
Abstract
Neutrophils are the most abundant type of leukocytes in the blood, traditionally regarded as the first immune responders to infections and inflammations. In the context of tumors, neutrophils have been shown to possess both tumor-promoting and tumor-limiting properties. A better understanding of the inter-cellular dynamics between the neutrophils and aggregated tumors could possibly shed light on the different modalities of neutrophil involvement in tumor progression. To study in-vitro the interactional dynamics of neutrophils and growing tumor aggregates, in this work, we engineered a novel, microfluidics-integrated, three-dimensional (3D) tumor-immune microenvironment (TIME)-on-Chip device, and we investigated the effect of neutrophils on the inception of collective 3D invasion of ovarian tumor cells. Herein, tumor spheroids generated and cultured on hydrogel based multi-microwell plates, and embedded within collagen matrix of defined thickness, were magnetically hybrid-integrated with a 3D bioprinting enabled microfluidic system fabricated on a porous membrane and carrying neutrophils. This setting recreated a typical TIME in-vitro to model dynamic neutrophil migration and 3D tumor invasion. Using this device, we observed that neutrophils respond to the growing tumor spheroids through both chemotaxis and generation of neutrophil extracellular traps (NETs). The formation of NETs stimulated the reciprocation of tumor cells from their aggregated state to collectively invade into the surrounding collagen matrix, in a manner more significant compared to their response to known tumor-derived stimulants such as transforming growth factor and Interleukin- 8. This effect was reversed by drug-induced inhibition of NETs formation, suggesting that induction of NETs by cancer cells could be a pro-migratory tumor behavior. Further, we additionally report a previously unidentified, location-dictated mechanism of NETosis, in which NETs formation within the stromal extracellular collagen matrix around the spheroids, and not tumor-contacted NETs, is important for the induction of collective invasion of the ovarian tumor cells, thus providing a rationale for new anti-tumor therapeutics research.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Tumor invasion into the surrounding stroma is a critical step in the process of metastasis, and it is necessary to clearly identify the role of the different microenvironmental factors involved in the regulation of tumor invasion [1]. The tumor-immune microenvironment (TIME) refers to the different subpopulations of the immune system and their interactions within a tumor microenvironmental niche that have a unique influence on the tumor initiation, development and response to therapies [2, 3]. Neutrophils constitute a significant portion of the immune cells present within a TIME [4–8], and their high numbers have, in general, been associated with poor prognosis and drug resistance in patients [9–11]. Conversely, chemotherapy induced neutropenia has also predicted improved survival rates among cancer patients [12–17], thereby raising important questions on the impact of neutrophils in tumor development. In the context of tumor metastasis, research has produced conflicting data in explaining whether neutrophils could promote the metastatic cascade [18–22], or support immune reactions inhibiting tumor growth [23–26]. A fuller understanding of the multifaceted neutrophil functionalities in tumorigenesis is still evolving [27], but there is a steadily growing appreciation that neutrophils can provide critical insights that are essential to tailor suitable anti-tumor and immunotherapeutic strategies, complementary to current therapeutic approaches [4, 28–30]. In this work, we aim to engineer a biomimetic TIME platform, and conduct in-vitro studies of the interactional dynamics between neutrophils and pre-metastatic tumor clusters, in a pursuit of identifying prospective neutrophil-based targets that could mediate tumor invasion.
Of particular interest, is the ability of neutrophils to generate neutrophil extracellular traps (NETs) in response to infectious stimuli [30–32]. Through a sequence of processes collectively termed as NETosis, activated neutrophils release NETs, comprising of decondensed chromatin, histones and DNA, into their surrounding extracellular matrix (ECM), forming three-dimensional (3D) protein web structures with associated cytotoxic enzymes. While NETs can serve as a host defense mechanism to sequester circulating tumor cells [20, 33], recently, through increasing evidence, NETs have been directly associated with the initiation and induction of tumor invasion, aiding metastasis [20, 33–36].
In a classical tumor metastasis model [37–39], the tumor disseminates as single cells from the primary site by invading into the ECM, and enters the bloodstream as circulating tumor cells before colonizing onto distant tissues and organs. However, emerging studies have also reported on a collective tumor migration mechanism [40–43], wherein tumors could invade into the ECM as cohesive clusters following a traction established within a tumor microenvironment. The collectively migrating tumor clusters have been detected even in the early stage of cancers [43], and are said to have greater invasive capacity and higher resistance to the therapeutic treatments than the individually migrating tumor cells. Whereas the NETs-mediated tumor metastasis model has been clearly established for disseminating ovarian tumor cells [20], limited information is available about the interactions between the NETs and aggregated tumor clusters that could explain the possible downstream modalities of metastatic aggression of collectively invading tumors. Within a dynamic TIME, neutrophils could respond to the tumor-derived chemotactic stimuli by extravasating from the blood vessels into the tumor tissue through chemotaxis [44], and migrate towards the growing tumor aggregates within the stroma, releasing NETs along the way [45] (as schematically depicted in figure 1(A)(i)). We hypothesized that the collective invasion dynamics of tumors could be at least in part dependent on the neutrophil influx into the TIME, and the consequent NETosis. We therefore sought to better understand the interactional dynamics between the neutrophils and tumors that could regulate tumor invasion.
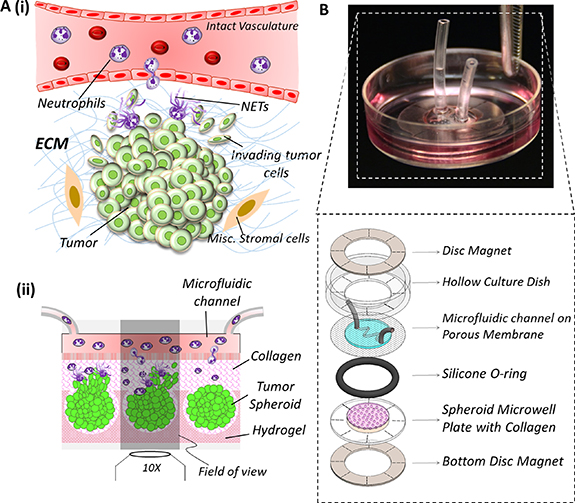
Figure 1. (A) (i) Schematic representation of a tumor-immune microenvironment (TIME) showing the typical interactional dynamics [44, 45] between pre-metastatic tumor and neutrophils extravasating across intact vasculature into the tumor tissue, generation of neutrophil extracellular traps (NETs), and the initiation of tumor invasion into the stromal region (ii) schematic conceptualization of TIME in-vitro: tumor spheroids housed within hydrogel microwells (such as polyacrylamide) are embedded within a collagen matrix, and integrated with a microfluidic channel through a porous membrane. Neutrophils loaded into the microfluidic channel (mimicking the vasculature) migrate across the porous membrane (mimicking the vascular lining) into the collagen region (mimicking he tumor stroma) towards the spheroids, and the tumor cells could invade three-dimensionally into the ECM. (B). Implementation of TIME-on-Chip on a 35 mm culture dish platform: using ring magnets, microfluidic channel printed on a porous membrane and attached to the modified culture dish is hybrid integrated with a microwell plate housing the spheroids, and the O-ring around the microwell plate provides a leak-proof assembly.
Download figure:
Standard image High-resolution imageTo address our hypotheses in-vitro, it is important to model the neutrophil-tumor interactions in a 3D setup because conventional two-dimensional (2D) culture platforms cannot replicate certain functionalities of a TIME that are specific to three-dimensionality, such as neutrophil infiltration into a 3D tumor cluster, or the collective 3D invasion behavior of the tumor cells. Therefore, in this work, we engineered a novel, microfluidics integrated TIME-on-Chip device that serves as a neutrophil migration and 3D tumor invasion platform. As schematically sectioned and shown in figure 1(A)(ii), herein, active tumor spheroid aggregates (henceforth addressed to as spheroids) generated within hydrogel microwells and embedded within collagen matrix with a precise spatial arrangement, are hybrid-integrated with 3D bioprinting-enabled microfluidic channels that are fabricated on a porous membrane and carrying neutrophils. This device design mimics the physiology of a typical TIME in a 3D setting wherein, neutrophils from the microfluidic channel could migrate towards the tumor spheroids in 3D through the collagen matrix, leading to NETosis and 3D tumor invasion into the stroma. Using representative ovarian cancer cells OVCAR-3, in this work, we firstly established the functionality of our TIME-on-Chip device by validating the collective 3D invasion behavior of aggregated cancer spheroids. We thereafter observed the neutrophil response to these tumor spheroids through chemotaxis and NETosis, and examined the impact of neutrophil recruitment on tumor invasion. The results established that NET formation induces collective invasion of OVCAR-3 cells into the collagen, and this behavior is reversed by drug-induced abrogation of the NETs. We further identified an interesting behavior of the neutrophil-tumor dynamics, that the NETs that are formed around the spheroids in the stromal extracellular collagen matrix, and not the tumor-contacted NETs, predominantly mediate the collective tumor invasion behavior, suggesting that tissue-level NETs-stromal interactions could be a potential target for cancer therapeutics.
2. Material and methods
Unless otherwise mentioned, all materials were procured from Thermofisher Scientific (Carlsbad, CA, USA), and all chemicals from Sigma Aldrich (St. Louis, MO, USA).
2.1. Tissue culture protocols
2.1.1. Tumor cell culture
Chemoresistant ovarian cancer cell line NIH:OVCAR-3 was used as the model tumor cell line in our experiments. OVCAR-3 cells were cultured in RPMI-1640 Medium (Gibco,USA) supplemented with 20% fetal bovine serum, 1% Penicillin-Streptomycin and 0.01 mg ml−1 Bovine Insulin. Culture media was changed every 2 d. Immediately before the experiments, the cells were detached from the culture plates using Trypsin-EDTA (0.25%), and re-suspended in complete growth medium to the desired concentration. For cell proliferation assays, cells were treated with 5 µM CellTrace CFSE dye (Thermofisher scientific, USA) for 15 min, and the media was replaced thereafter prior to trypsinization. OVCAR-3 cell lines used in preliminary imaging assays were modified to express GFP (generously gifted by Dr Imran Rizvi, UNC Chapel Hill) with an emission peak at 540 nm.
2.1.2. Preparation of collagen
To prepare collagen matrix, Type-I bovine collagen (Advanced Biomatrix, 5 mg ml−1) was diluted to 1.5 mg ml−1 in PBS, 1× RPMI and 10x MEM solution to obtain the final concentration. The acidic solution was neutralized with 1 M NaHC03 by titration based on the color of the indicator in RPMI. The pre-polymerized collagen was kept in ice until further use. Cold collagen gel solution was pipetted over the test devices and allowed to incubate at 37 °C for 45 min to gel. In some experiments, the collagen was doped with 100 ng ml−1 of transforming growth factor (TGF)-β or 100 ng ml−1 of Interleukin-8 (IL-8) to stimulate tumor invasion as required.
2.1.3. Preparation of polyacrylamide
The composition of polyacrylamide (PAAm) was adopted from previous works [46, 47]. PAAm pre-polymer solution is prepared in volumetric compositions of 30% of 5.3 M Acrylamide stock solution (Bio-Rad), 12% of 129.7 mM Bis-acrylamide stock solution (Bio-Rad), 47% of phosphate buffered saline (PBS), and 1% of tetramethylethylenediamine mixed in an Eppendorf tube. To initiate polymerization, 10% of freshly-prepared 1% w/v ammonium persulfate initiator was added to the solution. This PAAm formulation had an empirical shear modulus of ∼25 kPa.
2.2. Design and fabrication of TIME-on-Chip
The conceptualization of our TIME-on-Chip is shown in figure 1(B). The device consisted of a composite hydrogel-glass plate having arrayed microwells embossed on the hydrogel surface, to house the tumor spheroids. This setup was attached magnetically to microfluidic channels created on a porous track-etched membrane having 3 µm pores. The microfluidic channel replicates an intact vascular structure surrounding the tumor that have a junction gap of ∼2 µm [48]. Collagen layer filled in between the spheroids and the channel mimics the tumor stroma. This configuration allows in-vivo like 3D biochemical correspondence between the tumor cells, stroma and the vasculature. This device design not only facilitates the feasibility of replicating neutrophil infiltration into the tumor tissue across the porous membrane, but also enables the study of tumor invasion.
2.2.1. Fabrication of spheroid microwell plate
The hybrid hydrogel-glass microwell plate for tumor spheroid generation was fabricated through pattern transfer onto PAAm by replica molding from negative plastic molds [49, 50], as schematically shown in supplementary figure 1S (available online at stacks.iop.org/BF/13/035029/mmedia). The microwells were designed to have an inside diameter of 500 μm, and an opening of 200 μm, with a regular spacing of 600 μm between two adjacent features. The diameter of the PAAm base was 10 mm consistent with the inner diameter of the ring magnet used for hybrid integration. The microwell molds were 3D printed (Formlabs 3, USA) using standard resin, and PAAm (prepared as described in section 2.1.3) was cast on the molds. Surface-treated, silanized glass coverslips were attached with PAAm during polymerization in order to facilitate handling of the device. After carefully peeling off the PAAm containing embossed microwell features, the devices were immersed in PBS and gently agitated on a shaker plate for 1 h to remove excess unpolymerized chemicals. Prior to cell seeding, all the devices were stored immersed under PBS at 4 °C.
To create spheroids, the plates were firstly sterilized by exposure to germicidal UV light for 30 min, and thereafter placed on a flat surface under sterile conditions to gently pipette the tumor cell suspension (at a concentration of 5 × 106 ml−1) onto the PAAm surface. The setup was left undisturbed for 5 min to allow the cells to settle into the microwells by gravity. Given that untreated PAAm resists protein adhesion, cells present within the non-adhesive microwells spontaneously aggregated to form spheroidal tumor clusters. Thereafter, excess cells on the surface of the microwell plate were gently washed away using culture media (supplementary figure 1S). Prior to hybrid integration, the fabricated devices were stored inside multiwell plates under cell culture media for 24 h to allow for the initial spheroid compaction.
2.2.2. 3D bioprinting-assisted fabrication of microchannels
Microfluidic channels were fabricated using a combinatorial technique that involves printing of a sacrificial layer that acts as negative mold, and its subsequent removal upon casting a polymer bulk on top that acts as the enclosure to the channels [51]. To implement this approach, we leveraged the automated functionality of a 3D Bioprinter (Bio X, CellInk, VA, USA). Commercially available Pluronic gel (IK50000, CellInk, USA) was used as the sacrificial material to print the negative microchannel features, given its ability to gel at room temperatures even at relatively low concentrations, and reverse the gelation to transition into a dissolvable solution upon cooling below its lower critical gelation temperature [52]. UV curable polymer NOA63 (Norland Inc., USA) was used as the channel enclosure. Nucleopore track-etch membrane, 25 mm in diameter with 3 μm pores (Whatman Inc., USA) attached to the bottom of standard 35 mm culture dishes with a central 10 mm bore, was used as the platform for creating the microfluidic channels.
The process for microchannel fabrication is schematically depicted in supplementary figure 2S. The pluronic layer was printed on top of the membrane precisely along the desired coordinates relative to known reference alignment marks. For support, a thin layer of Gelatin-Methacrylate (GelMA-C, IK-351102, CellInk, USA) was printed on top of the pluronic layer. Microfluidic tubes (EW-06419-01, Cole Parmer, USA) were stably positioned over the desired inlet and the outlet locations. NOA63 was then printed on top of the membrane such that it filled up the space surrounding the channel. This step was followed immediately by exposure to 365 nm UV light for 5 min for curing the UV sensitive polymer. Printing of NOA and exposure to UV directly on the 3D printer bed helped control the amount of the dispensed NOA, and hence the curing time. This step also reduced manual handling of the device during fabrication.
After UV curing, the setup was then cooled down to 4 °C for 15 min using the temperature controlled print-table, and thereafter, the microfluidic tubes were flushed with cold PBS, to remove the sacrificial ink and create a perfusable channel on top of the porous membrane. For this work, all the fabricated channels were straight channels having a square cross-section with width and height of 500 µm, and length of 8 mm, unless otherwise specified.
2.2.3. Hybrid integration and assembly
Process of hybrid integrating the microfluidic assembly with the spheroids is schematically shown in supplementary figure 3S. For hybrid integration, Neodymium rare-earth ring magnets (R1090, SupermagnetMan Inc., USA) with inner diameter of 10 mm, outer diameter 22 mm and thickness of 0.4 mm were used. Firstly, a ring magnet was placed and aligned inside the culture dish (containing the microfluidic channel printed on the porous membrane) concentric with its central bore. The microwell plate, housing the spheroids, was placed atop a counterpart ring magnet and aligned carefully to the reference marks on the magnet. An O-ring with desired thickness is placed on top of this assembly and collagen gel was gently pipetted on top of the microwells to completely fill up the space inside the O-ring. The integration procedure simply involved bringing the culture dish close to the microwell plate assembly while maintaining it as upright as possible for enabling the attachment of the ring magnets. All the ensembles sandwich between the two ring magnets instantaneously as shown in figure 1(B). The O-ring prevented any fluid leakage from the system while providing the necessary height for the stromal collagen layer between the tumor cells and the microfluidic channel. Culture media was then flown into the microfluidic channel (replenished every 12 h) and the devices were incubated at 37 °C till further assays. For neutrophil tumor interaction assays, after culturing the spheroids for 24 h to allow for compaction, neutrophils were loaded into the microchannels, and thereafter quickly hybrid integrated with the spheroids–collagen assembly.
2.2.4. Finite element modeling
To optimize the geometric configuration of our TIME-on-Chip based on the diffusion and mass transport parameters, 2D finite element modeling (FEM) studies were conducted using COMSOL (v5.3, Burlington, MA, USA). The computational domain consisted of the cross-section of the device orthogonal to the direction of fluid flow showing rectangular microfluidic channel on top of a 7 μm thick porous membrane, with a collagen layer underneath the porous membrane that serves as the ECM layer between the channel and the spheroids. The distribution of the pores was calculated for a membrane porosity of 15%. The model was built based on the assumption that the diffusion of solute into the collagen happens across the porous interfaces from the microfluidic channel, with negligible mass transport across a fully cured UV glue surrounding the channel. FEA was carried out by solving the diffusion equation based on Fick's law of diffusion, derived from the continuity equation in a non-homogenous system. The calculation and validation of the diffusion parameters is detailed in supplementary information. Consistency of fluid and mass transport across the different interfaces was studied experimentally through the diffusion behavior of 10 kDa Fluorescein Isothiocyanate (FITC)-dextran from a straight microfluidic channel into the collagen layer (thickness is 250 μm). The average fluorescence intensity within a field of view (FOV) obtained from time-lapse images. The experimental rate of change of normalized fluorophore concentration was compared with the time dependent variation of the normalized concentration obtained from the computational model.
2.3. Neutrophil isolation and NETs formation
Neutrophils were isolated using a previously established protocol [53] that involves collection of whole blood drawn from the peripheral vein of healthy donors, separation of nucleated cells from the erythrocytes, rinsing the cells, and thereafter magnetically separating the red blood cells from the purified neutrophils. This neutrophil isolation method depletes all other cells from blood, leaving the neutrophils unstimulated. The isolated neutrophils were treated with 16.2 μM Hoechst 33258 dye (H21491, Invitrogen) at 37 °C for 15 min for nuclear staining, and prepared to a final concentration of 1 × 107 ml−1 for the experiments unless otherwise specified.
Whereas naïve neutrophils were allowed to form NETs by interacting with tumor cells, externally stimulated NETs were generated using 500 nM Phorbol-12-Myristate-13-Acetate (PMA) [35] wherever required. Depending on the assay, PMA was added either to the media containing the suspension of neutrophils prior to passing the cells into the channel, or added directly to the collagen. The generation of NETs from neutrophils was observed by fluorescent labeling using 2.5 μM Sytox Green (S7020, Invitrogen) added to the pre-polymerized collagen (described in section 2.1.2.) For experiments that required the inhibition of NET formation, neutrophils were re-suspended in RPMI containing 10 μM Sivelestat (HY-17443, Medchemexpress) prior to use in the experiments.
2.4. Immunostaining
For stimulated tumor migration assays, spheroids were labeled for E-cadherin and nuclei in culture, using standard indirect immunostaining techniques. The microwell plates containing the spheroids were initially washed in PBS and fixed in 4% (v/v) paraformaldehyde in PBS for 1 h at room temperature. Thereafter the devices were washed twice with PBS for 5 min each and permeabilized with 0.1% (v/v) Triton-X in PBS for 1 h at room temperature. After washing 2 times with PBS for 5 min each again, the spheroids were blocked against non-specific binding with 2.5% (v/v) goat serum in PBS for 1 h at room temperature. Devices were then incubated overnight at 4 °C with mouse anti-E cadherin monoclonal antibody (HECD-1) (13-1700, Invitrogen) in a 1:200 dilution of goat serum. 200 μl of the antibody solution was dispensed on top of the device and the setup was left undisturbed for 1 h at room temperature. The devices were washed twice once again with PBS for 10 min each and blocked with 2.5% (v/v) goat serum in PBS for 1 h. After blocking, samples were incubated at room temperature for 2 h with goat anti-mouse IgG (H + L) secondary antibody, Texas Red-X (T-6390, Invitrogen) in a 1:1000 goat serum dilution. The samples were rinsed for 10 min in PBS and counterstained with nuclear stain, 4',6-Diamidino-2-Phenylindole, Dilactate (DAPI, D3571, Invitrogen) (2 μg ml−1 in PBS) for 1 h at room temperature, and were thereafter washed twice with PBS. For immunofluorescent staining of citrullinated Histone H3, primary antibody Anti-Histone H3 antibody (ab5103, Abcam) was conjugated with AlexaFluor 647 using Zip AlexaFluor 647 Rapid Antibody Labeling Kit (Z11235, Invitrogen), and the conjugated antibody was added (1:1000) to the pre-polymerized collagen.
2.5. Imaging and statistical analysis
Both brightfield and fluorescent imaging were carried out using an inverted fluorescence microscope coupled with a CCD camera (Olympus IX-83). CellSens image analysis software (Olympus) was used for data acquisition. Image processing and analyses were done using Image-J (NIH, USA). The experimental data of the calculated spheroid parameters (fluorescence intensity or area) or neutrophil parameters (neutrophils count or % NETs) for different experimental conditions are expressed as mean ± standard error (SEM) unless otherwise specified. In general, comparative data analysis of populations was performed without pre-specifying a required effect size. Datasets were normally distributed, with similar variances between compared groups. All statistical analysis was conducted using student's t-test or two-tailed one-way ANOVA analyses with Tukey post-hoc pairwise comparisons (Prism; GraphPad Software, La Jolla, CA). p-values less than 0.05 were considered significant.
3. Results
3.1. TIME-on-Chip facilitates dynamic 3D tumor cultures
We were able to consistently generate 3D tumor spheroids using the method described in section 2.2 (supplementary figure 1S). Most of the spheroids appeared circular upon formation, though OVCAR-3 cells are generally known to form loose aggregates [54]. The tumor aggregates were confined within the microwells, and subsequent addition of collagen on top, or magnetic attachment to the microfluidic channel, did not distort the spheroids. In general, the magnetically enabled hybrid integration approach provided robustness to maintain the integrated assembly free of distortion from vibrations associated with handling the device (during transfer of culture vessel or repeated movement of the microscope stage for imaging). This method of hybrid integration also enabled on-demand attach/detachment of the setup during both pre-and post-processing steps, and provided maneuverability to precisely align the different subcomponents under the microscope. We successfully fabricated and tested different microfluidic channel configurations with controlled positioning with respect to the spheroids along all three coordinate axes, similar to the framework of the vasculature-like networks [55] (schematically shown in supplementary figure 4S).
To ensure that the magnetic hybrid integration method sustains consistent fluid and mass transport across the different interfaces, we studied the diffusion behavior of 10 kDa FITC-dextran from a straight microfluidic channel into collagen (thickness is 250 μm) and PAAm layers. This diffusion was quantified by measuring the fluorescence intensity across the spheroids located away from the FOV of the microchannel (figure 2(A)(i)). We compared the experimental diffusion behavior with the 2D Finite Element Model built on COMSOL (figure 2(A)(ii)). The average fluorescence intensity around a chosen spheroid was measured over time, and the rate of change of concentration was measured by calculating the change in the normalized fluorescence intensity over time, and comparing with the change in the normalized concentration variation obtained from the computational model. The results of the FEM showed a close agreement with the experiment within a 3 h timespan (figure 2(A)(iii)). This serves as a validation for the capability of our simple 2D computational model to predict the diffusion behavior, as required to modify the design configuration of our TIME-on-Chip (such as the collagen layer height, or microchannel widths), so as to achieve optimal fluid transport parameters in applications such as gradient generation or drug delivery. Thus, the magnetic hybrid integration ensures connectivity across the interfaces of the different layers of the system, and our initial results demonstrate the capability of the platform to enable 3D interstitial fluid transport around the spheroids to support long-term cultures (supported by supplementary figure 5S).
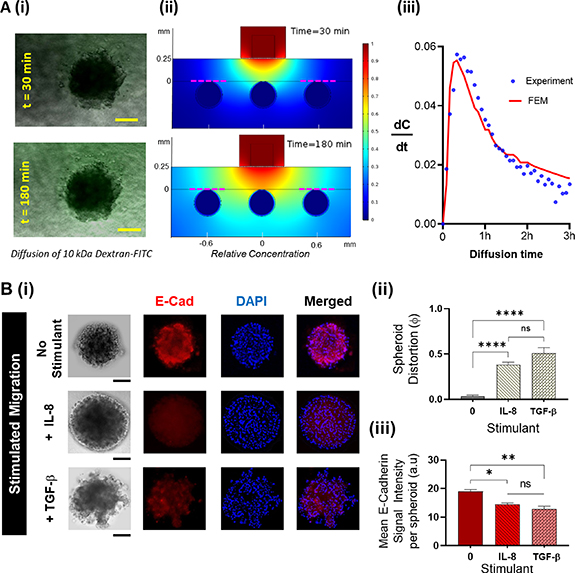
Figure 2. Functionality of the TIME-on-Chip: (A). (i) Experimental investigation of the feasibility of fluid transport across the hybrid integrated TIME-on-Chip device: microscopic images showing the evolution of FITC diffusion across a spheroid at different time points (scale bars represent 100 μm). (ii) Finite-element models based on a 2D geometry of the cross-section of the device simulating the diffusion based fluid transport from the microchannel into the collagen around the spheroids. Sections of the model representing the experimental diffusion condition around the spheroid located away from the microfluidic channel are marked in red dotted lines. (iii) Compared to the experimental data for variation in the rate of fluorophore diffusion across a spheroid, the 2D finite element simulations adequately predict the diffusion-based fluid transport behavior of the device as seen from the average rate of change of concentration. (B). 3D migration behavior of OVCAR-3: (i) representative fluorescence images upon immunostaining of the distorted OVCAR-3 spheroids for E-cadherin (red) and nuclear stain (DAPI blue). OVCAR-3 spheroids showed spontaneous migration when present within a 3D collagen matrix, the spheroid distortion was negligible within the first 24 h and thereafter marginally increased over the next 48 h. Upon stimulation with TGF-β and IL-8, OVCAR-3 cells migrate in clusters and fluorescence immunostaining shows qualitatively a reduced E-cadherin expression throughout the spheroids when stimulated with both IL-8 and TGF-β. Scale bars represent 100 μm. (ii) Compared to unstimulated conditions, spheroids undergo a significantly greater distortion over 24 h when stimulated by IL-8 (p < 0.0001) and TGF-β (p < 0.0001). (iii) The mean E-cadherin signal intensity per spheroid decreases during stimulated migration correlating with the spheroid distortion (data collected from n = 9 spheroids over N = 3 experiments, mean ± SEM. Significance was determined using one-way ANOVA with Tukey's post-hoc analysis * p < 0.05, **p < 0.01, ****p < 0.0001, ns = not significant).
Download figure:
Standard image High-resolution image3.2. Demonstration of collective 3D migration of OVCAR-3
Migrating OVCAR-3 cells could exist as single cells or as multicellular aggregates [56]. Therefore, we sought to verify the 3D migratory dynamics of the OVCAR-3 spheroids, and affirm the capability of our device to study tumor motility. A 3D collagen matrix migration assay is a versatile method to analyze the migration of cells within a physiological-like 3D environment [57, 58]. We hypothesized that aggregated OVCAR-3 cells undergo spontaneous migration when present in a 3D matrix [59]. We also tested spheroid motility upon stimulation with IL-8 [60] and TGF-β [61], both of which have been shown to induce epithelial to mesenchymal transition and to promote migration in OVCAR-3 cells. In separate assays, we doped the collagen layer with 100 ng ml−1 of TGF-β or IL-8 as the respective experimental condition, and observed tumor behavior over 24 h. To quantify the spheroid behavior, we defined a parameter called spheroid distortion (φ), taken as the ratio of change in the projected area of the migrated spheroids over the initial projected area of the intact spheroids. Additionally, we analyzed spheroid distortion in correlation with the down-regulation of E-cadherin, an important marker for cell-cell adhesion [62–64]. For each experimental condition, we were able to non-destructively dismantle the device after the assay to fix the cells with 4% Paraformaldehyde, and stain for E-cadherin (red), and nuclear stain (blue). We measured the mean E-cadherin per spheroid by calculating the ratio of integrated E-cadherin fluorescence per spheroid over the projected spheroid area.
At baseline, OVCAR-3 cells did not display spontaneous migration into the collagen matrix for at least 24 h. We observed a non-significant increase in spheroid distortion in 48 h, with further distortion at 72 h (supplementary figure 6S). We also found no significant difference in the mean E-cadherin signal intensity per distorted spheroid at different time points of the spontaneous migration (supplementary figure 6S). However, when exposed to the stimulants, TGF-β or IL-8, the resultant distortion of the spheroids was significantly higher (p < 0.001) than unstimulated spheroids (figures 2(B)(i) and B(ii)). The mean E-cadherin expression of the spheroids was significantly reduced under stimulated conditions (figure 2(B)(iii)). Upon stimulation, the cells migrated into the collagen matrix, and the final projected area of the distorted spheroids was always larger than the initial spheroid area (φ > 0), implying that in a 3D setup, OVCAR-3 cells migrated and invaded as a group rather than as single cells. The migratory cells at the periphery did not appear to change their morphology to mesenchymal-like, as observed in [56]. While it is possible that cells within a 3D matrix could undergo both proliferation and migration [58, 65], it has been reported that unstimulated OVCAR-3 could remain largely in a proliferation-quiescent state within a 3D matrix [64, 66]. Therefore, our results suggest that upon stimulation, the OVCAR-3 spheroids collectively invaded the collagen matrix.
Overall, these results validate the utility of our TIME-on-Chip to recreate and analyze the locomotion behavior of 3D tumor cultures, and carry out integrated labeling and post-assay analyses directly on chip.
3.3. OVCAR-3 spheroids elicit in-vivo like neutrophil response
To understand how neutrophils interact with OVCAR-3 spheroids, we experimentally simulated and visualized chemotaxis and NET formation behavior of naive neutrophils in response to the tumor, to recapitulate aspects of in-vivo neutrophil function in an in-vitro setting. Neutrophils were loaded into the microfluidic channel and their surveillance was presented with competing tumor targets, classified as S1, S2, and S3 based on the arrayed location of the spheroids inside the collagen matrix with respect to the channel (schematically shown as perspective view in figure 3(A)). For ease of visualization, neutrophils were stained with Hoechst blue (live nuclear stain), and we acquired end point z-stack images of all the spheroids in the microwell plate, allowing us to quantify neutrophil infiltration within a FOV. To recreate a physiologically-relevant environment, parallel experiments were carried out in which the fluid carrying the neutrophils inside the channel was actuated using an external syringe pump (supplementary figure 7S) at a flow rate of 2 μl h−1. Neutrophil response to tumor under both no-flow and flow conditions was analyzed over 6 h.
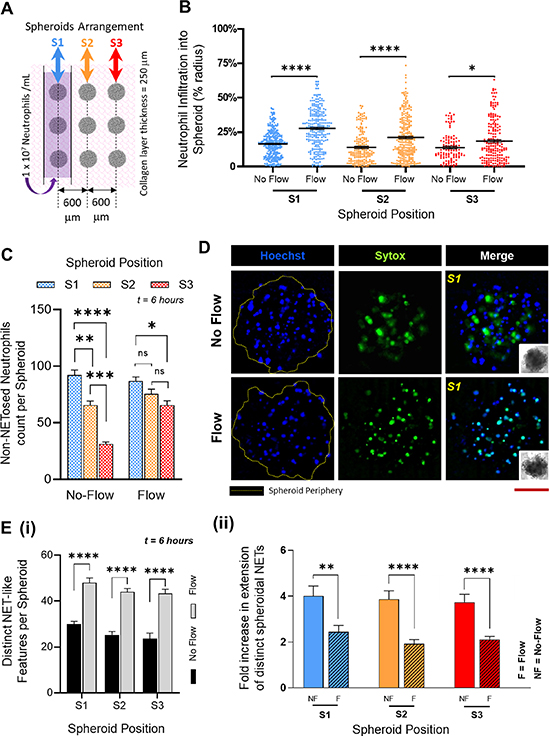
Figure 3. Neutrophils respond to OVCAR-3 spheroids: (A). Schematic top view of the TIME-on-Chip showing the arrayed spheroid arrangement with respect to the microfluidic channel carrying the neutrophils. Spheroid positions S1, S2 and S3 are denoted relative to the position of the microfluidic channel. (B). Irrespective of the spheroid position, the penetration of neutrophils into the spheroids i.e. the distance of neutrophil infiltration into the spheroids expressed in terms of the spheroid radius, is significantly higher (*p < 0.05, ****p < 0.0001) when the fluid in the microchannel is actuated with an external pump. (Data collected from n = 3 spheroids for each position over N = 3 experiments, mean ± SEM, t test between the comparing groups) C. Without flow, a greater percentage of neutrophils infiltrated the spheroids which had the closest initial proximity to the neutrophils, and neutrophil recruitment significantly decreased with an increase in the distance between the channel and the spheroids. With flow, however, no significant difference was observed in the recruitment of neutrophils to the adjacent arrays of the spheroids, though there was a decrease (p < 0.05) in the overall neutrophil recruitment between the S1 and S3 spheroids (data collected for n = 9 spheroids from N = 3 experiments, mean ± SEM. Significance was determined using one-way ANOVA with Tukey's post-hoc analysis *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant) (D). Representative images of spheroids for both flow and no-flow conditions showing the infiltration of neutrophils (Hoechst blue) and NETs (Sytox Green) generated by the tumor-infiltrating neutrophils. Red scale bar represents 100 μm. (E). (i) Irrespective of the spheroid position, the total number of distinct NET-like structures observed by counting the number of Sytox stained features, was significantly higher (p < 0.0001) when fluid in the microchannel was actuated. (ii) However, the NET structures formed within the spheroids were smaller, and had a significantly lesser spread area with flow in the microchannel (p = 0.008 for S1, p < 0.0001 for S2 and p < 0.0001 for S3) (data collected for n = 9 spheroids from N = 3 experiments, mean ± SEM, with t-test between the comparing groups, **p < 0.01, ****p < 0.0001).
Download figure:
Standard image High-resolution imageNeutrophils migrated to the spheroids through the collagen matrix (supplementary figure 8S(i)). The inception of neutrophil chemotaxis occurred as quickly as 30 min after loading of the cells into the channel. A count of the number of neutrophils that migrated shows that under no-flow conditions, neutrophil trafficking stabilized over ∼6 h (supplementary figure 8S(ii)) as expected [67], possibly running close to the typical ex-vivo lifetime of the neutrophils. Under both no-flow and flow conditions, not all the neutrophils in the vicinity migrated towards the spheroids even over a short migration range, and overall, a maximum of ∼10% of all neutrophils from the microchannel infiltrated into the tumor, with ∼6% influx observed within the stromal collagen layer. However, in the absence of spheroids, no random migration of the neutrophils into the collagen region was observed (supplementary figure 9S), thereby confirming that neutrophils respond to the tumor spheroids through chemotaxis. Also, for both no-flow and flow conditions, no significant difference was observed in the respective mean neutrophil penetration into the spheroids regardless of the spheroid location, implying that neutrophil infiltration into a tumor was not dependent on their migration range. However, we observed that under flow, neutrophils infiltrated significantly deeper (p < 0.0001 for S1 and S2, and p < 0.05 for S3) into the spheroids, with a mean penetration of ∼10% greater than the neutrophil infiltration under static conditions (figure 3(B)). We thereafter counted the total number of intact neutrophils that had infiltrated into the spheroids.
Spheroids closest to the microchannels recruited the maximum number of neutrophils, with the percentage recruitment dropping as the distance between spheroids and the channel increased (figure 3(C)). However, we also observed that with fluid flow in the channel, a significantly greater number of neutrophils were present in the farthest spheroids (S3) than what was seen under no-flow conditions. While the ability of the neutrophils to infiltrate a tumor could depend on the resultant degradation of inter-spheroidal cell-cell tight junctions, it is also possible that the shear stress caused by the fluid flow could prematurely activate the neutrophils [68] in the channel leading to altered tumor response. From these experimental results, while it is difficult to clearly identify the existence of a specific sub-group of tumor-infiltrating neutrophils (TINs) [27, 69, 70] among the heterogeneous population, the relevance of signaling modalities among these TINs and the effect of external biophysical factors on the behavior of TINs contributing to the development of tumors could be investigated further.
We then characterized the reciprocal effect of OVCAR-3 spheroids on the infiltrating neutrophils. Within 1 h, neutrophils extruded webs of DNA that could be detected by the Sytox stain. This was observed under both flow and no-flow conditions (figure 3(D)). In general, the Sytox stained features that were observed in the collagen layer surrounding the spheroids were markedly more spread out than those formed inside the tumors (supplementary figure 10S). We reasoned that this could be possibly due to the higher mechanical stiffness [71] around a neutrophil when present inside a condensed microenvironment of a spheroid that could restrict the spread of the extracellular mesh sprouting into the surrounding environment. Therefore, as a positive control, in separate experiments, we stimulated neutrophils embedded within collagen doped with 500 nM PMA, a potent inducer of lytic NETosis [35]. The structures of the extruded features from PMA stimulation were similar to those formed by the neutrophils migrating towards the spheroids (supplementary figure 11S), thereby confirming that OVCAR-3 cells induce the neutrophils to NETose. As another control to validate that the NETs induction was specific to the neutrophil interaction with tumor cells, we replaced the spheroids in the microwells with IL-8 doped collagen droplets. IL-8 is a known neutrophil chemokine, and while it stimulated neutrophil chemotaxis from the microchannel into the collagen, no spurious NET formation was observed.
To further understand the effects of fluid flow on the overall functionality of the tumor penetrating neutrophils, we quantified the inter-spheroidal NETosis through two parameters, namely, total number of distinct NET structures within the spheroids, and fold increase in the NETs spread area. Irrespective of the proximity of the spheroids to the channel, under flow, we found a significantly greater (p < 0.05) number of NETs structures consistently present in each of the spheroids (figure 3(E)(i)), albeit with a significantly lower spread area (figure 3(E)(ii)). To examine this further, the non-adherent neutrophils washed away from the channels under flow were collected and examined. We observed Sytox signaling in several of those cells (supplementary figure 12S), whereas under non-flow conditions, neutrophils that remained in the channel did not show any Sytox signaling. We reasoned that the shear stress from the fluid flow could rupture the cell membrane that could possibly lead to budding of the NETs [68]. These results suggest that fluid flow around the neutrophils activate them differently, and could affect their response to tumors.
Altogether, our results clearly establish that OVCAR-3 spheroids induce neutrophil response through both chemotaxis and NETosis.
3.4. NETs stimulate OVCAR-3 invasion
At the end of the 6 h neutrophil-tumor interaction assay, some spheroids (under both flow and no-flow conditions) showed a change in shape by losing their initial circularity. While this morphological disturbance could merely be due to the intrusion of additional cells into the spheroids, we also observed that the spheroids marginally elongated only at certain sites along their periphery (supplementary figure 13S(A)). Therefore, to quantify this change of shape, we defined spheroid circularity as the ratio of the perimeter of a spheroid to the ideal perimeter of a circle of an equivalent projected area [50]. The loss of circularity correlated with the cumulative total number of NETs and intact neutrophil cells present within the spheroids (supplementary figure 13S(B)), raising questions about the role that either neutrophils or the NETs could play in OVCAR-3 migration. Therefore, we looked at the counter-effects of neutrophil-tumor interactions on spheroid distortion over an extended duration (24 h). For this assay, neutrophils were loaded into microfluidic channels aligned directly over the spheroids with a separation of 250 μm, and no flow was introduced in the channel.
As shown in figure 2, OVCAR-3 cells have limited ability to migrate spontaneously in the absence of infiltrating neutrophils in the TIME. In the presence of neutrophil infiltration, however, significant spheroid distortion was observed in all the spheroids over 24 h. This distortion was much more pronounced and evident than the patterns observed following stimulation with TGF-β or IL-8 because the OVCAR-3 cells appeared to collectively invade into the collagen matrix in a highly directional manner (top row in figure 4(A)). Since NETs formation was always a complementary response of the naive neutrophils, we attempted to separate the action of NETs and non-NETosing neutrophils in inducing tumor migration. Therefore, we conducted comparative experiments wherein we pre-treated the neutrophils with 10 μM Sivelestat, a Neutrophil Elastase Inhibitor (NEI [72, 73]), to block NETs formation. The number of extensive NET structures clearly diminished due to NEI treatment (bottom row of figure 4(A)). While no significant difference was observed in the number of non-NETosed neutrophils that infiltrated the spheroids (figure 4(B)(i)), the inter-tumoral NETs density significantly reduced (p < 0.0001) upon treating the neutrophils with Sivelestat (figure 4(B)(ii)). Importantly, the resultant spheroid distortion also dropped significantly (p < 0.0001) compared to untreated neutrophil infiltration (figure 4(C)(i)). Thus, blocking NET formation impeded the neutrophils' ability to stimulate OVCAR-3 invasion into the collagen matrix. This also indicates that tumor cell-derived factors that stimulate neutrophil mobilization through chemotaxis do not necessarily also trigger NETosis.

Figure 4. NETs induce spheroid distortion (A). Representative images of the spheroids showing the infiltration of neutrophils (stained Hoechst blue) and the NETs generated within the spheroids (labeled with Sytox green), qualitatively showing that treatment of neutrophils with Sivelestat, a Neutrophil Elastase Inhibitor. Scale bar represents 100 μm. (B) (i) A count of the intact neutrophils that infiltrated into the spheroids are not statistically different (p = 0.1602) with or without pretreatment with Sivelestat, whereas (ii) the percentage of the spheroid covered by NETs was significantly lower (p < 0.0001) when neutrophils were pretreated with Sivelestat (for total n = 9 spheroids measured over 3 experiments, mean ± SEM, t test). (C) (i) Spheroid distortion is significantly reduced (p < 0.0001) by abrogation of NETs with NEI pre-treatment of the neutrophils. (ii) A comparison of the NETs distribution within the distorted spheroids shows that a greater proportion of the NETs (p = 0.0061) are present within the intact non-invasive domain of the spheroids (for total n = 9 spheroids measured over 3 experiments, mean ± SEM, t test). (D) (i) Brightfield image of the distorted spheroid showing the invasive domains, and the original spheroid periphery (marked in red) prior to tumor invasion. The invasive domain is distinct by its presence within the stromal collagen region seen out of plane from the rest of the spheroid. To calculate the spheroid polarization, the distorted spheroid was assumed to be an ellipsoid and an ellipse was fit passing through the outermost points of the spheroid as shown. The polarization (γ) was calculated as the ratio of the diameters along the major (b) and the minor (a) axes of the ellipse. (ii) Spheroid distortion due to the presence of NETs was significantly more polarized than the distortions observed with TGF-β (p < 0.05) and IL-8 (p < 0.001) (data collected for n = 9 spheroids from N = 3 experiments, mean ± SEM. Significance was determined using one-way ANOVA with Tukey's post-hoc analysis *p < 0.05, ***p < 0.001, ****p < 0.0001, ns = not significant).
Download figure:
Standard image High-resolution imageWhile it is known that the DNA mesh from the NETs could entrap the tumor cells at the site of their dissemination and migrate along with them, inflamed neutrophils sequestered at entrapped tumor cells via chemotactic confinement could also promote tumor migration [74]. We sought to understand if this dynamic could apply to cells migrating in clusters, wherein subpopulations of OVCAR-3 cells within the spheroids could acquire phenotypic variations [75] and neutrophils preferentially home to and infiltrate the more motile cells within the tumor aggregate to NETose. We therefore calculated the overall NETs distribution within the distorted spheroids, and found that the fraction of NETs present within the spheroid domain invading into the collagen was significantly lower than the NETs fraction present within the intact domain of the distorted spheroids (figure 4(C)(ii)). Also, the NETs present in the invasive domain were not seen specifically at the leading edge of the invading cells, suggesting that NETs do not necessarily trap the more motile OVCAR-3 phenotypes and drag them out of their intact domain to initiate the tumor invasion cascade.
The invasive domain is clearly identifiable (figure 4(D)(i)) not only due to its protrusion into the collagen matrix outside of the microwells, but also due to its directionality, i.e. inclination of the spheroids to migrate along a certain direction. To quantify this polarization, the approach that we adopted was to consider the distorted spheroid as an ellipsoid [76], and fit the projected boundary with an ellipse that approximately passes through its outermost points as shown in figure 4(D)(i). Using this strategy, we defined the polarization of spheroid distortion (γ) as the aspect ratio of the ellipse i.e. ratio of the diameters along its major and minor axes. As presented in figure 4(D)(ii), the average NET-induced spheroid polarization was significantly greater than those observed with TGF-β (p < 0.05) or IL-8 (p < 0.001). However, in 3D, while the cells invaded the collagen towards the microfluidic channel we did not observe any infiltration the tumor cells across the porous membrane even in the presence of NETs.
To confirm that the observed spheroid distortion is due to the tumor invasion and not proliferation [65], we stained the OVCAR-3 cells with CellTrace CFSE fluorescent proliferation marker [77] prior to the spheroid formation, As a negative control, we treated the spheroids with a combination of 0.6 µM Dasatinib and 30 nM Gefitinib drugs which are known to inhibit OVCAR-3 proliferation [78], and we compared the mean fluorescent intensity of the drug-treated spheroids with untreated spheroids and spheroids distorted in the presence of NETs. The results are as presented in the supplementary figure 14S. No significant difference in the mean signal intensity between the spheroids cultured simultaneously under different conditions. To further validate this finding, we quantified the fraction of cells that retained their fluorescent signal in the invasive region of the collagen, and we compared this with the cells in a regular 2D culture. The results show that most of the cells invading into the collagen retained their fluorescence even in the presence of NETs, and this number was significantly greater (p < 0.05) than the number of cells that expressed CFSE in the 2D culture over the same culture duration.
Thus, it is clear that NETs play an important role in OVCAR-3 cells acquiring a pro-migratory/invasive phenotype. NETs stimulate the invasion of OVCAR-3 cells into the collagen matrix and inhibiting NET formation abrogates tumor invasion.
3.5. Availability of stromal NETs positively correlates with collective invasion of OVCAR-3
For aggregated OVCAR-3 cells, given their tendency to migrate collectively, we hypothesized that the inter-spheroidal NETs may promote invasion by acting to concentrate NET-associated proteases to cleave or interfere with the cell–cell adhesion within the spheroid. Therefore, to verify this, we amplified the concentration of inter-tumoral neutrophils by pipetting them directly onto the spheroids, allowing them to adhere and washing away the unbound cells before adding collagen gel on top. We observed that after 24 h, even though a substantial population of these neutrophils produced NETs, (supplementary figure 15S), the resultant spheroid distortion was surprisingly negligible. Since it is possible that Sytox could stain dead tumor cells with compromised membranes, we immunostained for NETs with Anti-H3Citrulline, a NETs-specific marker [79], conjugated with Alexafluor647. While NETs production was identified by both Sytox and citrullinated histone H3 markers, no significant difference was observed in the spread area of the NET-like structures (supplementary figure 16S). Therefore, clearly, the products expelled out of NETs were not directly mediating the spheroid distortion.
However, if the NETs were to be involved in tumor invasion, then it is only possible that the tumor cells pick up NET-based cues available within the TIME either from the stroma or from the channel. In the present system, the only involvement of NETs generated from naive neutrophils outside the spheroid is within the collagen. But within a continuous system, it is not possible to create conditions wherein the neutrophils that infiltrate from the microchannel could be physically restricted to remain active only within the collagen without migrating all the way to the spheroids. Therefore, we firstly studied the effect of varying the stromal NET density on the spheroid distortion. This was achieved by testing different loading concentrations of neutrophils in the channel. As seen in figure 5(A)(i), the percentage of NET structures occupying the collagen layer (calculated from z-stack images) increased proportionally to the initial number of neutrophils present in the channel. While the elevated neutrophil concentration also increased the number of TINs, we reasoned that the non-TINs that initially migrate into the collagen could undergo NETosis within the stroma. But that count was also proportional to the initial neutrophil concentration.

Figure 5. The density of NETs available in the stromal collagen region correlates with the degree of spheroid distortion (A). (i) Increase in the initial concentration of neutrophils loaded into the microfluidic channel increases the availability of stromal NETs for a fixed collagen thickness (ii) stromal NET density varies with the height of the collagen layer between the spheroids and the microfluidic channel, depending on the number of neutrophils that undergo chemotaxis. (iii) Cumulative results of the above experiments show that the distortion of the OVCAR-3 spheroids increases with the corresponding density of NETs present in the collagen (data collected from at least n = 9 spheroids over N = 3 experiments for each experimental condition, mean ± SEM, ANOVA and Tukey's multiple comparisons; *p < 0.05, ** p < 0.01, ***p < 0.001, **** p < 0.0001, ns = not significant) (B). Representative z-stack images showing the distribution of neutrophils (Hoechst blue) and NETs (Sytox green) in the spheroids and the surrounding collagen region, and the periphery of the distorted spheroid (red scale bar represents 100 μm).
Download figure:
Standard image High-resolution imageWe also tested the variation in the stromal NET density by altering the height of the collagen layer for a fixed neutrophil concentration in the channel (1 × 107 ml−1). As expected, the maximum neutrophil infiltration occurred when the distance to the spheroids was minimal, beyond a certain minimum thickness of the collagen layer, and this also corroborated with the NETs density within the collagen region. The percentage recruitment dropped with increase in collagen thickness (figure 5(A)(ii)), possibly due to the increased migration distance for the neutrophils from the channel, thereby eliciting lesser chemotactic response. While this behavior is consistent with the observations of relay signaling mechanisms between neutrophils that helps recruit more neutrophils toward a target during chemotaxis [80], this data is not sufficient to establish the applicability of a similar mechanism for NETs. Even with the highest neutrophil concentration (1 × 107 ml−1), the maximum NETs density in the collagen region was not greater than 12%. But collectively, these results further validated that whenever there was a higher availability of NETs within the stromal collagen region, the resultant spheroid distortion was also greater (figure 5(A)(iii)). This result is qualitatively presented in figure 5(B) through representative images of the NETs formation in collagen, and the resultant spheroid distortion (classified as low, medium and high).
We further wanted to investigate how the tumor spheroids responded to NETs produced in the microfluidic channel. For this assay, we leveraged the advantage of our magnetic hybrid integration technique for on-demand attachment and detachment of the ensembles, as schematically shown in figure 6(i). We firstly cultured neutrophils separately in the microfluidic channel, and stimulated them for NETs with 500 nM PMA for 6 h. The conditioned media that seeped out of the porous channel was collected and added back to the collagen layer on top of the spheroids and incubated for 1 h, before attaching the channel with the spheroid-collagen assembly. More than 60% of the neutrophils NETosed within the channel upon PMA stimulation (figure 6(ii)). But irrespective of the NET density or the proximity of these NETs to the spheroids, the resultant spheroid distortion observed after 24 h was negligible. No significant difference was observed in the distortion of spheroids due to either direct stimulation with PMA or by stimulation of neutrophils in the channel with PMA (figure 6(iii)).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. To test whether NETs produced outside the collagen region induced tumor invasion within a TIME, we separately stimulated the neutrophils in the microchannel with 500 nM PMA. (i) Schematic illustration of the different steps shows that the TIME-on-Chip easily enables on-demand attachment and detachment of the components to initially induce the neutrophils in the microchannel to produce NETs, add the conditioned media to the spheroids, and re-attach the device to enable NETs-spheroid co-culture for analysis. (ii) More than 60% of the neutrophils in the microchannel NETosed upon PMA stimulation for 6 h. But even after 24 h of co-culture, the spheroids do not appear significantly distorted as seen in the brightfield images (scale bars represent 150 μm). (iii) No significant difference was observed in the distortion of the spheroids with the PMA-stimulated NETs present in the channel or with direct stimulation of the spheroids with PMA (data collected for n = 3 spheroids, mean ± SEM, t test, ns = not significant).
Download figure:
Standard image High-resolution image{kind=link}
Thus, taken together, our data clearly suggest that stromal NETs, and not the tumor-contacted NETs or the vascular NETs, play a significant role in mediating the collective invasion of OVCAR-3 from an aggregated state.
4. Discussion
The interactions of neutrophils and tumors are highly dynamic, expected to swing depending on the state of the disease (reviewed in [4]). This high degree of heterogeneity in the association of neutrophils with tumors make them exciting targets for therapeutic interventions or for exploring immunotherapeutic opportunities for adoptive cell therapy, thereby creating a plethora of opportunities for prospective research leitmotifs. Developing a better understanding of the modalities by which neutrophils interact with tumors, particularly during early-stage tumor formation, could possibly shed light on the characteristics of tumor growth, or relapse after initial treatment, a phenomenon commonly reported in this disease model [81]. In this work, we demonstrated that neutrophils respond to a growing tumor aggregate through two mechanisms, namely chemotaxis and NETosis, and further we found that the interaction with neutrophils also elicited a complementary response from the tumor cells through migration and invasion into the stroma.
When present within a 3D collagen matrix, tumor spheroids formed by the aggregates of OVCAR-3 ovarian cancer cells displayed a conserved migratory behavior that required additional instigation to proliferate. Our data shows that OVCAR-3 cells migrated cumulatively rather than as individual cells disseminating from the original aggregate. In general, the migration of OVCAR-3 was dependent on the downregulation of E-Cadherin. Two stimulants were initially used to test the tumor proliferation, namely TGF-β and IL-8. Both of these have also been reported to be secreted by activated neutrophils [82, 83]. Therefore, theoretically it is possible for the neutrophils to induce tumor migration if they were to produce sufficient amounts of TGF-β or IL-8 within the TIME. Detection of neutrophils in biopsies of ovarian cancer patients and quantification of the cell density required to produce the required concentration of the stimulants could be may be a more accurate method of validating the clinical relevance of such a modus operandi.
We also demonstrated that without external NET-inducing agents such as the PMA, tumor-associated inflammatory factors by themselves trigger naive neutrophils to produce NETs. Most in-vitro assays stimulate neutrophils with PMA for the production of NETs. However, PMA could also have counter-effects upon direct interaction with tumors [84, 85], which may lead to spurious results in a co-culture system. Therefore, in-vitro assays allowing the heterogeneous population of neutrophils to interact with the tumors in their naivety could enable studies of their wholesome behavior that drive distinct tumor responses. Till date, the cues from a Tumor Microenvironment (TME) that induce neutrophils to form NETs are poorly understood. While we do not identify any single cytokine responsible for NETosis, previous works have shown that depletion of a few tumor-derived factors [20] such as IL-8, G-CSF, GROα, or GROβ could diminish NET formation.
NETs are known to play an active role in tumor metastasis. NETs have been detected in patients with advanced-stage metastatic breast cancer, and found in the omentum of ovarian cancer patients as well. The availability of NETs at the destination site is also reflective of the metastatic potential of certain tumors. However, the dynamics of NET induction and the mechanisms by which NETs facilitate tumor metastasis are unclear. The demonstrations from our experimental tumor proliferation assays suggest that NETs that are formed in the surrounding stroma of a TIME, and not the NETs generated by the TINs, regulate the collective migration and invasion of cancer cell aggregates. NET-induced collective tumor invasion is specific to the interaction of the NETs with the collagen matrix surrounding the spheroids. Moreover, the stromal NETs appear to stimulate the collective invasion of the tumor cells into the tissue, and the expansion of the aggregated cell colonies.
Much like the tumor-associated factors, the current results do not attribute the role of any single NET-associated autocrine or paracrine to induce tumor migration. In general, the NETs generated in the stromal collagen region showed a greater spread than the NETs produced within the spheroid. While it is known that neutrophils could release NETs selectively based on the size of the pathogens [86], it could be interesting to study whether the amount of proteases or cytotoxic factors released during NETosis depend upon the spread area, and the resultant biochemically mediated tumor proliferation. But in that case, one would also expect a more aggressive tumor invasion in response to the conditioned media collected from the extensive NET formation, when artificially stimulated using PMA within the microfluidic channel of our device. It has been reported that intravascular NETs can increase local vascular permeability [87, 88], which would permit cancer cells to extravasate more easily. However, our data do not support a role for NETs even within 'leaky vasculature'-like microfluidic channels in enhancing the extravasation of the tumor cells into the channel. On the contrary, the OVCAR-3 spheroids displayed negligible distortion when exposed to a rich, PMA-stimulated NETs microenvironment outside the collagen region. The manner of distortion was not distinguishable from the patterns of weak spontaneous migration of the tumor cells within the 3D matrix. Therefore, we believe that the collective invasion of tumor spheroids seen in our experiments is specific to the NETs-collagen interactions, and it is simply not due to the tumor-chemotactic factors or other components ejected out of the neutrophils during NETosis.
Stromal-tumor interactions are highly dependent on the physical properties of the ECM [89], and it is possible that the regulation of collective OVCAR-3 migration by the stromal NETs is mediated through biophysical transduction. Given the nature of the components released during NETosis, the proteins [90] that are typically associated with the NET structures (elastase, cathepsin G, myeloperoxidase, and histones) could interact and bind directly with the collagen and other proteins present within the ECM, to form complex interlinked structures that result in altered properties of the matrix architecture. Consequently, it is possible that the physical parameters of the collagen matrix such as stiffness, pore size, and fiber re-organization could differ [91], leading to modified chemical cues, adhesive ligand densities, or the availability of binding sites to integrins and other matrix receptors. Each of these features could potentially impact tumor motility and gene expression. NETs could stimulate ECM remodeling [36, 92] and to promote integrin-mediated NETs-tumor interactions [92, 93]. However, till date, not much attention has been paid to the mechanobiological modalities through which the NETs accumulating around a tumor, could influence the stroma and subsequently influence tumor development. While DNAse treatment effectively degrades the NETs and thus limits the complementary effects on tumor progression, research shows that the degradation process leaves behind fragments of NETs which could still be biologically active [94]. This observation further strengthens the argument that amidst other multifarious dynamics, NETs could regulate the initiation of tumor invasion through biophysical tailoring of the tissues in and around a TIME. Dismantling the architecture of the NETs with DNAse treatment could reduce tumor invasion. However, extensive incorporation of the mechanobiology of NETosis within tumor metastasis research will require a better understanding of the mechanical evolution of NETs associated with specific tumor states.
Lastly, our work emphasizes the necessity for studying tumors on a 3D platform that offer unique insights into tumor behavior than regular 2D cultures. While dimensionality based functional differences could also exist in neutrophil response [95], conventional laboratory-based cancer models built on planar, 2D culture vessels could be way too simplified [96, 97] to faithfully mimic the tissue-specific diversities of neutrophil dynamics, and thus may not accurately represent the pathophysiology of tumor progression within a TIME. Spheroids [98, 99] are, in general, considered similar to an early vascular stage of tumors. Tissue-engineered 3D tumor models [100, 101], consisting of spheroids or organoids grown within synthetic ECM constructs, have enabled more rigorous tissue level studies of the TIME, and restore important architectural and stromal cues [102]. Microfluidics integrated Tumor-on-Chips are touted to better represent the in-vivo tumor conditions, and thus are expected to bridge the gap between 2D and animal models for potential applications as cost-effective drug screening platforms [103] in translational research, given their capabilities to control the cellular microenvironment with higher spatiotemporal precision and to present cells with biochemical signals in a more physiologically relevant context similar to a TIME.
However, increasing the complexity of in-vitro tumor models may be a successful strategy only if the crucial components of a TIME are identified, replicated, and appropriately controlled for. Engineering the dynamics of a TIME necessitates the incorporation of multiple tissues, inclusion of active vascular conduits, accountability of cell/tumor-specific differences in the ability to form 3D spheroids, and arrangement of each of these ensembles in an intricate and biochemically-relevant tumor-specific microenvironment without affecting the functionalities of each of the sub-components. Thus, the success of Tumor-on-Chips not only depends on the novelty of their applications, but also in their ease of implementation. Soft lithography and conventional microfluidics-based Tumor-on-Chips suffer tremendously from this limitation, further compounded by the involvement of expensive cleanrooms for the microfabrication processes that are not readily accessible to non-technical personnel [104]. Tumor-on-Chips are more heterogeneous than the conventional 2D cultures with more complex analysis parameters, and could therefore require higher resolution probing of the cells or tissue components for the confirmation of functional read-outs. Most of the Tumor-on-Chip platforms with enclosed microfluidic channels are irreversibly bonded to render them leak-proof. Consequently, non-destructive retrieval of the cells or tissue samples from the different micro-compartments of such systems, and simultaneous maintenance of compatibility with existing analysis and imaging systems for closer post-assay operations can be extremely challenging [105]. These drawbacks highlight the need for innovative bioengineering to create more user-friendly and mass-producible Tumor-on-Chip platforms, for reliable usage by a broad spectrum of research experts.
Our TIME-on-Chip model is capable of recreating in-vivo like neutrophil extravasation and NETosis functionalities within a tumor microenvironment, and this work lays the foundation for conducting future immune-tumor dynamics studies in 3D relevance. As seen from the results of this work, our device easily enabled the biomimicry of certain exclusive NETs-associated functionalities and their reciprocal effects on tumor development, which are otherwise impossible to implement on conventional in-vitro assay platforms. The device design proposed in this work enables a rapid, reproducible method to integrate functional 3D spheroids with tissue-engineered perfusable microfluidic channels to mimic the tumor-stromal-vascular organization. The versatility of the device also facilitates rapid incorporation of additional biophysical functionalities of in-vivo relevance (for e.g. interstitial fluidics or hemodynamics) without the need for drastic design modifications. Moreover, the hybrid integration method proposed herein facilitates easy dismantling and reassembly of all the sub-components for post-processing procedures such as immunostaining and imaging. This hybrid integration with magnetic attachment can be implemented on a variety of macro-culture vessels, including multiwell plates and Transwell inserts (supplementary figure 17S).
Overall, our TIME-on-Chip device enables sophisticated recapitulation of certain critical in-vivo biological events in a physiologically-relevant 3D-microenvironment, making this an extremely valuable tool for translational research. This multi-functional device has a strong potential for adaptation into applications that involve increased analysis throughput for conducting complex ex-vivo biomimetic assays, such as drug screening or evaluation of cytotoxic properties of biochemical molecules. The biofabrication techniques used in this work are ideal for rapid prototyping of physiologically relevant in-vitro tumor models, with a feasibility of automation of the entire process.
5. Conclusion
This work reinforces the necessity to carefully analyze the role of neutrophils within a tumor microenvironment. Distinct neutrophil responses and their functional modalities could be novel therapeutic targets in specific tumor types. The results of our work underline the importance of incorporating three-dimensionality to Tumor Microenvironment as a critical factor for gaining unique insights into significant biological events related to tumor growth and development. Our Tumor Immune Microenvironment-On-Chip model is simple to fabricate and could be implemented in several potential applications in a broad range of scientific research.
Acknowledgment
The authors thank Dr William Polacheck, Dr Imran Rizvi, and Dr Mustafa Kemal Ruhi from University of North Carolina, Chapel Hill, USA and Dr Roni F Rayes, from McGill University, Montreal, Canada for useful discussions on this work. The work was supported by start-up funding to Dr Chandrasekaran from the College of Engineering, NC A&T State University and National Institute of Health Support of Competitive Research Grant (1SC2-GM136523). The authors thank Ms. Taiya Good at the Student Health Center of the NC A&T State University for facilitating the availability of fresh human blood for neutrophil isolation experiments. The work was conducted per the guidelines of the Institutional Biosafety Committee (IBC#19-10), NC A&T State University.