

Abstract
Graphene oxide (GO) is constituted of various oxygen-containing functionalities, primarily epoxides and hydroxyl groups on the basal plane, with a very low amount of carbonyl, quinone, carboxylic acid, phenol, and lactone functions at the edges. The high chemical reactivity of these oxygenated groups makes functionalization difficult to control as different reactions can occur concomitantly. In this study we have investigated the reactivity of GO towards orthogonal reactions to selectively functionalize the hydroxyl groups, which are present in a high amount. We explored both the esterification and the Williamson reaction. Our strategies present the main advantage to occur in mild conditions, thus preserving the intrinsic properties of GO, whereas most reactions reported in literature require relatively harsh conditions, leading to (partial) reduction, and/or are not chemoselective. We have also extended our study to the ketones and examined their derivatization by the Wittig reaction. This work has allowed developing two facile methods for the covalent derivatization of the hydroxyl groups in mild conditions, while GO was not reactive towards the Wittig reaction, probably due to the low amount of ketones. Overall, this work leads to a better understanding of the reactivity of GO for controlled derivatization. This opens promising perspectives for multi-functionalization of GO in order to design graphene-based nanomaterials endowed of multiple properties.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Graphene-based nanomaterials have been raising a great interest within the scientific community due to a wide range of unique outstanding physical and electrical properties [1]. Graphene and its derivatives can be used in myriad applications ranging from nanoelectronics [2, 3], composite materials [4, 5], energy conversion [6, 7], and medicine [8, 9]. In the past decade the oxidized form of graphene, graphene oxide (GO), has been one of the most intensely studied nanomaterials owing to its 2D structure, water dispersibility, low cost, and possibility to be reduced to graphene [10]. Chemical functionalization of the graphene surface allows to modulate its physicochemical properties and interfacial characteristics for designing advanced functional graphene-based materials [11–14]. The surface of GO is composed on both sides of a variety of oxygen-containing functionalities. GO contains primarily epoxides and hydroxyl groups on the basal plane, and a relatively low number of carbonyls, quinones, carboxylic acids, phenols, and lactones at the edges [15–17]. The presence of many oxygenated functional groups associated to their high chemical reactivity renders functionalization difficult to control. Indeed, multiple reactions of these functions can occur simultaneously. In this context, it is highly important to design well-defined and thoroughly characterized graphene-based conjugates for any type of applications, in particular in nanomedicine [18]. In a previous work, we investigated the reactivity of GO towards amine derivatives [19]. We demonstrated that treating GO in conditions used to perform amidation leads instead to epoxide opening because of the high reactivity of epoxy groups for nucleophiles and the very low amount of carboxylic acids, as assessed by solid-state NMR spectroscopy. But, the majority of the studies reported in literature omits the epoxide opening, resulting in false and/or incomplete structure of functionalized GO. Still, a detailed study investigating the reactivity of GO towards orthogonal reactions to selectively functionalize one oxygenated functional group over another is necessary and will open promising perspectives for multi-functionalization of GO to design graphene-based nanomaterials with multiple properties [20, 21]. Apart from reactions involving amine derivatives and epoxides, GO has been derivatized through the hydroxyl groups in different conditions, most of them being non-selective and/or requiring relatively harsh conditions that can affect the structure of GO. Derivatization with isocyanate derivatives has been one of the first method used to obtain functionalized GO [22]. The treatment of graphite oxide with isocyanates results in the functionalization of both the hydroxyls and carboxylic acids via the formation of carbamate esters and amides, respectively, with concomitant exfoliation of the graphene sheets. Silanization is a strategy that has been investigated by many research groups [23–25]. This reaction involves the hydrolysis of silane derivatives, followed by dehydration condensation with the hydroxyl and epoxy groups on GO, resulting in the formation of a siloxane bond. GO has also been derivatized by nucleophilic addition of n-butyllithium and subsequent etherification and esterification with 2-ethylhexyl bromide [26]. In this case, GO is concurrently alkylated due to the addition of n-BuLi and 2-ethylhexyl moiety on the basal plane of GO. All these strategies are not selective for the derivatization of the hydroxyl groups and involve other oxygenated moieties and/or bonds. Only a few approaches are selective for the derivatization of the hydroxyls. For instance, Claisen rearrangement has been carried out on graphite oxide and GO by exploiting the allylic character of the hydroxyl groups. But, this strategy requires strong conditions as it is performed at temperatures between 60 and 150 °C, resulting in the reduction of GO [27, 28]. Alternatively, the hydroxyl functions of graphite oxide have been reacted with the carboxyl moieties of a fullerenoacetic acid derivative at 90 °C by Fisher esterification [29] or with methacrylic anhydride [30]. Heat treatment of GO induces reduction to different extents depending on temperature [31]. Therefore, performing reactions in mild conditions is required to preserve the structure and chemical composition of GO. There is thus a strong need to develop efficient methods to derivatize the hydroxyl groups, which are present in a high amount on GO, by chemoselective reactions. Hence, in this work we have explored esterification of the hydroxyl functions of GO and investigated the reactivity of the OH groups through etherification by the Williamson reaction. Regarding esterification reactions, most approaches involve the COOH groups of GO [32, 33]. But, as we demonstrated in our previous work, the carboxylic acids are present in a small amount, contrary to hydroxyl groups, thus leading to low levels of functionalization [19]. We have also extended our study to the ketones and examined their derivatization by the Wittig reaction. To the best of our knowledge, the reactivity of the ketones of GO has not been explored yet. This study has allowed developing two facile methods for the covalent derivatization of the hydroxyl groups in mild conditions, thus preserving the intrinsic properties of GO. The modified GO samples have been characterized by x-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), magic-angle spinning (MAS) solid-state NMR, and transmission electron microscopy (TEM). Overall, this work leads to a better understanding of the reactivity of GO for controlled derivatization.
Methods
Materials and characterization
GO samples were provided by NanoInnova (Spain) (batches: NIT.GO.Z.10.4, NIT.GO.R.10.1, and NIT.GO.M.10.200) and Grupo Antolin (Spain). The chemicals and solvents were obtained from commercial suppliers and used without purification. The solvents used for synthesis were analytical grade. Water was purified using a Millipore filter system MilliQ®. When stated, suspensions were sonicated in a water bath (20 W, 40 kHz). For dialysis MWCO 12 000–14 000 Da membranes were purchased from Spectrum Laboratories, Inc. XPS was performed on a Thermo Scientific K-Alpha x-ray photoelectron spectrometer with a basic chamber pressure of 10−8–10−9 bar and an Al anode as the x-ray source (1486 eV). More information is given in supplementary information (stacks.iop.org/TDM/5/035037/mmedia). TGA was performed on a TGA1 (Mettler Toledo) apparatus from 30 to 900 °C with a ramp of 10 °C·min−1 under N2 using a flow rate of 50 ml·min−1 and platinum pans. MAS NMR experiments were performed on an AVANCE 750 MHz wide bore spectrometer (Bruker™) operating at a frequency of 188.5 MHz for 13C NMR. More information is given in Supplementary Information. The syntheses of tert-butyl (4-iodobutyl)carbamate 1, Boc-protected 6-aminocaproic acid 4, and (4-((tert-butoxycarbonyl)amino)butyl)triphenylphosphonium iodide 7 are described in supplementary information. In case the filtration of the GO suspensions was too long, 0.45 µm pore size polytetrafluoroethylene (PTFE) Millipore® membranes were used.
Protocols for the derivatization of GO
Williamson reaction
To a suspension of GO (20 mg) in dry DMF (20 ml), sonicated in a water bath for 10 min, were added K2CO3 (400 mg, 2.89 mmol) and tert-butyl (4-iodobutyl)carbamate 1 (237 mg, 0.79 mmol). The reaction mixture was stirred under argon atmosphere for 2 d and then filtered over a PTFE Millipore® membrane with 0.1 µm pore size. The solid recovered on the filter was dispersed in water, sonicated in a water bath until thorough dispersion of GO, and filtered again over a PTFE membrane. This sequence was repeated twice with water, DMF, methanol, and dichloromethane. The resulting solid was dispersed in water using sonication and the suspension was dialyzed in water. Freeze drying of the suspension allowed obtaining GO 2.
Boc deprotection
To a suspension of GO 2 (10 mg) in 1,4-dioxane (5 ml), sonicated in a water bath for 10 min, was added a solution of 4 m HCl in 1,4-dioxane (5 ml). The reaction mixture was stirred overnight. After filtration (0.1 µm PTFE Millipore® membrane) the solid was dispersed in DMF, sonicated in a water bath for a few minutes, and filtered again. This filtration sequence was repeated twice with DMF, methanol, and dichloromethane. The solid was dried under vacuum to obtain GO 3.
Control reactions
Control reactions were performed in the same conditions as GO 2, but without adding either K2CO3 (named GO-CONT-I 2) or tert-butyl (4-iodobutyl)carbamate 1 (named GO-CONT-II 2). Both samples were further treated in acidic conditions following the protocol used for Boc deprotection, leading to GO-CONT-I 3 and GO-CONT-II 3.
Esterification
To a suspension of GO (10 mg) in dry DMF (10 ml), sonicated in a water bath for 10 min, were added 6-((tert-butoxycarbonyl)amino)hexanoic acid 4 (37 mg, 0.16 mmol), N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide (EDC) (15 mg, 0.1 mmol) and 4-dimethylaminopyridine (DMAP) (6 mg, 0.05 mmol). The reaction mixture was stirred for 2 d and then filtered over a PTFE 0.1 µm membrane. The solid recovered on the filter was dispersed in DMF, sonicated in a water bath for a few minutes, and filtered again over a PTFE membrane. This sequence was repeated twice with DMF, methanol, and dichloromethane. The resulting solid was dispersed in water using sonication and the resulting suspension was dialyzed in water. Freeze drying of the suspension allowed giving GO 5. GO 6 was obtained in the conditions used to remove the Boc group.
Control reaction
The control reaction was performed in the same conditions as GO 5, but without adding the coupling agents (EDC and DMAP). The sample was further treated in acidic conditions following the protocol used for Boc deprotection, leading to GO-CONT 6.
Wittig reaction
Potassium bis(trimethylsilyl)amide (KHMDS) 0.7 M in THF (50 µl, 0.032 mmol) was added to a solution of (4-((tert-butoxycarbonyl)amino)butyl)triphenylphosphonium iodide 7 (20 mg, 0.036 mmol) in dry THF (2.5 ml) under argon atmosphere at 0 °C. The reaction mixture was stirred for 30 min at room temperature. Then, a suspension of GO (10 mg) in dry THF (2.5 ml) under argon, previously sonicated in a water bath for 1 min, was added to the solution of 7 and KHMDS cooled at 0 °C. The reaction mixture was sonicated for 10 min at 0 °C and then stirred at 0 °C. The temperature was allowed to slowly reach room temperature. After 24 h, the reaction mixture was diluted with 6 ml of H2O/methanol mixture (1:5) and then filtered over a PTFE Millipore® membrane with 0.1 µm pore size. The solid recovered on the filter was dispersed in DMF, sonicated in a water bath until thorough dispersion of GO, and filtered again over a PTFE membrane. This sequence was repeated twice with DMF, methanol, and dichloromethane. The resulting solid was dispersed in water using sonication and the resulting suspension was dialyzed in water. Freeze drying of the suspension allowed obtaining GO 8. GO 9 was obtained in the conditions used to remove the Boc group.
Control reaction
The control reaction was performed in the same conditions as GO 8, without adding (4-((tert-butoxycarbonyl)amino)butyl)triphenylphosphonium iodide 7. The sample was further treated in acidic conditions following the protocol used for Boc deprotection, leading to GO-CONT 9.
Results and discussion
Williamson reaction
We first investigated the selective derivatization of the hydroxyl groups by the Williamson reaction. As GO is a generic term to name various types of oxidized graphite products, we aimed to compare the chemical reactivity of two commercially available GO samples from NanoInnova (named GO-N) and Grupo Antolin companies (named GO-A). GO-N has been synthesized by the Hummers' method, while GO-A has been prepared by a different process from rolled carbon nanofibers that are cut and exfoliated. The amount of the different types of oxygenated groups vary between both GO samples. In addition, GO-N contains aggregated sheets and is ~20 nm thick, whereas GO-A is mainly constituted of monolayers, along with some irregular particles of 3–10 nm thickness [34]. It is noteworthy that thickness may induce differences in term of chemical reactivity.
The Williamson reaction allows the formation of an ether bond from an organohalide and a deprotonated alcohol (alkoxide ion) via a substitution reaction. Discovered in 1850 and widely used in both academy and industry, it remains the simplest and most commonly used strategy to form ethers from free hydroxyl groups [35, 36]. In this work, the Williamson ether synthesis was done in the presence of an iodinated derivative (compound 1) and potassium carbonate in DMF (scheme 1). As the Williamson reaction is often performed upon heating, the reaction was initially carried out at 80 °C on GO-N yielding functionalized GO (f-GO)-N-80 2 [36]. Compound 1 was prepared from 4-amino-1-butanol in two steps (see supplementary information for details). The Boc protecting group on f-GO-N-80 2 was removed using a solution of HCl in 1,4-dioxane, leading to f-GO-N-80 3. The amount of amino groups introduced on GO was assessed by UV/Vis spectroscopy using the Kaiser test, which is a colorimetric test commonly used in peptide synthesis and applied to carbon-based nanomaterials to assess the quantity of amine functions [37, 38]. The amount of  resulted 70 µmol per gram of f-GO-N-80 3. The presence of amine functions on GO allows further derivatization with molecules of interest [39]. In parallel, we performed a two-step control experiment without adding potassium carbonate in the first step to assess if compound 1 has an affinity for the GO surface and adsorbs by hydrophobic and electrostatic interactions (this sample is named GO-N-80-CONT-I 3). The Kaiser test performed after heating GO-N and 1 in DMF at 80 °C and subsequent HCl treatment was negative. This result is a proof that the positive Kaiser test loading obtained in the presence of the base is due to covalent grafting and not to unspecific non-covalent interactions of compound 1 with GO.
resulted 70 µmol per gram of f-GO-N-80 3. The presence of amine functions on GO allows further derivatization with molecules of interest [39]. In parallel, we performed a two-step control experiment without adding potassium carbonate in the first step to assess if compound 1 has an affinity for the GO surface and adsorbs by hydrophobic and electrostatic interactions (this sample is named GO-N-80-CONT-I 3). The Kaiser test performed after heating GO-N and 1 in DMF at 80 °C and subsequent HCl treatment was negative. This result is a proof that the positive Kaiser test loading obtained in the presence of the base is due to covalent grafting and not to unspecific non-covalent interactions of compound 1 with GO.

Scheme 1. Derivatization of GO by the Williamson reaction. For the sake of clarity, only one hydroxyl group is derivatized.
Download figure:
Standard image High-resolution imageThe f-GO-N-80 3 sample was characterized by XPS and TGA. According to XPS, starting GO-N is composed of 69.2% C and 30.8% O (figure S1(a)). After derivatization using the Williamson reaction, a peak corresponding to the N1s envelope appeared at a value close to 399.8 eV, confirming the incorporation of nitrogen (figure S1(a)) [40]. TGA allowed comparing the thermal profile of GO before and after functionalization. The experiments were performed under inert atmosphere at a heating rate of 10 °C min−1. As already reported in the literature, it is complicated to interpret the TGA data as GO is thermally unstable [41, 42]. In fact, GO displays a thermogram with three main weight loss slopes (figure S1(b)) [43]. The weight loss below 100 °C is due to desorption of water and some unstable oxygenated functionalities. The weight loss at around 200 °C is attributed to the decomposition of other labile oxygen-containing groups. Finally, the small and continuous weight loss above 250 °C can be ascribed to the removal of more stable oxygenated moieties. The thermogravimetric profile of f-GO-N-80 3 was different in comparison to GO-N, displaying higher thermal stability. For better comparison, we prepared a second control sample by treating GO in the same conditions (two steps), but without adding compound 1 in the first step to assess if the base (potassium carbonate) could affect the structure of GO (this sample is named GO-N-80-CONT-II 3 in figure S1(b)). In fact, the control treated GO also showed higher thermal stability. It seems that the treatment at 80 °C used to perform the reaction induced the removal of some labile functions [44]. Thus, in the temperature range from 250 to 900 °C, the higher weight loss for f-GO-N-80 3 compared to the control treated GO can be attributed to the pyrolysis of the functional groups attached to the surface of GO by the Williamson reaction, confirming that the reaction occurred.
The conventional Williamson reaction is generally conducted at 50 °C–100 °C [36]. But, at this temperature GO is partially reduced [31, 44], as we confirmed by TGA. Indeed, the C/O ratio calculated by XPS increases from 2.2 for starting GO-N to 3.5 after derivatization. Therefore, we tried to optimize the protocol for the Williamson reaction in order to avoid heating and hence prevent the reduction of GO. The reaction was performed at different temperatures, namely at 60 °C, 40 °C, and room temperature (corresponding sample names are f-GO-N-60 3, f-GO-N-40 3, and f-GO-N-RT 3). The reaction at room temperature was also conducted using cesium carbonate as alternative base to K2CO3 (sample name: f-GO-N-RT-Cs 3), as Cs2CO3 has been used for the O-alkylation of phenols in mild conditions [45]. Furthermore, cesium base-promoted phenol alkylation has been extensively applied [46]. The level of functionalization for each condition reaction was assessed by the Kaiser test (table 1). We can observe that the amine loading increases when the temperature decreases. The highest loading was obtained by performing the reaction at room temperature using potassium carbonate (f-GO-N-RT 3). Regarding the percentage of nitrogen obtained by XPS, the tendency is inverted. Indeed, the %N decreases upon reducing the temperature, from 1.77% at 80 °C to 0.65% at room temperature. These decreasing values can be explained by the fact that there is a loss of CO and CO2 from GO upon increasing temperature. As a consequence, besides the introduction of nitrogen there is also the elimination of carbon and oxygen atoms at high temperature [43]. This is the reason why the Kaiser test is more appropriate to determine the level of functionalization.
Table 1. Amine loading assessed by the Kaiser test and %N obtained by XPS for the Williamson reaction performed in different conditions.
| Name | Temperature | Base | Amine loading (µmol g−1) | %N (XPS) |
|---|---|---|---|---|
| GO-N | — | — | — | — |
| f-GO-N-80 3 | 80 °C | K2CO3 | 69 | 1.77 |
| f-GO-N-60 3 | 60 °C | K2CO3 | 92 | 1.02 |
| f-GO-N-40 3 | 40 °C | K2CO3 | 123 | 0.87 |
| f-GO-N-RT 3 | RT | K2CO3 | 132 | 0.65 |
| f-GO-N-RT-Cs 3 | RT | Cs2CO3 | 106 | 0.68 |
The different samples of GO-N functionalized by the Williamson reaction were characterized by high resolution XPS and TGA (figure 1). Both techniques confirmed the partial reduction of GO upon heating. Indeed, there is a temperature-dependent reduction of intensity of the XPS peak at ~287 eV, attributed to the C–O bond, compared to the peak at ~285 eV corresponding to the C–C bond (figure 1(a)). Furthermore, GO that reacted at 60 °C and especially at 80 °C shows a higher thermal stability according to TGA, which is consistent with partial reduction (figure 1(b)). Even though cesium was demonstrated to have a high sorption capacity onto GO [47], no residual cesium was detected in f-GO-N-RT-Cs 3 by XPS. However, the use of cesium carbonate did not allow reaching a higher functionalization degree compared to potassium carbonate.
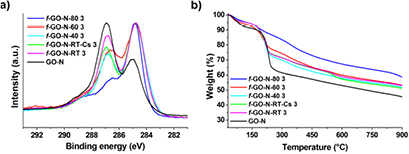
Figure 1. (a) C1s high resolution XPS and (b) TGA of GO-N before and after functionalization by the Williamson reaction in different conditions.
Download figure:
Standard image High-resolution imageTwo control reactions were performed in the optimized conditions, namely by treating GO in DMF at room temperature in the presence of compound 1 (GO-N-CONT-I 3) or K2CO3 (GO-N-CONT-II 3) to verify that 1 does not adsorb on the surface of GO and that the base does not affect GO, respectively. Kaiser test of GO-N-CONT-I 3 resulted negative, confirming that compound 1 has not been immobilized on GO at room temperature via non-covalent interactions that would have led to misleading conclusions. This result was confirmed by XPS as no nitrogen was detected for GO-N-CONT-I 3 (data not shown). We also analyzed the other control sample (GO-N-CONT-II 3) by XPS and we noticed a change in the relative intensity of the two peaks in the C1s spectrum (figure S2). Indeed, the C–O bond peak at ~287 eV is higher than the C–C peak at ~285 eV in GO-N and GO-N-CONT-I 3, while it has a similar intensity in the case of GO-N-CONT-II 3. Nevertheless, we found that there was no significant reduction of GO by K2CO3 as %O was similar for GO-N-CONT-II 3 compared to starting GO-N and GO-N-CONT-I 3 (29.2%, 30.8%, and 30.5%, respectively). We think the relative increase of the C–C XPS peak could be explained by the metastability of GO resulting in the loss of CO and CO2 [48–50].
The control samples are also necessary to interpret the TGA data of functionalized GO samples [19]. Indeed, TGA showed that both control samples display a slightly higher thermal stability compared to the starting GO, due to the loss of some labile oxygenated functions that occurs even at room temperature, as we previously observed in the study of the epoxide opening on GO (figure 2(a)) [19]. Nevertheless, the weight loss difference is much less important in this case in comparison to the control sample treated at 80 °C (figure S1(b)). By comparing the TGA of both control GO samples with the thermal profile of f-GO-N-RT 3, it was not possible to confirm the positive outcome of the Williamson reaction. This result emphasizes however the difficulty to interpret the TGA data due to the thermal instability of GO and the low molecular weight of the functionalities introduced on GO by the Williamson reaction.
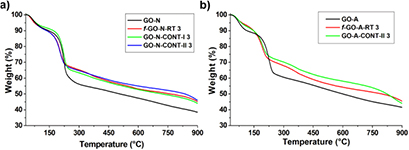
Figure 2. (a) TGA of GO, f-GO-RT 3 and control samples for GO from NanoInnova and (b) GO from Grupo Antolin.
Download figure:
Standard image High-resolution imageThe optimized conditions were used then to functionalize GO-A leading to f-GO-A-RT 3. In this case, a clearer difference was observed by comparing the TGA curve of the functionalized GO and the control sample GO-A-CONT-II 3 (figure 2(b)). Indeed, the weight loss was higher after the Williamson reaction in comparison with the control sample. It seems that the level of functionalization is more important in the case of GO-A compared to GO-N. This result is in agreement with both the Kaiser test and XPS (figure 3), showing a higher loading for f-GO-A-RT 3 (190 versus 132 µmol g−1 for f-GO-N-RT 3) and a superior %N (1.25% versus 0.65% for f-GO-N-RT 3), respectively. Moreover, the peak attributed to the C–N bond was identified in the carbon high resolution spectra, confirming the introduction of the amino chain on GO (figure 3, right).
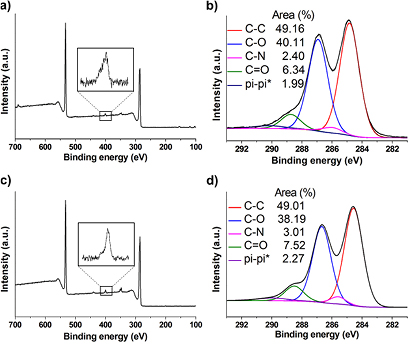
Figure 3. (a) and (b) XPS survey (left) and high resolution (right) C1s spectra of f-GO-RT 3 for GO from NanoInnova and (c) and (d) GO from Grupo Antolin.
Download figure:
Standard image High-resolution imageTEM was also used to characterize the different starting GO and f-GO 3 (figure S3). Both GO-N and GO-A samples display a lateral dimension of a few micrometers and have wrinkled sheets with folded edges. The morphology of the GO sheets seems to be unaffected by the conditions used for the Williamson reaction and the subsequent acidic treatment to remove the protecting group.
Taken together, these data demonstrate that the hydroxyls of GO can be functionalized by the Williamson reaction in mild conditions. The characterization of f-GO 3 by TGA, Kaiser test, and XPS indicates a slightly higher level of functionalization of GO-A compared to GO-N.
Esterification
We then explored the reactivity of GO towards esterification of the hydroxyl groups. The esterification reaction was performed on both GO samples at room temperature using Boc-protected aminocaproic acid 4 in the presence of EDC and DMAP as coupling agents, leading to f-GO 5 (scheme 2). Boc cleavage was performed using HCl in 1,4 dioxane, leading to f-GO 6. The amine loading assessed by the Kaiser test resulted 165 and 136 µmol g−1 for f-GO-N 6 and f-GO-A 6, respectively. A control reaction was performed without adding the coupling agents to verify if compound 4 has a tendency to adsorb onto GO (GO-CONT 6).
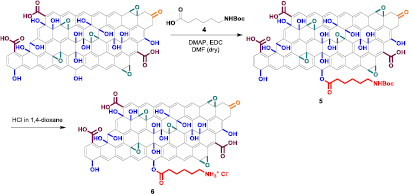
Scheme 2. Derivatization of GO by esterification. For the sake of clarity, only one hydroxyl group is derivatized.
Download figure:
Standard image High-resolution imageAs already observed before, the control reaction performed at room temperature shows a higher thermal stability compared to the starting GO, most probably due to the elimination of some labile functional groups from GO, even at room temperature (figure S4). The thermogravimetric curves of both f-GO-N 6 and f-GO-A 6 are rather similar to their respective control samples, probably due to the low molecular weight of carboxylic derivative 4, making the TGA data inconclusive.
XPS confirmed the introduction of nitrogen on GO (2.0 and 2.4% for GO-N and GO-A, respectively), while no nitrogen was detected for the control samples, further confirming the covalent functionalization of GO (figures 4 and S5). In addition, in the carbon high resolution spectra we could identify the signal of the C–N bond at ~286 eV for both f-GO 6 samples.

Figure 4. (a) and (b) XPS survey (left) and high resolution (right) C1s spectra of f-GO 6 for GO from NanoInnova and (c) and (d) GO from Grupo Antolin.
Download figure:
Standard image High-resolution imageTEM showed that the esterification conditions and the acidic treatment did not alter the morphology of the GO sheets (figure S6).
In the case of these esterified GO samples we decided to extend the characterization using solid-state NMR. Both f-GO 6 samples were analyzed by 13C MAS solid-state NMR and compared to respective starting GO to confirm the covalent introduction of derivative 4 by esterification. There are three main peaks in the 13C multiple contact-cross polarization (MC-CP)/MAS NMR of starting GO-N at ~60, 70, and 130 ppm that are attributed to the epoxides, C–OH, and C=C bonds, respectively (figure 5(a), red spectrum) [19]. In the case of starting GO-A, the peak at ~130 ppm is less intense (figure 5(b), red spectrum).

Figure 5. (a) 13C MC-CP/MAS NMR spectra of GO (red) and f-GO 6 (black) for GO from NanoInnova and (b) GO from Grupo Antolin.
Download figure:
Standard image High-resolution imageWe also performed a quantitative 13C direct polarization (DP)/MAS NMR analysis and observed that this peak is as intense as in the case of GO-N (figure S7, red spectra; table S1). This result could be explained by the fact that the distribution of protonated carbon atoms is different in both GO samples. Indeed, in the case of GO-N, the protonated carbon atoms seem to be well distributed on the graphene basal plane. Hence, the protons are capable of transferring magnetization to the carbons in close proximity. On the contrary, in the case of GO-A, there would be fewer protonated carbons, maybe in the form of clusters. As a consequence, only the protons at the cluster border can transfer magnetization to the adjacent carbons, thus explaining the low intense peak at ~130 ppm in the case of GO-A in comparison to GO-N.
After esterification new peaks were identified at ~25 and 40 ppm that can be assigned to the methylene groups of compound 4 (figure 5, black spectra). An additional peak with lower intensity also appeared at ~160 ppm in the case of GO-A and it was assigned to the ester function (figure 5(b)) [51]. In the case of GO-N this peak was not visible, probably due to its very low intensity (figure 5(a)). The peaks of the C–O bonds at ~60 and 70 ppm corresponding to epoxides and hydroxyls are unchanged, which confirms that the mild conditions did not affect the chemical composition of GO. In the quantitative 13C DP/MAS NMR spectra the peaks assigned to the methylene groups of derivative 4 are visible and intense, confirming the successful derivatization of GO (figure S7). Nevertheless, as the spectra are noisy, it is not possible to distinguish the peak attributed to the ester bond in the quantitative 13C DP/MAS NMR spectra.
Overall, the hydroxyl groups of GO were functionalized by the Williamson reaction and by esterification. The reactivity was rather similar for the two types of GO used in this study and for both reactions, thus highlighting the efficiency of the two derivatization strategies.
Wittig reaction
We also examined the reactivity of the ketones towards the Wittig reaction. For this purpose, GO was reacted with (4-((tert-butoxycarbonyl)amino)butyl)triphenylphosphonium iodide 7 and potassium bis(trimethylsilyl)amide (KHMDS) as a base to form the ylide in situ, leading to the formation of f-GO 8 (scheme 3). KHMDS was employed because of its steric hindrance to avoid undesired side reactions between the base and the oxygenated groups of GO. The Boc group was removed using HCl in 1,4-dioxane, resulting in the formation of f-GO 9. A control reaction was performed without derivative 7 to verify that the base did not provoke changes in the structure of GO (GO-CONT 9).

Scheme 3. Derivatization of GO by the Wittig reaction. For the sake of clarity, only one ketone is derivatized.
Download figure:
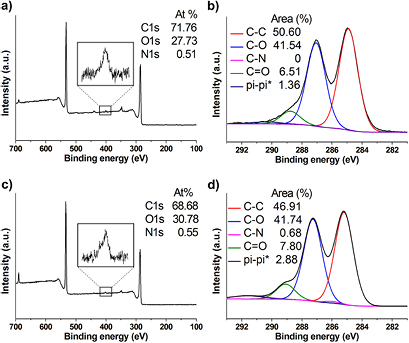
Standard image High-resolution imageThe amine loading calculated by the Kaiser test after Boc deprotection was 22 and 26 µmol g−1 for f-GO-N 9 and f-GO-A 9, respectively. These loading values are indicative of a negligible amount of ketones on the surface of both GO samples and/or a low reactivity towards the Wittig reaction. Based on MAS solid-state NMR and the negligible amount of carbonyls (figures 5 and S7), the first hypothesis seems to be the most probable. The low level of functionalization was confirmed by XPS and TGA. Indeed, ~0.5% of nitrogen was detected by XPS for both f-GO 9 (figure 6), while no nitrogen was found for the control samples (figure S8). Moreover, no peak (or a very low peak) corresponding to the C–N bond could be identified upon deconvolution of the carbon high resolution spectra.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. (a) and (b) XPS survey and high resolution C1s spectra of f-GO 9 for GO from NanoInnova and (c) and (d) GO from Grupo Antolin.
Download figure:
Standard image High-resolution image{kind=link}
The samples were finally characterized by TGA (figure S9). GO-CONT 9 samples have similar thermal profiles compared to previous control samples GO-CONT-I 3, GO-CONT-II 3, and GO-CONT 6. This similarity confirms that KHMDS does not reduce GO, as also confirmed by XPS (figure S8). The curve of f-GO-N 9 displays a slightly higher weight loss compared to the control sample (figure S9(a)), whereas f-GO-A 9 and GO-A-CONT 9 have a comparable thermal profile (figure S9(b)). But, as already mentioned before, TGA is not the appropriate characterization technique to assess the level of functionalization of GO derivatized with low molecular weight molecules. Overall, based on characterization by Kaiser test, XPS, and TGA, we can conclude that the derivatization of GO in conditions to perform the Wittig reaction was not successful. This is certainly due to the absence or to a negligible amount of ketones.
Conclusions
In summary, we have explored different chemical approaches to selectively functionalize the hydroxyls and ketones of GO from two commercial sources. The hydroxyl groups were derivatized by the Williamson reaction with an amino-terminated linker, resulting in the formation of ether bonds. The presence of a primary amine allows envisaging further derivatization of GO with molecules of interest. The reaction takes place in mild conditions at room temperature, thus avoiding reduction of GO, while most strategies reported in literature for the derivatization of hydroxyl groups are performed under heating. We also successfully derivatized the hydroxyls by esterification with aminocaproic acid at room temperature. The presence of the ester bond was confirmed by MAS solid-state NMR. Overall, the reactivity of the hydroxyls for both the esterification and the Williamson reaction was rather similar. Finally, we investigated the reactivity of the ketones towards the Wittig reaction. But, the reaction was unsuccessful, presumably due to the absence of a significant amount of ketones on GO.
This study extends the possibility to selectively derivatize the hydroxyl groups, which are present in a high amount on GO. The mild conditions allow preserving the structure of GO and avoiding reduction. This work enlarges the knowledge on the chemistry of GO, thereby enabling to better understand and control the reactivity of GO and ultimately its properties.
Acknowledgments
This work was supported by the Centre National de la Recherche Scientifique (CNRS) and by the International Center for Frontier Research in Chemistry (icFRC). The authors gratefully acknowledge financial support from EU H2020-Adhoc-2014-20 GrapheneCore1 (no. 696656) and EU H2020-MSCA-RISE-2016 project CARBO-IMmap (no 734381), and from the Agence Nationale de la Recherche (ANR) through the LabEx project Chemistry of Complex Systems (ANR-10-LABX-0026_CSC). Ngoc Do Quyen Chau and Fanny Richard are gratefully acknowledged for help with characterization by XPS. The authors wish to thank Cathy Royer and Valérie Demais for help with TEM analyses at the 'Plateforme Imagerie in vitro' at the Center of Neurochemistry (Strasbourg, France). Bhimareddy Dinesh and Rajendra Kurapati are gratefully acknowledged for fruitful discussion.
Supplementary material description
Materials and methods. Experimental procedures for the synthesis of tert-butyl (4-iodobutyl)carbamate 1, 6-((tert-butoxycarbonyl)amino)hexanoic acid 4, and iodine salt of tert-butyl (4-(triphenyl-λ4-phosphanyl)butyl)carbamate 7. TEM images of GO and f-GO. Complementary XPS, TGA, and MAS solid-state NMR data.