of the bond length, for a series of 82 MAX and o-MAX phases [92] plus 9 RE-containing i-MAX [93]. The grey lines are guide to the eyes. (a) Adapted from [92] with permission of The Royal Society of Chemistry. (b) Reprinted figure with permission from [93], Copyright (2020) by the American Physical Society.
Download figure:
Standard image High-resolution imageTheoretically, the static exfoliation energy can be seen as the energy needed to transform a MAX phase into isolated 2D MXene sheets and aluminum atoms. The energies of the MAX, MXene, and bulk Al, are computed for the optimized ground-state structures. The static exfoliation energies of about 90 systems are plotted in figure 4(b) as a function of the M-A bond length. Since no experimental exfoliation data are present in the literature, the best we can do is to suggest a threshold energy. Among the successfully etched MAX phases, V2AlC has the largest predicted exfoliation energy of 0.205 eV/Å2 [92]. It is therefore expected that MAX phases with an exfoliation energy lower than this threshold could, a priori, be successfully etched into 2D MXenes [93].
Based on the evaluation of the exfoliation energies and the force constants, Khazaei et al [92] has shown that 37 MAX or o-MAX phases can potentially be transformed into 2D MXenes. Similarly, we predicted the potential exfoliation of nine rare-earth containing i-MAX phases into 2D RE-i-MXenes [93]. Given that all RE-i-MAX phases exhibit exotic magnetic characteristics, their exfoliation potential represents a promising breakthrough for future use in applications where 2D magnetic materials are desired.
4. Properties of MXenes and possible derived applications
Since the discovery of MXenes in 2011, several experimental and theoretical studies have been conducted to shed light on their chemical and physical properties. The MXene properties are unique and combine high electrical and thermal conductivities, tunable electronic band gap, various magnetic ordering, and high Young's modulus. In this review article, we focus on the electronic, magnetic, vibrational, mechanical, and electrochemical properties of MXenes, predominantly predicted or confirmed by a first-principles approach.
4.1. Electronic and transport properties
The electronic and transport properties of 2D MXenes have been intensively studied from a theoretical point of view. Several works have already reported and reviewed the wide range of electronic properties accesssible by playing on the composition [18, 19, 41, 72, 77, 78, 100]. In general, most structural and electronic properties of MXenes were computed within the GGA framework [18, 101, 102], while hybrid functionals were sometimes used to obtain an accurate estimation of the electronic band gap [103–106]. In some works, due to the presence of strongly correlated d electrons in transition metal elements, a Hubbard correction ranging from 2 to 5 eV was applied [102, 106, 107].
4.1.1. Metals and semiconductors
Similar to the MAX phases, the pristine MXenes are predicted to be metallic. As depicted in figure 5(b), the states in the vicinity of the Fermi level correspond to M−d states and are expected to give rise to electrical conductivity in the MXene systems. The electron density of states (DOS) near the Fermi level is predicted to be higher in the bare MXenes than in their parent MAX phases [16].

Figure 5. (a) MXene atomic structure, unit cell, and Brillouin zone, including Γ, M, and K points, with Γ-M and Γ-K respectively along the armchair and zigzag direction. Electronic band structure and DOS of (b) pristine V2C and (c) functionalized V2CF2 computed within GGA, and (d) projected band structure of Ti2CO2. The Fermi level is set as the reference of zero energy. (a)−(c) Reprinted figure with permission from [85], Copyright (2018) by the American Physical Society. (d) Reprinted figure with permission from [85], Copyright (2018) by the American Physical Society.
Download figure:
Standard image High-resolution imageUpon functionalization, the electronic properties of MXenes vary from metallic to semiconducting, depending on the nature of the M, X, and T groups [18]. While the majority of terminated MXenes retain their metallic character (figure 5(c) for V2CF2), Sc2CT2 [18], Ti2CO2 [18], Zr2CO2 [18], Hf2CO2 [18], Cr2CF2 [103], Cr2C(OH)2 [103], and (M'2/3M"1/3)2CO2 (M' = Mo, W; M" = Sc, Y) [108] are predicted to be semiconductors, due to a shift of the Fermi level (figure 5(d) for Ti2CO2). The band gap energies of M2XT2 MXenes, computed within the generalized gradient approximation (GGA) [109] and within the nonlocal Heyd-Scuseria-Ernzerhof (HSE06) hybrid functional [110, 111], are reported in table 1. All semiconducting MXenes have an indirect band gap, except Sc2C(OH)2 that has a direct band gap at Γ. Given that both −F and −OH terminal groups can only accept one electron from the surface, they often affect the electronic properties in a similar way. In contrast, = O terminations accept two electrons from the surface [18]. A comprehensive study of the electronic properties of a dozen of M2XT2 systems can be found in [18]. Moreover, a materials database, aNANt, containing the structural and electronic band structure of more than 23,000 MXenes in their pristine and functionalized forms, has recently been released [112, 113].
Table 1. Electronic and magnetic properties of M2XT2 MXenes. For semiconducting systems, the third column corresponds to the electronic band gap; for half-metallic systems, to the band gap found for the minority spin channel; and for topological insulator (TI), to the gap opened upon considering SOC. The fourth column corresponds to the functional used in the DFT calculations. The fifth column describes the magnetic behavior (NM: non-magnetic, FM: ferromagnetic, AFM: antiferromagnetic).
| Metallic | Band gap | Computational | |||
|---|---|---|---|---|---|
| Compounds | behavior | (eV) | details | Magnetism | references |
| Sc2C | Metallic | 0 | GGA | NM | [18] |
| Sc2CF2 | Semiconducting | 1.03 (1.85) | GGA (HSE06) | NM | [18, 104] |
| Sc2C(OH)2 | Semiconducting | 0.45 (0.85) | GGA (HSE06) | NM | [18, 104] |
| Sc2CO2 | Semiconducting | 1.80 (2.90) | GGA (HSE06) | NM | [18, 104] |
| Ti2C | Near half-metallic | 0.04 | GGA | FM | [101] |
| Ti2CF2 | Metallic | 0 | GGA | NM | [18] |
| Ti2C(OH)2 | Metallic | 0 | GGA | NM | [18] |
| Ti2CO2 | Semiconducting | 0.24 (0.92) | GGA (HSE06) | NM | [18, 104] |
| Ti2N | Near half-metallic | 0.04 | GGA | FM | [101] |
| Ti2NF2 | Metallic | 0 | GGA | AFM | [102] |
| Ti2N(OH)2 | Metallic | 0 | GGA | AFM | [102] |
| Ti2NO2 | Metallic | 0 | GGA | FM | [102] |
| V2C | Metallic | 0 | GGA | NM | [18] |
| V2CF2 | Metallic | 0 | GGA | NM | [18] |
| V2C(OH)2 | Metallic | 0 | GGA | NM | [18] |
| V2CO2 | Metallic | 0 | GGA | NM | [18] |
| V2N | Metallic | 0 | GGA | NM | [102] |
| V2NF2 | Metallic | 0 | GGA | AFM | [102] |
| V2N(OH)2 | Metallic | 0 | GGA | AFM | [102] |
| V2NO2 | Metallic | 0 | GGA | AFM | [102] |
| Cr2C | Half-metallic | 2.85 | HSE | FM | [103] |
| Cr2CF2 | Semiconducting | 3.49 | HSE | AFM | [103] |
| Cr2C(OH)2 | Semiconducting | 1.43 | HSE | AFM | [103] |
| Cr2CO2 | TI (semimetal) | 0 | HSE06 | NM | [105] |
| Cr2N | Metallic | 0 | GGA+U | AFM | [106] |
| Cr2NF2 | Metallic | 0 | GGA+U | AFM | [106] |
| Cr2N(OH)2 | Metallic | 0 | GGA+U | AFM | [106] |
| Cr2NO2 | Half-metallic | 2.79 (3.88) | GGA+U (HSE) | FM | [106] |
| Mn2C | Metallic | 0 | GGA+U | FM | [107] |
| Mn2CF2 | Half-metallic | 0.94 (2.52) | GGA+U (HSE06) | FM | [107] |
| Mn2C(OH)2 | Metallic | 0 | GGA+U | FM | [107] |
| Mn2CO2 | Metallic | 0 | GGA+U | FM | [107] |
| Mn2N | Metallic | 0 | GGA+U | FM | [102] |
| Mn2NF2 | Half-metallic | 4.0 | GGA+U | FM | [102] |
| Mn2N(OH)2 | Half-metallic | 2.6 | GGA+U | FM | [102] |
| Mn2NO2 | Half-metallic | 3.3 | GGA+U | FM | [102] |
| Zr2C | Metallic | 0 | GGA | NM | [18] |
| Zr2CF2 | Metallic | 0 | GGA | NM | [18] |
| Zr2C(OH)2 | Metallic | 0 | GGA | NM | [18] |
| Zr2CO2 | Semiconducting | 0.88 (1.70) | GGA (HSE06) | NM | [18, 104] |
| Nb2C | Metallic | 0 | GGA | NM | [18] |
| Nb2CF2 | Metallic | 0 | GGA | NM | [18] |
| Nb2C(OH)2 | Metallic | 0 | GGA | NM | [18] |
| Nb2CO2 | Metallic | 0 | GGA | NM | [18] |
| Mo2C | Metallic | 0 | GGA | NM | [18] |
| Mo2CF2 | Semiconducting | 0.27 (0.86) | GGA (HSE06) | NM | [60, 104] |
| Mo2C(OH)2 | Semimetal | 0 | GGA | NM | [60] |
| Mo2CO2 | TI (semimetal) | 0 | HSE06 | NM | [105] |
| Hf2C | Metallic | 0 | GGA | NM | [18] |
| Hf2CF2 | Metallic | 0 | GGA | NM | [18] |
| Hf2C(OH)2 | Metallic | 0 | GGA | NM | [18] |
| Hf2CO2 | Semiconducting | 1.02 (1.66) | GGA (HSE06) | NM | [18, 104] |
| Hf2N | Metallic | 0 | GGA | NM | [18] |
| Ta2C | Metallic | 0 | GGA | NM | [18] |
| W2C | Metallic | 0 | GGA | NM | [105] |
| W2CO2 | TI | 0.19 (0.47) | GGA (HSE06) | NM | [105] |
4.1.2. Topological insulators
Interestingly, some MXenes (and o-MXenes) with group VI transition metals (Cr, Mo, W) are predicted to be 2D topological insulators (TI), i.e. to present an insulating gap in the bulk and gapless states at the edges. In these systems, the spin–orbit coupling (SOC) is found to significantly affect the electronic properties. For instance, without the SOC, M2CO2 (M = Mo, W, Cr) [105], M'2M"C2O2 (M' = Mo, W; M" = Ti, Zr, Hf) [114–116], and Ti3N2F2 [117] are semiconductors with a zero energy gap or semimetals with compensated electron and hole Fermi pockets. As depicted in figure 6(a) for Mo2HfC2O2, the topmost valence band and the lowest conduction band only touch at the Γ point, around which the bands have a parabolic dispersion. These bands mostly correspond to the d-states of the M element. In addition, the edge states presented in figure 6(c) only touch at the M point and connect the bulk valence and conduction bands. The inclusion of the SOC lifts the degeneracy of the bands at the Fermi level and the above-mentioned systems become insulators (figure 6(b)), with band gaps ranging from 0.05 to 0.47 eV within HSE06. In general, the energy band gap is larger as the SOC is larger [114]. A summary of the electronic characteristics of the non-trivial TI MXenes is proposed in table 2. Large band gap TI are appropriate systems to observe quantum spin Hall effect at room temperature (RT) and above and are therefore potential candidates for electronic and spintronic device applications. It should be noted that other MXenes such as Sc2C(OH)2 and M3N2F2 (M = Zr, Hf) become TI by applying an electric field [118] or a tensile strain of a few percents [117], respectively.

Figure 6. Electronic band structure of Mo2HfC2O2 computed (a) without SOC and (b) with SOC. Upon including SOC, a gap opens at the Γ point. (c) Edge states connecting the bulk valence and conduction bands and only touching at the M point. Reprinted with permission from [115]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution imageTable 2. Electronic characteristics of the topological insulating MXenes. The situations with and without SOC are distinguished and the electronic band gap observed with SOC, computed with GGA and HSE are reported.
| With SOC | ||||
|---|---|---|---|---|
| Compounds | Without SOC | Eg GGA (eV) | Eg HSE (eV) | references |
| Cr2CO2 | compensated SM | − | − | [105] |
| Mo2CO2 | compensated SM | − | − | [105] |
| W2CO2 | zero-gap SM | 0.194 | 0.472 | [105] |
| W2TiC2O2 | zero-gap SM | 0.136 | 0.290 | [114] |
| W2ZrC2O2 | zero-gap SM | 0.170 | 0.280 | [114] |
| W2HfC2O2 | zero-gap SM | 0.285 | 0.409 | [114] |
| Mo2TiC2O2 | zero-gap SM | 0.041 | 0.119 | [114] |
| Mo2ZrC2O2 | zero-gap SM | 0.069 | 0.125 | [114] |
| Mo2HfC2O2 | zero-gap SM | 0.153 | 0.238 | [114] |
| Ti3N2F2 | compensated SM | 0 | 0.05 | [117] |
| Zr3N2F2 | compensated SM | 0.05 if stretched | [117] | |
| Hf3N2F2 | semiconductor | 0.10 if stretched | [117] | |
In order to observe semiconducting and topological insulating states in MXenes, more work is needed on the experimental side to control the surface termination of MXenes and find new routes to create homogeneously-functionalized MXenes. So far, only the semicondutor-like behavior of Ti2CO2 has been observed experimentally, with a band gap of 80 meV [119], which is much smaller than the theoretical prediction.
4.1.3. Electronic transport
Regarding the electronic transport in 2D MXenes, coherent transport calculations within the non-equilibrium Green's functions (NEGF) formalism have demonstrated that the metallic MXenes are highly conductive [120, 121]. It was also determined that the electrical character and conductivity of MXenes strongly depend on the surface terminations. For instance, the presence of nearly free electron (NFE) states in the electronic band structure of several OH-terminated MXenes has been evidenced [120]. In particular, in Ti2C(OH)2, Zr2C(OH)2, Zr2N(OH)2, Hf2C(OH)2, Hf2N(OH)2, Nb2C(OH)2, and Ta2C(OH)2 MXenes, the NFE states are close to the Fermi level and their partial occupation contributes to a higher electron conductivity, without atomic scattering with the surface vibrations [120]. In contrast, the NFE states in graphene, h-BN, and MoS2 are located at high energies above the Fermi level, and remain therefore unoccupied. Hu and coworkers [122] reported on the intrinsic electronic conductivity of stacked multilayered Ti3C2(OH)2 MXenes. The theoretical calculations of the electronic band structure, band dispersions, and Fermi surface indicate that the charge carriers are preferentially transferred in the plane rather than out-of-plane. Said otherwise, the electronic conduction in stacked Ti3C2(OH)2 MXenes is highly anisotropic.
4.1.4. Thermoelectrics
Semiconducting materials are usually good candidates for electronic devices or thermoelectric applications. In this regard, the thermal and thermoelectic properties of a series of semiconducting MXenes have been studied.
The thermoelectric efficiency can be evaluated through the figure of merit zT given by:

where S, σ,  are respectively the Seebeck coefficient, electrical conductivity, and thermal conductivity with the electronic κe
and lattice κl
contributions. Using the Boltzmann theory, Khazaei et al [18] reported the Seebeck coefficient of the semiconducting Ti2CO2 and Sc2C(OH)2 systems, being as high as 1140 and 2200 µVK−1 at 100 K. The thermoelectric performance of Sc2C-based MXenes has been investigated by Kumar et al [62], based on the explicit calculation of σ, κe
, and κl
, using the Boltzmann transport equation for electrons and phonons, respectively. At RT, Seebeck coefficients of 1022, 1036, and 372 µVK−1 are obtained, respectively for O-, F-, and OH-terminated Sc2CT2 systems. Their lattice thermal conductivities are of 59, 36, and 10 Wm−1K−1, respectively, while the electronic thermal conductivities range from 3 to 6 Wm−1K−1. The quite low thermal conductivity in the Sc2C(OH)2 system with respect to other semiconducting MXenes leads to a maximal figure of merit of 0.1 at 300 K up to 0.5 at 900 K. The value of the figure of merit could be significantly improved by further reducing the thermal conductivity down to values around 1 to 5 Wm−1K−1, which are typical values of efficient thermoelectric materials reported in the literature [123].
are respectively the Seebeck coefficient, electrical conductivity, and thermal conductivity with the electronic κe
and lattice κl
contributions. Using the Boltzmann theory, Khazaei et al [18] reported the Seebeck coefficient of the semiconducting Ti2CO2 and Sc2C(OH)2 systems, being as high as 1140 and 2200 µVK−1 at 100 K. The thermoelectric performance of Sc2C-based MXenes has been investigated by Kumar et al [62], based on the explicit calculation of σ, κe
, and κl
, using the Boltzmann transport equation for electrons and phonons, respectively. At RT, Seebeck coefficients of 1022, 1036, and 372 µVK−1 are obtained, respectively for O-, F-, and OH-terminated Sc2CT2 systems. Their lattice thermal conductivities are of 59, 36, and 10 Wm−1K−1, respectively, while the electronic thermal conductivities range from 3 to 6 Wm−1K−1. The quite low thermal conductivity in the Sc2C(OH)2 system with respect to other semiconducting MXenes leads to a maximal figure of merit of 0.1 at 300 K up to 0.5 at 900 K. The value of the figure of merit could be significantly improved by further reducing the thermal conductivity down to values around 1 to 5 Wm−1K−1, which are typical values of efficient thermoelectric materials reported in the literature [123].
Eventually, Zha et al [63] computed the thermal conductivity, thermal expansion coefficient, electronic band gap, and charge carrier mobility of M2CO2 (M = Ti, Zr, Hf) systems, by means of DFT, density functional perturbation theory (DFPT), and the Klemens theory. In all three systems, the thermal conductivity is found to be highly anisotropic. Among the three compounds, Hf2CO2 possesses the highest thermal conductivity of about 86 Wm−1K−1 and 36 Wm−1K−1 at 300 K for a 5 µm-large crystal, along the armchair and zigzag direction, respectively [63]. Additionally, it has a low thermal expansion coefficient which guarantees its good structural stability at all temperatures. Given the low thermal expansion coefficient and high thermal conductivity values, Hf2CO2 is considered as a promising 2D materials for nanoelectronics.
4.2. Magnetic properties
A magnetic ground state has been established for a large number of MAX phases based on a subtle combination of theoretical and experimental studies [124, 125]. Almost all magnetic MAX phases contain Cr and/or Mn elements, including Cr2AlC [126–135], Cr2GeC [124, 132, 136–138], Cr2GaC [132, 139, 140], Cr2AlN [135], Cr2GaN [139, 141], Fen + 1ACn (n = 1, 2, 3, and A = Al, Si, Ge) [142], Mn2AlC [128, 135], Mn2GaC [143–147], (Cr,Mn)2AlC [36, 134], (Cr,Mn)2GeC [132], Cr4AlN3 [148], (Cr2Ti)AlC2 [34, 36, 149]. The latest additions to the list of magnetic MAX phases concern the in-plane ordered (M2/3Sc1/3)2AlC (M = Cr, Mn) [40], (Mo2/3RE1/3)2AlC (RE = Ce, Pr, Nd, Sm, Gd, Tb, Dy, Ho, Er, Tm, and Lu) [38], and (Mo2/3RE1/3)2GaC (RE = Gd, Tb, Dy, Ho, Er, Tm, Lu, and Yb) [39] MAX phases, with various magnetic characteristics. The possibility to play on the chemical ratio between two M elements and on the chemical ordering suggests a high tuning potential of the magnetic properties, beneficial for future applications.
To date, unfortunately, none of the above magnetic MAX phases has been etched into 2D MXenes, except (Cr2Ti)AlC2 [41]. Nevertheless, the magnetic properties of MXenes have been intensively studied from first-principles calculations, and are summarized in table 1. In general, the magnetic ground state of MXenes has been obtained from spin-polarized calculations considering a series of magnetic configurations, including a collinear ferromagnetic (FM) configuration, a few antiferromagnetic (AFM) configurations with different spin orientations between the transition metal layers, and the non-magnetic (NM) configuration [81, 102, 106, 107, 150]. Spin-polarized calculations were often performed within the GGA+U framework, with a U correction ranging from 2 to 5 eV. Given that the choice of the U correction is somehow arbitrary, either a series of U values was tested, or the U value was chosen based on previous works treating similar elements. In some cases, it has been observed that the inclusion of a Hubbard correction could alter the favored spin orientation [151]. Although the majority of the pristine MXenes are non-magnetic, some of them such as Ti2C, Ti2N, Cr2C, Mn2C, and Mn2N have ferromagnetic ground-states, while V2C and Cr2N are antiferromagnetic. Because of the surface terminations that are inevitably introduced during the synthesis process, none of the magnetic pristine MXenes has been realized in experiment. Interestingly, some functionalized MXenes have been predicted to preserve a magnetic moment, including Ti2NO2 [102], Cr2NO2 [106], and all Mn2CT2 [107] and Mn2NT2 [102] systems, regardless of surface functionalization. Magnetic moments up to 3 µB have been predicted in these systems that might retain their magnetism up to near RT. Notwithstanding, large magnetic moments may be induced in 2D MXenes via tensile/compressive strain or via doping with Cr/Mn elements [17, 152, 153]. Eventually, NM, AFM, and FM magnetic behaviors are also found for o-MXenes systems, depending on their composition [154, 155].
Recently, half-metallicity has been predicted in magnetic MXenes. Half-metallicity involves that one spin-channel is metallic, while the other one is insulating [156], resulting in a conductivity solely ensured by a single spin charge carrier channel and a 100% spin-polarization of the electrons at the Fermi level [102]. Si and coworkers [103] first predicted half-metallicity in Cr2C, with a substantial minority spin gap of 2.85 eV (figure 7), computed within HSE. Subsequently, near-half-metallicity has been predicted in Ti2C and Ti2N systems, for which true half-metallicity is only observed under biaxial strain [101]. In all three systems, half-metallicity disappears upon functionalization, together with the ferromagnetism. Upon surface passivation with oxygen, Cr2N becomes a half-metallic ferromagnetic system, with a computed minority spin gap of 2.79 eV (3.88 eV) within GGA (HSE06) [106]. Similarly, half-metallicity is also predicted in Mn2CF2 [107]. It should be noted that all these materials, i.e. Cr2C, Cr2NO2, and Mn2CF2, exhibit half-metallicity for none or specific surface terminations, which is currently difficult to achieve experimentally. In contrast, a half-metallic behavior was found in Mn2NT2 systems for all kind of surface terminations (−F, −OH, and = O) [102], suggesting great promise for these systems to be used in spintronic devices such as spin filters, spin injectors, and magnetic sensors [102].

Figure 7. Electronic band structure of Cr2C MXene highlighting its half-metallicity, with a metallic majority spin channel (left pannel) and an insulating minority spin channel with a gap of 2.85 eV (right pannel). Black and green squares represent the contributions of Cr-d and Cr-p orbitals respectively. Reprinted with permission from ACS Applied Materials & Interfaces 7, 2015 [103]. Reprinted with permission from [103]. Copyright (2015) American Chemical Society.
Download figure:
Standard image High-resolution imageBesides the symmetrically-terminated MXenes, a new class of asymetrically-functionalized MXenes, called Janus MXenes, has been intensively studied [157]. As depicted in figure 8(a), Janus MXenes have distinct terminations on their opposite surfaces. Both intrinsic FM and AFM ordering have been predicted in Janus MXenes, highlighting the possibility to effectively tune the magnetic behavior based on the selected pair of chemical terminations. For instance, it is predicted that all Cr2CTT' (T,T' = H, F, Cl, Br, OH) systems are AFM semiconductors with Neel temperatures of ∼400 K [157]. More precisely, as evidenced in figure 8(a), they are bipolar magnetic semiconductors with zero-magnetization, as the valence and conduction bands are made up of opposite spin channels that are fully compensated [157]. This makes the most noticeable difference between symmetrically-functionalized Cr2CF2 with a symmetrical distribution and the asymmetrically-functionalized Cr2CFCl, as illustrated in figures 8(b) and (a), respectively. Similar bipolar magnetic features are also predicted in V-, Mn-, and Ti-based Janus MXenes [17]. Eventually, interesting magnetic characteristics have been predicted in pristine and functionalized Janus MXenes where the asymmetry comes from the presence of distinct transition metal elements on opposite surfaces, rather than distinct terminal groups [158].

Figure 8. Atomic structures and electronic DOS of (a) Cr2CFCl janus MXene and (b) Cr2CF2 MXene, deposited on SiC(0001). Both systems are AFM, with the spin-polarized charge density represented in cyan and yellow for the spin up and spin down, respectively. Reproduced from [157] with permission of The Royal Society of Chemistry.
Download figure:
Standard image High-resolution image4.3. Vibrational properties
The computation of the phonon dispersion spectrum of a system contributes to the verification of its thermodynamic stability and provides in-depth understanding of its thermodynamic properties. The absence of negative and imaginary frequencies in the phonon spectrum is frequently used as a theoretical criterion for phase stability. Experimentally, phonon spectra can be obtained by inelastic neutron scattering technique. Complementarily to the phonon band structure, the phonon DOS describes the number of phonon modes at each energy.
The phonon spectra of various pristine and terminated M2XT2 (M = Sc, Ti, Zr, Mo, Hf; X = C, N; T = F, O) systems have been computed considering the four termination configurations discussed in section 2 (figure 2), confirming some specific stabilities [159]. The phonon spectrum and DOS of pristine V2C are presented in figure 9(a) [85]. Due to the similar crystal structure and chemical bonding of M2C systems, many features are common to all of them, and are even shared by higher-order M3C2 and M4C3 MXenes [160]. The phonon dispersions have three acoustic modes; two of them exhibit a linear dispersion near Γ and correspond to in-plane rigid-body motions. In contrast, the third acoustic mode corresponding to out-of-plane vibration has a quadratic dispersion close to Γ and a lower energy in the rest of the spectrum. This quadratic dependence is analogous to the one observed in graphene [161] and in MoS2 [162] and was originally demonstrated by Lifshitz [R1]. Additionally, a wide phonon band gap is observed separating the low-frequency vibrations of the M element from the high-frequency vibrations of the lighter C element. Upon functionalization, this band gap is filled by additional optical modes (figure 9(b)).

Figure 9. Phonon band structure and DOS of (a) pristine V2C and (b) functionalized V2C(OH)2 in their high-symmetry configuration. Reprinted with permission from [85]. Copyright (2018) American Chemical Society.
Download figure:
Standard image High-resolution imageBasics of solid state physics inform us that 3 N phonon modes are observed in a system made of N atoms. In addition to the 3 acoustic modes, there are therefore 3 N−3 optical modes. Based on the crystal structure of the M2X MXene including 3 atoms, the optical modes at the zone center of the Brillouin zone can be classified with the following irreducible representation:

where the E-symmetry modes (Eg and Eu ) are doubly-degenerate. In the same way, the optical modes of M3X2 and M4X3 systems with respectively 5 and 7 atoms in their unit cells, have the following irreducible representations:


Upon functionalization, the addition of 2 atoms in the unit cell (for F2 and O2 terminations) involves the addition of 6 optical modes, namely  , and the addition of 4 atoms via (OH)2 terminations results in 12 additional optical modes. Making good use of the group theory, the Raman and infrared (IR) activity of the modes can be predicted. Phonon modes with symmetry Eg
and
, and the addition of 4 atoms via (OH)2 terminations results in 12 additional optical modes. Making good use of the group theory, the Raman and infrared (IR) activity of the modes can be predicted. Phonon modes with symmetry Eg
and  are found to be Raman-active, while Eu
and
are found to be Raman-active, while Eu
and  are IR-active. In the case of pristine M2X, in general, and V2C, in particular, the Raman-active modes correspond to the low-frequency optical modes (at 224 and 359 cm−1), which exclusively involve the vibrations of the M element (figures 10(a)−(c)). The IR-active modes correspond to the upper branches in the phonon spectrum (at 610 and 701 cm−1) and predominantly involve C-vibrations (figures 10(d)−(f)). The Raman- and IR-active mode frequencies have been reported for several MXene systems, including [159] and [160].
are IR-active. In the case of pristine M2X, in general, and V2C, in particular, the Raman-active modes correspond to the low-frequency optical modes (at 224 and 359 cm−1), which exclusively involve the vibrations of the M element (figures 10(a)−(c)). The IR-active modes correspond to the upper branches in the phonon spectrum (at 610 and 701 cm−1) and predominantly involve C-vibrations (figures 10(d)−(f)). The Raman- and IR-active mode frequencies have been reported for several MXene systems, including [159] and [160].

Figure 10. Schematic representations (top and side views) of the Raman-active (a)−(b) degenerate Eg
mode at 224 cm−1 and (c)  mode at 359 cm−1; and infrared-active (d)
mode at 359 cm−1; and infrared-active (d)  mode at 610 cm−1 and (e)−(f) degenerate Eu
mode at 701 cm−1, of pristine V2C MXene. Reprinted with permission from [85]. Copyright (2018) American Chemical Society.
mode at 610 cm−1 and (e)−(f) degenerate Eu
mode at 701 cm−1, of pristine V2C MXene. Reprinted with permission from [85]. Copyright (2018) American Chemical Society.
Download figure:
Standard image High-resolution imageIn addition, we reported the experimental Raman spectrum of multilayered V2CTz MXene system and compared the peak positions with the predicted Raman mode frequencies [85]. Our study pointed out the importance to consider mixed terminations in the calculations in order to obtain a satisfactory agreement with the experimental spectrum (figure 11). All the experimental peaks were predicted theoretically, with a maximal deviation of 15% in the low-frequency range, and only 3% in the high-frequency range. The in-depth understanding and accurate definition of the peak positions and associated atomic vibrations is of high importance in order to gain insight into the composition and quality of samples, by the mean of Raman spectroscopy.

Figure 11. Raman spectrum of the exfoliated V2C-based sample collected at room temperature. The calculated Raman-active frequencies of the V2C, V2CF2, V2C(OH)2, and V2CFOH monosheets are included under the experimental spectrum for comparison. The matching between the predicted normal-mode frequencies and the experimental spectrum confirms the presence of heterogeneous terminal groups at the V2C surface. Reprinted with permission from [85]. Copyright (2018) American Chemical Society.
Download figure:
Standard image High-resolution image4.4. Mechanical properties
Kurtoglu and coworkers [163] first reported on the elastic constants (c11) of a series of pristine MXenes. In average, the elastic constants of 2D MXenes are twice larger than those of the corresponding MAX phases. In addition, in most cases, nitrides are stiffer than the carbide counterparts, as presented in figure 12(a) [164]. This might be explained by the additional electron provided by N atoms with respect to C atoms, and hence the formation of stronger MM-X bonds in nitride compounds [159, 165]. Based on the c11 values, 2D transition metal carbides tend to get stiffer as the atomic mass of the M element increases (figure 12(b)). Further computational studies have investigated the effect of terminations on the elastic properties of MXenes [104, 163]. The presence of terminal groups drastically reduces the c11 values of all MXenes, as evidenced in figure 12(b). The general trend is that the smaller lattice parameters in the O-terminated systems result in stronger MXenes with respect to those terminated with −F or −OH groups [104]. The thickness dependence of the elastic constants has also been studied through DFT and MD calculations, both predicting M2X systems stiffer than their M3X2 and M4X3 counterparts (figure 12(a)) [164, 166]. Experimentally, the elastic response of a single layer Ti3C2Tz has been measured through nanoindentation technique with an AFM tip [167]. A Young's modulus of 330± 30 GPa was found, which is lower than the one of graphene (1000± 100 GPa) and h-BN (870± 70 GPa), but is the highest one reported for a solution-processed 2D material, e.g. MoS2: 270± 100 GPa and graphene oxide: 210± 20 GPa [167]. Despite a lower Young's modulus than other 2D materials, the higher bending stiffness, hydrophilicity, and high negative zeta potential of MXenes make them promising candidates in composites with polymers, oxides, or carbon nanotubes. Experimentally, many polymer-MXene composites have been realized, with enhanced tensile strength, elastic moduli, thermal and electrical conductivities, and electrochemical capacitance, thanks to the presence of 2D MXene [69, 168].

Figure 12. Comparison of the theoretical elastic constants c11 of (a) pristine Tin + 1Xn carbides and nitrides with n = 1, 2, 3, and (b) pristine and functionalized M2CT2 systems with T = O, F, and OH. Reproduced from [104]. © IOP Publishing Ltd. All rights reserved.
Download figure:
Standard image High-resolution image4.5. Electrochemical properties
The development of safe and powerful devices is becoming increasingly important for use in a wide variety of applications including smart electronics, electric, and hybrid cars, and storage of renewable energy [24, 169]. In the field of energy storage, two main families of materials arise. On one hand, the electrical double-layer capacitors (EDLCs) store electrical energy via the formation of an electrical double layer at the electrode/electrolyte interface. Their capacitance is proportional to the electrode's surface area available for the ion adsorption [57]. Batteries, on the other hand, rely on electron transfer to metal centers that is made possible by the intercalation of ions such as Li+ and Na+ [170]. Electrochemical capacitors (including pseudocapacitors and supercapacitors), which occupy a middle ground between EDLCs and batteries, use either ion adsorption or fast surface redox mechanisms that allow them to store much more energy than EDLCs, within a charging time of seconds to minutes, i.e. much faster than batteries do [170].
In this context, 2D MXenes have shown great promise in energy storage applications, which can be explained by their high electronic conductivity, their redox active surface generated during the etching process, and their 2D morphology optimal for fast ion transport and intercalation [57]. About 50% of the publications on MXenes are on energy-related topics and mostly include experimental works. Nevertheless, the success of MXenes in electrodes for batteries and electrochemical capacitors has also been investigated theoretically, considering MXene systems in both their pristine and functionalized forms.
4.5.1. Mono and multivalent ion batteries
The energy stored in the battery is the product of its operating voltage and its electrochemical capacity [171]. The electrochemical capacity, expressed in mAhg−1, basically depends on the amount of charges exchanged between the electrodes and their formula weight:

where nA is the number of adsorbed metal adatoms, ZA is the valence state of the metal adatoms, F is the Faraday constant, Mmaterial is the molar weight of the electrode material, and MA is the molar weight of metal adatoms. In common commercial Li ion batteries (LIBs), the cathode consists of LiCoO2 spinel compound, while the anode is made of graphite (LiC6) which has a limited storage capacity of 330 mAhg−1 (372 mAhg−1, theoretically). The most promising MXenes in battery applications thus correspond to those with suitable anode voltage (0.2−1.0 V) and high gravimetric capacity (> 372 mAhg−1). In the initial report of 2D MXene, a Li capacity of 320 mAhg−1 was predicted for the pristine Ti3C2 system [14]. Soon afterwards, theoretical simulations of Li storage predicted a diffusion barrier of 0.07 eV in pristine Ti3C2 [52], which is much lower than the diffusion barrier in graphite of 0.3 eV [172]. However, in functionalized Ti3C2T2, the presence of −F, −OH, and = O groups blocks the Li transport and the computed diffusion barriers become as large as 0.36 [52], 1.02 [52], and 0.62 eV [173], respectively. Since then, the capacities of several 2D MXenes have been predicted theoretically [54, 174] and are presented in figure 13(a), revealing several trends. First, M2X systems with light transition metal elements exhibit the highest gravimetric capacities, resulting from their low molar weight (equation (5)). The theoretical Li capacity values of pristine M2X (M = Sc, Ti, V, Cr) systems are greater than 400 mAhg−1. The surface terminations can affect the electrochemical properties of MXenes; O terminations being the most favorable in terms of capacity [174]. From equation (5), a method to improve the capacity is by transferring more than one electron per ion. This can be achieved by using multivalent cations (Mg2 + , Ca2 + , Al3 + , etc) instead of monovalent ones (Li+, Na+, K+). The theoretical capacities of multivalent metal ions are shown in figures 13(a) and (b), and clearly outperform the values for carbon-based electrodes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 13. (a) Theoretical gravimetric Li and Mg capacities of various M2CT2 systems in their pristine, O-, and F-terminated forms, and (b) theoretical gravimetric capacities of monovalent (Li, Na, K) and multivalent (Mg, Ca, Al) metal ions in oxygen-terminated MXenes. The data in (a) are obtained from equation (5) considering a full surface coverage (adsorption of the ions), while data in (b) take partial coverage into account, resulting from a systematic study of the ion adsorption energies. Reprinted with permission from [54]. Copyright (2014) American Chemical Society.
Download figure:
Standard image High-resolution image{kind=link}
Experimentally, the predicted trends are not always verified due to the complex nature of ion storage, and the highest Li capacity has been reported for V2CTz systems (280 mAhg−1 at a cycling rate of 1 C and 125 mAhg−1 at 10 C). To get in-depth understanding of the ion storage process, Xie and coworkers [54] computed the ion adsorption energies of Ti2C, V2C, Nb2C, and Ti3C2 MXenes, considering various metallic ions (Li+, Na+, K+, Mg2 + , Ca2 + , and Al3 + ). For pristine systems, the full coverage was favorable for most considered metallic ions (negative adsorption energies), exept for K+ (2/3 surface coverage) and Ca2 + ions (1/2 surface coverage for all systems but Nb2C). The O-terminated MXenes exhibit a favorable full surface coverage with respect to all considered ions, except for Al3 + which shows a 2/3 surface coverage on Ti- and V-based MXenes, while physisorption of Al3 + is even expected for Nb2CO2 (a positive adsorption energy is found at all coverage percentages). This partial coverage of the MXene surface is taken into account to compute the gravimetric capacities presented in figure 13(b) [54].
4.5.2. Electrochemical capacitors
The 2D morphology of MXenes allows for electrochemical intercalation of large organic molecules or metal ions, which can participate in energy storage. To date, the best volumetric capacitance of carbon-based electrodes is around 300 Fcm−3 [175], while volumetric capacitances exceeding 900 Fcm−3 have been reported for free-standing Ti3C2Tz MXene electrodes [24, 55, 57]. MXenes also exhibit excellent cyclability, with no change in capacitance reported after 10,000 cycles for Ti3C2Tz electrodes [24]. Experimentally, several factors can affect the volumetric capacitance of MXenes, including their surface chemistry, hence the synthesis process, and the nature of the electrolyte solution (basic, neutral, or acidic), resulting in different charge-discharge mechanisms. Recent experimental works also pointed out the beneficial effect of divancy ordering in Mo1.33CTz with a volumetric capacitance of ∼1100 Fcm−3, compared to 700 Fcm−3 obtained in Mo2CTz [35]. A capacitance up to 1500 Fcm−3 was even revealed in the ordered (Mo2/3Y1/3)2CTz MXene used as an electrode in a supercapacitor with basic KOH electrolyte [43]. Promising performances as supercapacitors were finally predicted in MXene-based hybrid materials, polymer-MXene composites, and hydrogels, with volumetric capacitances exceeding 1550 Fcm−3 [43, 57, 168, 176–180].
4.6. Other properties and applications
Even though energy storage has been the first and most studied application for MXenes, there are several other applications where MXenes have shown great promise. Anasori et al [19] reported a comprehensive survey with most of the energy- and non-energy-related applications for MXenes investigated so far. In the search for new potential applications, first-principles calculations are adequate tools, usually less expensive and time-demanding than experimental works.
Thin films of Ti3C2Tz are transparent and have a transmission of 97% of visible light per nanometer thickness [25, 66, 181], which is close to the 97.7% transmission of monolayer graphene [182]. Thin films of M2CTz (M = Ti, V) are twice as transparent and, given their high electrical conductivity, they are considered as promising materials for transparent conductive electrodes [68, 183]. Similarly to most of the MXenes properties, their optical behavior is affected upon surface termination [121] and can be tuned by electrochemical ion intercalation [65, 184]. Using DFT calculations, the transmittance, absorption, and reflectivity of a few MXenes have been predicted, revealing their potential use in photocatalytic, optoelectronic, photovoltaic, and transparent conductive electrode devices [185–188].
Another exciting physical property of MXenes is their high electromagnetic interference (EMI) shielding [64]. Thin films of Ti3C2Tz , Mo2TiC2Tz , and Mo2Ti2C3Tz showed EMI shielding capacities higher than graphene and other carbon-based materials with comparable thickness [64, 189]. Combined with their good flexibility and lightweight, MXenes are thus promising 2D materials for EMI electronic devices.
MXenes have also shown suitable affinity with various gasses, including CO2, ethanol, ammonia, etc [190, 191]. In addition, first-principles calculations have predicted efficient physisorption capability of Ti2C with respect to several gas molecules. Consequenly, their potential use as gas sensors has also been pointed out [74].
MXenes performances have been tested in several other applications, such as hydrogen storage media [46–49], sensors and membranes [74–76], biomedical engineering [75, 192–194], thermoelectric materials [60–63], structural composites [69], catalysts [195], electrocatalysts [70, 71], photocatalysts [70, 196], water purificatants [197, 198], organic photovoltaics [199], flexible photovoltaics [200], and many more.
5. Perspectives
Since the discovery of the first MXene less than 10 years ago, remarkable progress has been made in the synthesis process, in the characterization of the structural and physical properties, and in the search for potential applications. Additionally, MXenes have been investigated intensively from first-principles calculations which play an important role in understanding and predicting the MXenes properties. However, several challenges must be overcome, on both experimental and theoretical sides, before promoting their use in next-generation applications.
The experimental challenges can be summarized into three main points: (i) only 20% of the existing MAX phases have been exfoliated, which leaves a lot of space for future experimental works. Besides, many MXenes have been investigated theoretically for which the MAX precursors have not been produced yet. Therefore, the synthesis of new MAX phases is an important research direction to increase the number of available 2D MXenes. Additionally, the mechanism that governs MAX phase etchability still needs to be clarified. In this context, ab initio calculations could help to gain insight into the exfoliation process and find new routes for efficient etching of non-Al elements. (ii) The lack of large defect-free monolayer MXenes prevents the experimental measurement of their intrinsic mechanical response. Indeed, most reports of the mechanical properties of MXenes are theoretical works. Similarly, the electrical, thermoelectric, and magnetic properties have been intensively studied from first-principles approaches and await experimental verification. Given that most of the exotic electrical (semiconductor, topological insulator) and magnetic properties (half-metallicity, large magnetic moment) have been predicted for pristine or homogeneously-terminated systems, an essential challenge consists in the synthesis of MXenes with none or uniform surface terminations. In this context, it is highly desirable to develop new etching routes or post-processing techniques with better control of the surface functionalization. Alternatively, more efforts should be focused on the study of the chemical vapor deposition (CDV) growth of MXenes. This could be further used to combine MXenes with other 2D materials and produce van der Waals heterostructures. (iii) Finally, most experimental works have been conducted on the first MXene, i.e. Ti3C2Tz , while very few other MXenes have been characterized to date. Among others, the freshly discovered o-MXene, i-MXene, and ordered divacancy systems await for experimental characterization of their electronic, magnetic, and transport properties.
Theoretically, understanding the role played by the chemistry and structure on the transport, magnetic, and electrochemical properties is fundamental and would ultimately allow to build a material with the desired properties for a targeted application. In this context, the theoretical approach should be as realistic as possible. To date, an accurate modeling of the experimental conditions is still lacking, since most works are dedicated to pristine or homogeneously-functionalized MXenes, which is not the case experimentally. It is highly desirable to consider heterogeneous terminations in the simulations in order to match better with the real situation and recover the experimental observations. Besides, most theoretical works use DFT calculations, and few AIMD simulations have been reported. In the future, more robust tools should be used or developed with the aim to more accurately simulate the chemical etching process with various etching solutions and to study the transport properties, including electronic band gap, thermoelectric power factor, and thermal transport. In general, the rich chemistry and versatility of MXenes offer a wide area for theoretical works. With the constant discovery of new MAX phases, the amount of potential predictive works continuously increases, and can be extended to the investigation of their 2D counterparts. Notwithstanding, the recent discovery of RE-i-MAX phases with various magnetic ordering, and the possibility to etch them into 2D RE-i-MXenes, could potentially enlarge the palette of intrinsic magnetic 2D crystals. Besides, the influence of doping and strain proposed in several works to tune the electronic and magnetic character of MXenes should be further investigated. More generally, the optical, electrochemical, and thermal properties of all MXenes have not been well described yet. Finally, the stability of various van der Waals heterostructures combining different MXene layers or 2D materials, should be investigated. In this context, the inclusion of vdW interactions in the simulations is required to accurately predict both electronic and elastic properties. With the significant role played nowadays by machine learning in the discovery of new systems and the prediction of their properties, it is reasonable to expect it to be used in the field of MAX and MXenes in the near future.
6. Conclusions
In conclusion, this work not only provides an overview of the electronic, magnetic, vibrational, mechanical, and electrochemical properties of 2D MXene crystals, but also sheds light on some potential applications for MXenes. Since the discovery of the first MXene in 2011, about 30 MXenes have been successfully produced. From the above sections, it has been evidenced how theoretical calculations play an important role to predict the exfoliation of MAX phases into MXenes. Considering the versatility in both elemental composition and surface termination of MXenes, hundreds of them could potentially be formed.
Although most theoretical works are based on conventional DFT calculations, attractive electronic, magnetic, and electrochemical properties have been predicted. In the search for new MXenes, and in the evaluation of their properties, it is of the highest importance to consider stable and energetically-favored configurations. As a simple approach, the dynamic stability can be assessed through the computation of phonon spectra. Similar to MAX phases, most pristine MXenes are electrically conductive. However, upon specific functionalization or considering SOC effects, some MXenes become semiconducting or topologically insulating. However, these predictions still await experimental verification. Calculations of the magnetic properties predict NM, FM, and AFM ground states depending on the intrinsic composition and surface termination. For the magnetic systems, large magnetic moments are predicted which could be retained up to RT, making MXenes good candidates for spintronics devices. In addition, MXene Young's modulus of ∼330 GPa is the highest one reported for a solution-processed 2D system. Theoretically, higher elastic moduli are predicted for MXenes with lighter M elements and fewer layers (n = 1). Such good elastic properties combined with good electrical conductivity and hydrophilicity, give MXenes great promise to find use in composite materials. Additionally, MXene morphology is suitable to intercalate various organic molecules and metal cations, enabling the use of MXenes in energy-storage applications. Although the electrochemical properties have mostly been studied from an experimental point of view, the gravimetric capacitance of various MXenes has been computed. Most MXenes with light M elements exhibit Li-capacitance higher than the one of graphite (LiC6), suggesting their potential use as anode material in LIBs. Gravimetric capacities greater than 700 mAhg−1 have even been predicted for multivalent ions such as Mg2 + , Ca2 + , and Al3 + . Finally, extremely high volumetric capacitances of ∼1500 Fcm−3 have been measured in MXenes, which have consequently been integrated in supercapacitors.
Considering that the first MXene has been produced less than 10 years ago, the amount of information already available in the literature, regarding the synthesis and properties of MXenes, is remarkable. However, proportionally to the amount of predicted MXenes and accessible properties, there is still a large space for future experimental and theoretical works.
Acknowledgments
A C and J-C C acknowledge financial support from the Fédération Wallonie-Bruxelles through the Action de Recherche Concertée (ARC) on 3D nanoarchitecturing of 2D crystals (No. 16/21-077), from the European Union's Horizon 2020 researchers and innovation program (Graphene Flagship Core 1 – No. 696656 and Core 2 – No. 785219), and from the Belgium FNRS. A C and J -C C are also indebted to the Flag-ERA JTC 2017 project entitled 'MORE-MXenes'. Computational resources were provided by the supercomputing facilities of the UCLouvain (CISM) and the Consortium des Equipements de Calcul Intensif en Fédération Wallonie-Bruxelles (CECI) funded by the Fonds de la Recherche Scientifique de Belgique (F R S - FNRS) under convention No. 2.5020.11.