



Abstract
In this work, we investigate the effect of the coordination of cross-links (i.e., the number of monomers participating in one cross-link) on the mechanical performance of a single polymeric chain. The framework provided by the reactive empirical bond order potential is used to generically describe the ability of certain monomers to form cross-links of different coordination. A systematic investigation of the influence of the coordination of cross-links on the mechanical properties of single polymeric chains is presented by comparing systems that contain cross-links in the classical form between two monomers (dimer) and such where the cross-links are formed by three monomers (trimer). The results show that the mechanical performance crucially depends on the coordination of cross-links. The overall shape of the load-displacement curves as well as mechanical parameters like stiffness, strength and work-to-straighten the molecule are different for the different systems. While the load-displacement curve shows an overall more continuous shape for the system containing trimers compared to the system including dimers only, the mechanical parameters are consistently lower for the first system. On the other hand, in contrast to the dimer case a trimer remains stable upon detachment of one of the monomers and the bonds are more mobile. This will be of importance in the case of fiber bundles, where the loading situation is even more complicated than in the single chain system due to the presence of inter-chain cross-links.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
(Reversible) cross-linking is a common strategy to specifically tailor the mechanical properties of natural and synthetic polymeric structures [1]. In technological materials, one of the prime examples is the permanent cross-linking of natural rubber in the process of vulcanization to improve its stiffness, strength and wear properties [2, 3]. Cross-links are also present in many natural materials like the actin skeleton of the cell [4, 5] and in a variety of load-bearing materials like bone [6], wood [7], silk [8–11] and the mussel byssus [12]. In biological materials, the cross-links are often weaker than the covalent bonds that provide the overall integrity of the structure and they may reform after the load is released (i.e., they are reversible) [13]. Upon loading, the weak cross-links rupture before the strong covalent backbone of the structure fails. The rupture of cross-links reveals parts of the polymer that were previously shielded from loading and, thus, allows for a straightening of the previously hidden length. This process provides a very efficient energy dissipation mechanism increasing considerably the toughness of the structure [14]. Consequently, in the context of natural materials, these (weak and reversible) cross-links are often called sacrificial bonds [15].
A lot of theoretical and experimental effort has been put in the detailed understanding of the influence of cross-links on the mechanical performance of polymeric materials. The theoretical models can be roughly separated into two groups: in the first, the structure of the polymer as a repetition of many monomers is kept (at least partially) by using a full atomistic approach [16, 17] or a coarse-grained bead-spring model [18]. In the second, the polymer is described as a continuous contour in the framework of the worm-like chain model [19–24] or simply as stiff rods [25–27]. In these works cross-links are modeled by introducing additional bonds between two parts of the same or of different polymers. Thus, in a microscopic view the cross-links form between two monomers. Nevertheless, it is a well known fact that cross-links may also form between more than only two monomers. One important type of such cross-links is metal coordination bonds, where several ligands may bind to one metal ion [28]. Prominent examples are the histidine–Zn and DOPA–Fe
and DOPA–Fe complexes found in the mussel byssus [29–31] that is a fibrous structure serving as a holdfast adhering the mussel to rocky substrates. Depending on the ion and the ligands these complexes show a large geometric variety, one of the most important cases being three ligands that bind to the ion [32, 33]. It is believed that these bonds are responsible for the superior mechanical properties that have been reported for the byssus. Among these are its high stiffness and toughness, its large extensibility up to
complexes found in the mussel byssus [29–31] that is a fibrous structure serving as a holdfast adhering the mussel to rocky substrates. Depending on the ion and the ligands these complexes show a large geometric variety, one of the most important cases being three ligands that bind to the ion [32, 33]. It is believed that these bonds are responsible for the superior mechanical properties that have been reported for the byssus. Among these are its high stiffness and toughness, its large extensibility up to  , its hard outer coating, the ability to self-heal and to adhere in wet environments [12, 34–36].
, its hard outer coating, the ability to self-heal and to adhere in wet environments [12, 34–36].
These remarkable properties of sacrificial bonds in general and of the mussel byssus in particular make it especially promising to transfer these basic concepts into technological applications. Possible applications include the development of mussel-inspired chemistry to create polymer networks with self-healing properties [37–39], to create adhesives for medical applications [40, 41], to build separators for high-power Li-ion batteries [42] and to use the concept of sacrificial bonds in general to enhance the mechanical properties of hydrogels [43–45], to toughen elastomers [46, 47] or to build bioinspired platelet reinforced polymer films [48].
Although cross-links of coordination larger than two are abundant in natural and recently also in technological materials no detailed theoretical investigation on the influence of cross-link coordination on the mechanical properties of polymeric structures is currently available. The current paper aims at closing this gap by explicitly modeling cross-links of varying coordination using computer simulations. In the context of this paper, coordination stands for the number of monomers participating in one cross-link. Consequently a two-fold coordinated cross-link is a dimer/bis-complex consisting of two monomers, while a three-fold coordinated cross-link is a trimer/tris-complex made of three monomers. In particular the mechanics of a single polymer chain containing cross-links that are energetically most stable when consisting of three monomers is compared to the mechanical behavior of systems where cross-links consist of two monomers only. Because no detailed information on the energetics of cross-links is available, a generic approach is chosen by modeling the cross-links using the framework provided by potentials of reactive empirical bond order (REBO) type [49, 50]. This kind of potential includes three-body contributions and allows to choose the parameters such that cross-links of a certain coordination are prevailing. The results show that the coordination has a profound impact on the mechanical behavior. Cross-links of a higher coordination lead to a much smoother load-displacement curve over the entire deformation range and the main mechanical parameters like stiffness, strength and work to straighten the polymer are reduced. Nevertheless, the individual bonds are more mobile and allow for re-arranging during deformation.
The paper is organized as follows. In the next section the model is introduced. Special emphasis is put on the explanation of REBO-type potentials used to describe the energetics of cross-links. In the next section, results and discussion, first the potential is characterized. The binding energies of different complexes are compared and the deformation of a single tris-complex is discussed. Then the mechanical performance of more complex systems containing more than only one tris-complex is analyzed. Special emphasis is put on the comparison of systems containing trimers and such where cross-links are strictly two-fold coordinated. The mechanical performance is obtained for systems of varying sticky-site density and distribution of sticky sites. Finally the results are summarized and an outlook of future work is given.
2. The model
The linear polymer is described using a bead-spring model (see figure 1). The polymer consists of N beads. To avoid physical overlap and self-intersection of the chain each bead was modeled as a hard sphere with radius R (that serves as the unit of length in the simulations). The covalent backbone of the chain is described using a Morse potential between neighboring beads. Excluded volume interaction UEV between beads i and j and the covalent energy Ucov are given by
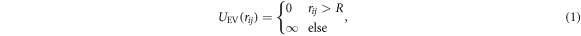
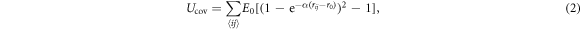
where the sum runs over all covalent bonds and E0 is the equilibrium binding energy, r0 is the equilibrium bond length, α is a measure of the width of the potential and rij is the distance between the beads i and j (for numerical values of the parameters see table 1). The contour length of the polymer is given by  .
.

Figure 1. Schematic of the model used in the simulations. The polymer is described as a linear chain of hard spheres, where each bead is covalently linked to its two neighbors (black lines). The two end beads that define the end-to-end distance are shown in gray, while non-sticky inner beads are labeled in black. Sticky monomers that have the ability to form a cross-link are depicted in red and bonds corresponding to a cross-link are shown as gray dashed lines. For reasons of clarity the schematic shown is two-dimensional. Nevertheless, the model is fully three-dimensional and the beads can move in all three directions of space. In the upper left part of the figure a sketch of a metal coordination bond is shown, also indicating the involved metal ion.
Download figure:
Standard image High-resolution imageTable 1. Simulation parameters describing covalent bonds and reversible cross-links. The unit of length is set by the hard sphere radius R. The parameters for RCLs forming solely dimers are chosen such that the binding energy of the dimer is 1.25 eV (the same as for the trimer) and that the equilibrium bond length is equal to the covalent bond length. The equation number after each parameter labels the equation where the parameter is defined.
| Covalent bonds | RCL trimer | RCL dimer | ||||
|---|---|---|---|---|---|---|
| E0 | 5.0 eV | Equation (2) | A | 9177.7 eV | 27533.1 eV | Equation (4) |
| R | — | Equation (1) | B | 134.9 eV | 371.0 eV | Equation (4) |
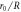
|
3.0 | Equation (2) |
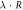
|
3.33 | 3.33 | Equation (4) |
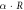
|
2.0 | Equation (2) |
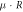
|
1.66 | 1.66 | Equation (4) |
| n | 4 | 0 | Equation (6) | |||
| β | 1.0 | — | Equation (6) | |||
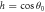
|
0.5 | — | Equation (6) | |||
| c | 8.0 | — | Equation (6) | |||
| d | 2.0 | — | Equation (6) | |||
| R | 1.4 | 1.4 | Equation (5) | |||
| S | 1.8 | 1.8 | Equation (5) |
To model the effect of cross-links, Ns of the beads are defined as sticky giving a cross-link number density  . It is these sticky beads that have the ability to form reversible cross-links. In the upper left part of figure 1 a sketch of a metal coordination bond is shown that is the inspiration for the cross-links investigated in this work. A metal ion binds to three ligands (monomers). One prominent example of such a bond is the DOPA–Fe
. It is these sticky beads that have the ability to form reversible cross-links. In the upper left part of figure 1 a sketch of a metal coordination bond is shown that is the inspiration for the cross-links investigated in this work. A metal ion binds to three ligands (monomers). One prominent example of such a bond is the DOPA–Fe complex found, e.g., in the mussel byssus [30, 37]. In the simulations presented these bonds are described in a coarse-grained manner, where the ion is replaced by effective interactions directly between the involved monomers (gray dashed lines in figure 1). This approach correctly reproduces the geometry of ligands in space, but averages out the actual bonding patterns as in reality the ligands would be bound to the central metal ion. The framework to perform the coarse-graining procedure is given by the REBO potential [49, 50] as described below. This potential was initially developed for describing non-closed packed covalently bonded systems such as carbon and silicon. The special feature of this potential is that the strength of an individual bond depends not only on the distance but also on the coordination of the involved atoms. Furthermore, REBO like potentials are reactive, meaning they allow for changes in bond coordination and, thus, take the reversibility of cross-links naturally into account. The energy corresponding to the cross-links is given by
complex found, e.g., in the mussel byssus [30, 37]. In the simulations presented these bonds are described in a coarse-grained manner, where the ion is replaced by effective interactions directly between the involved monomers (gray dashed lines in figure 1). This approach correctly reproduces the geometry of ligands in space, but averages out the actual bonding patterns as in reality the ligands would be bound to the central metal ion. The framework to perform the coarse-graining procedure is given by the REBO potential [49, 50] as described below. This potential was initially developed for describing non-closed packed covalently bonded systems such as carbon and silicon. The special feature of this potential is that the strength of an individual bond depends not only on the distance but also on the coordination of the involved atoms. Furthermore, REBO like potentials are reactive, meaning they allow for changes in bond coordination and, thus, take the reversibility of cross-links naturally into account. The energy corresponding to the cross-links is given by
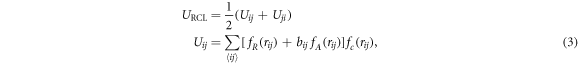
where the sum runs over all pairs of sticky sites. The different terms in the preceding equation are given by Morse-type repulsive and attractive contributions
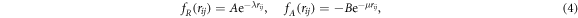
a cut-off function going smoothly from 1 to 0 for distances between S and R
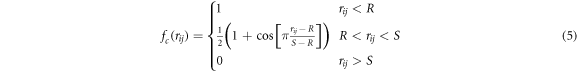
and the bond-order parameter bij that contains information on the local environment of the involved atoms (i.e, the angle  between the sites i, j and k)
between the sites i, j and k)
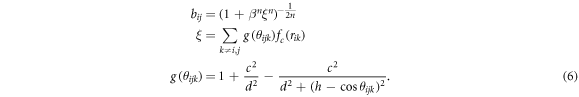
The sum in ξ runs over all sticky sites k inside the cut-off region of site i. The form of the potential is identical to the one given by Tersoff in [49].
Due to the long-range nature and the strength of the electrostatic interaction stemming from the metal ion, an ab initio treatment of these bonds is difficult [51] and there is only a limited number of first principles calculations on these complexes [52–54] and an even fewer number of works explicitly investigating the mechanical performance of these bonds [55]. Consequently, the exact energetics of the bonds are still not well characterized, meaning that the parameters needed for the REBO potential are not available yet. However, several experimental investigations indicate that metal-coordination bonds have a binding energy of about 20%–30% of a covalent bond [56–58]. This leads to the following constraints in choosing the parameters needed to describe the cross-links using a REBO potential: first, the binding energy of a cross-link made of three monomers should correspond to a quarter of the binding energy of the covalent bonds  . Second, a cross-link comprising three sticky sites is more stable than cross-links comprising any other number of sticky sites. Third, the equilibrium bond length of the cross-link is equal to the covalent bond length. Table 1 shows a collection of parameters that fulfills the aforementioned conditions and that was used throughout this work. The results of these simulations were compared to the results obtained from a different parameter set. This second parameter set was chosen such that the cross-links present in the system are purely two-fold. Within the REBO formalism, a purely two-fold coordinated structure can be achieved by setting n = 0. In this case we have
. Second, a cross-link comprising three sticky sites is more stable than cross-links comprising any other number of sticky sites. Third, the equilibrium bond length of the cross-link is equal to the covalent bond length. Table 1 shows a collection of parameters that fulfills the aforementioned conditions and that was used throughout this work. The results of these simulations were compared to the results obtained from a different parameter set. This second parameter set was chosen such that the cross-links present in the system are purely two-fold. Within the REBO formalism, a purely two-fold coordinated structure can be achieved by setting n = 0. In this case we have
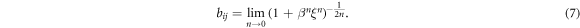
As long the sticky sites are two-fold coordinated, we have  and consequently
and consequently
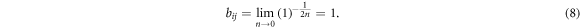
Whenever a third particle enters inside the cutoff region, it is found that  leading to
leading to
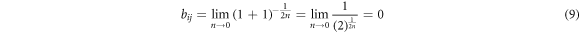
which means that the attractive part of the potential is set to zero for structures higher than two-fold coordinated. The remaining parameters were chosen such that the complex energy of the dimer equals the complex energy of the trimer and that the equilibrium bond length of the dimer equals the bond-length of the trimer (see table 1 for numerical values of the parameters). This choice of parameters allows to directly compare the results to previous investigations of purely two-fold cross-linked systems having the same binding energy. Nevertheless, it should be noted that this normalization also means that the energy per bond is a factor of three smaller for a three-fold coordinated structure compared to a two-fold coordinated one. Correspondingly, mechanical properties like strength and stiffness are equally reduced for the trimers compared to dimers.
The presented load-displacement curves are obtained by Monte-Carlo simulations in the Helmholtz-ensemble [59]. By pinning the first and last bead of the chain, this ensemble corresponds to a displacement controlled experiment. In contrast to simulations in the Gibbs-ensemble (fixed load) working in the Helmholtz ensemble allows the force to drop and, thus, enables to resolve also subsequent smaller force peaks after a larger force peak in the beginning. In the Gibbs ensemble such features in the load-displacement curve would not be resolved and the system would slip until the system is strong enough to sustain the applied load. In each simulation step the displacement is defined by the distance L between the pinned beads and the load is obtained by averaging over different configurations. These configurations are obtained using a standard Metropolis algorithm [60]. The Metropolis algorithm gives the probability of accepting a new trial configuration by  . Here
. Here  denotes the energy difference of the trial and the actual configuration and
denotes the energy difference of the trial and the actual configuration and  defines the temperature. If not stated otherwise, throughout the manuscript the temperature is set to the ambient value
defines the temperature. If not stated otherwise, throughout the manuscript the temperature is set to the ambient value  .
.
Starting from a small end-to-end distance  load-displacement curves are obtained by gradually increasing
load-displacement curves are obtained by gradually increasing  in steps of 0.002 until the chain is fully straightened at
in steps of 0.002 until the chain is fully straightened at  . 42 million Monte Carlo steps (i.e. jump trials per bead) are carried out at each deformation step to obtain a good estimate of the corresponding load. The starting configurations are obtained by unloading a straight chain without sticky sites from
. 42 million Monte Carlo steps (i.e. jump trials per bead) are carried out at each deformation step to obtain a good estimate of the corresponding load. The starting configurations are obtained by unloading a straight chain without sticky sites from  to the starting length
to the starting length  . Then Ns beads are labeled as sticky and the cross-links are allowed to form. Labeling of sticky sites is performed under the constraint that two sticky sites are not allowed to be direct neighbors along the chain.
. Then Ns beads are labeled as sticky and the cross-links are allowed to form. Labeling of sticky sites is performed under the constraint that two sticky sites are not allowed to be direct neighbors along the chain.
3. Results and discussion
3.1. Characterization of the potential
Because potentials of REBO type do intrinsically contain many-body contributions, their spatial dependence shows a much richer and complex behavior than a purely pair-wise potential. The many body effects are condensed in the bond-order parameter bij that contains a sum involving the bond angle  of all triplets
of all triplets  inside the specified cut-off (see equation (6)). bij is equal to one for a dimer and decreases with increasing coordination of the involved particles. Consequently, the influence of the attractive part of the potential is reduced with higher coordination (see equation (3)). For a given coordination the bond-order parameters takes its maximum value for bond-angles equal to
inside the specified cut-off (see equation (6)). bij is equal to one for a dimer and decreases with increasing coordination of the involved particles. Consequently, the influence of the attractive part of the potential is reduced with higher coordination (see equation (3)). For a given coordination the bond-order parameters takes its maximum value for bond-angles equal to  (e.g.,
(e.g.,  for a complex of three particles making up angles of 60°). Figure 2 shows the binding energies per particle of some complexes calculated for the parameters corresponding to the trimer complex (see table 1). The parameters were chosen to ensure that the trimer has a binding energy of 1.25 eV corresponding to
for a complex of three particles making up angles of 60°). Figure 2 shows the binding energies per particle of some complexes calculated for the parameters corresponding to the trimer complex (see table 1). The parameters were chosen to ensure that the trimer has a binding energy of 1.25 eV corresponding to  per particle. The dimer having a binding energy of 0.25 eV per particle is also reasonably stable meaning that a cross-link still provides mechanical stability after one monomer detaches due to loading. Finally, also a tetramer consisting of four monomers in tetrahedral geometry having bond angles of 60° has a considerable binding energy of
per particle. The dimer having a binding energy of 0.25 eV per particle is also reasonably stable meaning that a cross-link still provides mechanical stability after one monomer detaches due to loading. Finally, also a tetramer consisting of four monomers in tetrahedral geometry having bond angles of 60° has a considerable binding energy of  per particle. Despite this quite large binding energy such a complex was never found in the simulations (although depending on the type of ion and ligand such complexes may be found in reality [61]). This is due to several reasons: first, it is highly improbable that four monomers meet in such a fashion that a tetramer is formed directly. Second, forming a tetramer from a trimer is hindered by a considerable energy barrier. Third, if a tetramer has formed, whenever a fifth particle comes close it is energetically favorable for the complex to decompose into a tri- and a dimer, respectively.
per particle. Despite this quite large binding energy such a complex was never found in the simulations (although depending on the type of ion and ligand such complexes may be found in reality [61]). This is due to several reasons: first, it is highly improbable that four monomers meet in such a fashion that a tetramer is formed directly. Second, forming a tetramer from a trimer is hindered by a considerable energy barrier. Third, if a tetramer has formed, whenever a fifth particle comes close it is energetically favorable for the complex to decompose into a tri- and a dimer, respectively.

Figure 2. The binding energy per particle for certain complexes obtained with the REBO parameters as given in table 1. The figure shows that the finite complexes dimer Z = 2, trimer Z = 3 and tetramer Z = 4 are much more stable than the infinite complexes. The latter are the honeycomb lattice Z = 4, the diamond lattice Z = 5, the triangular and simple cubic lattice Z = 7, the body centered cubic lattice Z = 9 and the face centered cubic lattice Z = 13, respectively. The coordination Z denotes the number of monomers participating in one cross-link. In the case of the lattices this is given by the number of nearest neighbors increased by one.
Download figure:
Standard image High-resolution imageTo gain a first understanding of the deformation behavior, a simple system containing only one tris-complex was studied. The starting configuration was set as shown in the bottom part of figure 1 with a polymer made of N = 50 monomers. This structure is loaded by changing the end-to-end distance L of the pinned outer beads. For the limiting case of zero temperature and infinitely stiff covalent bonds, the corresponding load-displacement curve can be determined directly as the derivative of the potential given in equations (3)–(6). The deformation mechanism is such that L defines the distance between beads 1 and 3, while the position of bead 2 is determined by the condition that it experiences no net-force (see figure 1). In the left part of figure 3 load-displacement curves for this simple setting including one trimer are shown. The gray line corresponds to the result obtained directly from the REBO potential for the case of zero temperature. This curve shows a complex structure including several peaks. Qualitatively it resembles the curves obtained with ab initio methods for metal coordination bonds [55]. The multi-peak structure comes from the fact that the dissociation of a trimer is a multiphase process. The first and second peak slightly above  and
and  with heights of 0.47 eV R−1 and 0.2 eV R−1, respectively, correspond to the detachment of the first ligand. The first peak corresponds to elongating monomers 1 and 3. The second peak shows the final detachment of the ligand, where it is equally probable that monomer 1 or 3 detaches. The remaining dimer fails at an elongation of
with heights of 0.47 eV R−1 and 0.2 eV R−1, respectively, correspond to the detachment of the first ligand. The first peak corresponds to elongating monomers 1 and 3. The second peak shows the final detachment of the ligand, where it is equally probable that monomer 1 or 3 detaches. The remaining dimer fails at an elongation of  . The small additional peaks close to the main peaks are due to the cut-off. Blue symbols correspond to a simulated load-displacement curve of a N = 50 bead system at the low temperature
. The small additional peaks close to the main peaks are due to the cut-off. Blue symbols correspond to a simulated load-displacement curve of a N = 50 bead system at the low temperature  . The curve coincides well with the theoretical zero temperature case. The blue curve is only slightly shifted to the right due to the finite stretching of the covalent bonds in the N = 50 system. Furthermore, the influence of the cut-off is already completely washed out. At the more realistic value of
. The curve coincides well with the theoretical zero temperature case. The blue curve is only slightly shifted to the right due to the finite stretching of the covalent bonds in the N = 50 system. Furthermore, the influence of the cut-off is already completely washed out. At the more realistic value of  (red symbols) the rather complex behavior observed at low temperatures is lost. The only remaining features are two single peaks corresponding to the rupture of the trimer and later the remaining dimer. The force peaks shift towards smaller values of
(red symbols) the rather complex behavior observed at low temperatures is lost. The only remaining features are two single peaks corresponding to the rupture of the trimer and later the remaining dimer. The force peaks shift towards smaller values of  , the entropic background increases (i.e. the nature of loading shifts from enthalpic to entropic) and the height of the peak decreases, i.e. the effective strength becomes considerably smaller than the theoretical strength of the trimer and dimer given by 0.47 eV R−1 and 0.41 eV R−1, respectively. As was already shown for systems cross-linked by bis-complexes only, this means that entropic effects play a dominant role in the deformation behavior of such systems [62]. In particular, the load-displacement curve obtained at ambient temperature cannot be directly taken to correspond to the molecular enthalpic energetics given by the configurational potential.
, the entropic background increases (i.e. the nature of loading shifts from enthalpic to entropic) and the height of the peak decreases, i.e. the effective strength becomes considerably smaller than the theoretical strength of the trimer and dimer given by 0.47 eV R−1 and 0.41 eV R−1, respectively. As was already shown for systems cross-linked by bis-complexes only, this means that entropic effects play a dominant role in the deformation behavior of such systems [62]. In particular, the load-displacement curve obtained at ambient temperature cannot be directly taken to correspond to the molecular enthalpic energetics given by the configurational potential.

Figure 3. Load-displacement curve (left) and corresponding free energy contributions (middle) and coordination plots (right) of a system with a single tris-complex. In the load-displacement curve, the gray line denotes the theoretical deformation of a tris-complex at zero temperature obtained directly from the potential (equations (3)–(6)). Blue and red symbols depict a simulated load-displacement curve of a polymer chain of length N = 50 at low ( ) and ambient temperature (
) and ambient temperature ( ), respectively. The middle part of the figure shows the change of free energy of the system as well as its enthalpic and entropic constituents
), respectively. The middle part of the figure shows the change of free energy of the system as well as its enthalpic and entropic constituents  . Solid lines correspond to the free energy
. Solid lines correspond to the free energy  , dashed lines to the change in energy
, dashed lines to the change in energy  and dotted lines to the (negative) change in entropy
and dotted lines to the (negative) change in entropy  . Blue and red lines correspond to the low and ambient temperature case, respectively. The right part of the figure shows the mean coordination of sticky sites, i.e., the mean number of neighbors (sites are labeled as defined in figure 1).
. Blue and red lines correspond to the low and ambient temperature case, respectively. The right part of the figure shows the mean coordination of sticky sites, i.e., the mean number of neighbors (sites are labeled as defined in figure 1).
Download figure:
Standard image High-resolution imageThis behavior is confirmed by the behavior of the free energy as shown in the middle panel of figure 3. The change in free energy  is given as the integral of the load-displacement curve shown in the left panel of figure 3. The energetic contribution is given as the average of the potential energy calculated for the independent configurations obtained in the course of the simulations. Knowing
is given as the integral of the load-displacement curve shown in the left panel of figure 3. The energetic contribution is given as the average of the potential energy calculated for the independent configurations obtained in the course of the simulations. Knowing  and
and  it is possible to calculate the entropic contribution via
it is possible to calculate the entropic contribution via  . As the figure shows the two contributions to the free energy change drastically when the temperature is raised. At low temperature, the entropic contribution dominates the free energy until the cross-link is loaded at an elongation slightly above
. As the figure shows the two contributions to the free energy change drastically when the temperature is raised. At low temperature, the entropic contribution dominates the free energy until the cross-link is loaded at an elongation slightly above  . Then the energetic contribution rises and remains above the entropic contribution until the polymer is fully straightened. This behavior is completely reversed at ambient temperature. The entropy clearly dominates the free energy for the entire deformation range. The opening of the complex can be clearly seen by an increase in the energy and the entropy (a decrease in
. Then the energetic contribution rises and remains above the entropic contribution until the polymer is fully straightened. This behavior is completely reversed at ambient temperature. The entropy clearly dominates the free energy for the entire deformation range. The opening of the complex can be clearly seen by an increase in the energy and the entropy (a decrease in  ).
).
The right part of figure 3 shows the average coordination, i.e. the average number of bonds, of each sticky site as a function of elongation obtained at ambient temperature. An intact bond is formed between two sticky sites having a distance closer than  (see equation (5)). When the trimer is intact, all beads have a coordination of two. During stretching, first the monomer 3 detaches. This can be seen in the figure by the decline of the coordination of sites 1 and 2 to one and site 3 to zero. Eventually, during further elongation the bond formed between 1 and 2 changes to a bond formed between 2 and 3. This shows that at ambient conditions the bonds are highly mobile and the corresponding force in the load-displacement plot is lost in the entropic background. The bonds remain mobile as is confirmed by a further re-formation of the bond shortly before the final rupture of the complex indicated by the decline to zero of the coordination of all sites for elongations above 0.95.
(see equation (5)). When the trimer is intact, all beads have a coordination of two. During stretching, first the monomer 3 detaches. This can be seen in the figure by the decline of the coordination of sites 1 and 2 to one and site 3 to zero. Eventually, during further elongation the bond formed between 1 and 2 changes to a bond formed between 2 and 3. This shows that at ambient conditions the bonds are highly mobile and the corresponding force in the load-displacement plot is lost in the entropic background. The bonds remain mobile as is confirmed by a further re-formation of the bond shortly before the final rupture of the complex indicated by the decline to zero of the coordination of all sites for elongations above 0.95.
3.2. Load-displacement curves
Figure 4 shows the comparison of typical load-displacement curves and corresponding coordination plots obtained for a system with sticky sites having the ability to form trimers (black symbols) and dimers (gray symbols), respectively. A system forming solely bis-complexes is formally achieved by setting n = 0 in the parameters of the potential (table 1). This means that the bond order parameter bij jumps directly to zero when a third particle enters the cut-off of two particles forming a dimer. Consequently, the dimer is the only stable cross-link that may form. The parameters are chosen such that the binding energy of a dimer equals the binding energy and bond length of a trimer. The length of the polymers is set to N = 50 and the sticky site densities shown correspond to a low ( ), a medium (
), a medium ( ) and high (
) and high ( ) density. The sticky sites are distributed randomly along the chain having the constraint that two sticky sites are not allowed to be direct neighbors along the chain. The presented curves are averages over 48 independent runs. Furthermore, the mechanical performance explicitly due to the cross-links is separated from the trivial covalent contributions by subtracting from each load-displacement curve the corresponding curve from a system with no cross-links. This ensures that the load-displacement curves drop to zero when they are completely straightened at
) density. The sticky sites are distributed randomly along the chain having the constraint that two sticky sites are not allowed to be direct neighbors along the chain. The presented curves are averages over 48 independent runs. Furthermore, the mechanical performance explicitly due to the cross-links is separated from the trivial covalent contributions by subtracting from each load-displacement curve the corresponding curve from a system with no cross-links. This ensures that the load-displacement curves drop to zero when they are completely straightened at  , because at this elongation all cross-links are open.
, because at this elongation all cross-links are open.
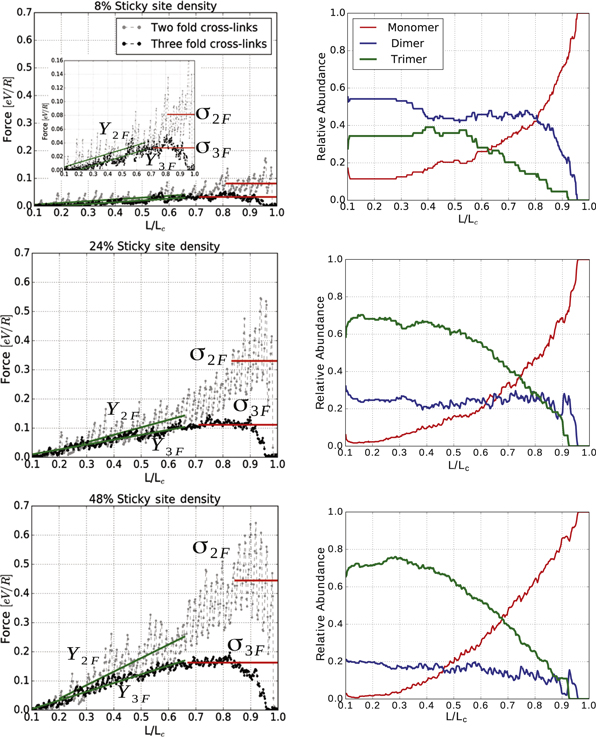
Figure 4. Load-displacement curves (left column) and coordination plots (right column) of polymeric systems for different sticky site densities ρ and different coordination of cross-links. In the simulations N = 50 and the temperature is set to  . The sticky site density is
. The sticky site density is  , respectively, and the sticky sites are distributed randomly along the chain. Black symbols correspond to the case where the trimer is the most stable cross-link configuration, gray symbols denote the results obtained for cross-links forming dimers only. From each curve the load-displacement curve of a non-cross-linked chain of equal length was subtracted to separate the contributions stemming from the cross-links and the covalent backbone to the mechanical performance. Solid lines indicate the stiffness Y (the slope of the curve) and strength σ (its highest plateau) in the corresponding curves, respectively. The scaling of the figures is kept the same for all different sticky site densities. For a better visualization of the
, respectively, and the sticky sites are distributed randomly along the chain. Black symbols correspond to the case where the trimer is the most stable cross-link configuration, gray symbols denote the results obtained for cross-links forming dimers only. From each curve the load-displacement curve of a non-cross-linked chain of equal length was subtracted to separate the contributions stemming from the cross-links and the covalent backbone to the mechanical performance. Solid lines indicate the stiffness Y (the slope of the curve) and strength σ (its highest plateau) in the corresponding curves, respectively. The scaling of the figures is kept the same for all different sticky site densities. For a better visualization of the  case the inset shows a zoom of the load-displacement curve. The adjacent column shows the corresponding coordination plots of the system containing trimers. The graphs show the fraction of mono-, bis-, and tris-complexes in the system as a function of elongation. The results shown are the averages over 48 independent simulation runs.
case the inset shows a zoom of the load-displacement curve. The adjacent column shows the corresponding coordination plots of the system containing trimers. The graphs show the fraction of mono-, bis-, and tris-complexes in the system as a function of elongation. The results shown are the averages over 48 independent simulation runs.
Download figure:
Standard image High-resolution imageFigure 4 clearly depicts that the mechanical behavior of the system crucially depends on the coordination of the cross-links involved. The system with cross-links forming tris-complexes shows a much smoother behavior over the entire deformation range compared to the system containing dimers only. As a tris-complex consists of three bonds instead of only one in the case of a dimer, there are more bonds in the first system than in the latter. Consequently, these bonds rupture already at lower elongations resulting in the more continuous rise of the force. Furthermore, as the complex energies are the same, the energy per bond (and correspondingly the peak-force) is smaller for the tris- than the bis-complex. The smaller peak-force results in a washing out of the peaks in the entropic background. The coordination plots presented in the lower row of figure 4 indicate that the observed behavior is mainly due to the deformation of tris-complexes, as the number of tris-complexes decays over the entire deformation range, while the number of bis-complexes stays constant over a wide range and decays only in the end.
Although, the parameters of the potential are designed such that the three-fold coordinated cross-links are the most stable and energetically favorable structures in the system, the coordination plot clearly shows that not all sticky sites are three fold coordinated. This is true even at low elongations, where it is not expected that the bonds rupture due to loading. Furthermore, this effect is more pronounced for low sticky site densities. At  the fraction of dimers is larger than the fraction of trimers over the entire deformation range. The reason for this behavior is that bond formation is a stochastic process and usually proceeds via the formation of a dimer that is transformed into a trimer by further attachment of a third monomer. On the other hand, transformation of two dimers into one trimer and one monomer (although energetically favorable) is hindered by the energy barrier resisting the dissociation of one dimer into two monomers. Thus, if it happens that most of the sticky sites in the system are two-fold coordinated, the formation of trimers is effectively hindered. This is more likely to happen at low sticky site densities, because at
the fraction of dimers is larger than the fraction of trimers over the entire deformation range. The reason for this behavior is that bond formation is a stochastic process and usually proceeds via the formation of a dimer that is transformed into a trimer by further attachment of a third monomer. On the other hand, transformation of two dimers into one trimer and one monomer (although energetically favorable) is hindered by the energy barrier resisting the dissociation of one dimer into two monomers. Thus, if it happens that most of the sticky sites in the system are two-fold coordinated, the formation of trimers is effectively hindered. This is more likely to happen at low sticky site densities, because at  there are only four sticky sites in the system and the probability that these form two dimers in the first place is rather high. Eventually during stretching some of the cross-links rupture resulting in an increasing number of free monomers. These are now able to coordinate with a dimer forming a trimer. This process is responsible for the slight increase in the number of three-fold coordinated structures with elongation observed for all sticky site densities.
there are only four sticky sites in the system and the probability that these form two dimers in the first place is rather high. Eventually during stretching some of the cross-links rupture resulting in an increasing number of free monomers. These are now able to coordinate with a dimer forming a trimer. This process is responsible for the slight increase in the number of three-fold coordinated structures with elongation observed for all sticky site densities.
Previous investigations identified the number and the distribution of sticky sites as a major factor determining the mechanical behavior of a polymer cross-linked with dimers [62–64]. Therefore, the influence of the same parameters on the mechanical performance is also investigated in the current study and shown in figure 5. The investigated parameters are the sticky site density (i.e., the number of sticky sites) and the distribution of sticky sites. The sticky site density is varied from  –
– which is the maximum possible density, respecting the constraint that two sticky sites are not direct neighbors and that first and last bead are non-sticky. The distribution of sticky sites is varied between the extremes of a random and an ordered arrangement. In the latter one the same number of non-sticky sites separates each sticky site from the next. The mechanical properties analyzed are the work to straighten the polymer W (the area under the load-displacement curve from
which is the maximum possible density, respecting the constraint that two sticky sites are not direct neighbors and that first and last bead are non-sticky. The distribution of sticky sites is varied between the extremes of a random and an ordered arrangement. In the latter one the same number of non-sticky sites separates each sticky site from the next. The mechanical properties analyzed are the work to straighten the polymer W (the area under the load-displacement curve from  to 1, which is closely related to the toughness of the structure), the stiffness Y (the slope of the load-displacement curve) and the strength σ (the highest plateau found in the load-displacement curve). In figure 4 the stiffness and strength of the depicted load-displacement curves are indicated by solid lines.
to 1, which is closely related to the toughness of the structure), the stiffness Y (the slope of the load-displacement curve) and the strength σ (the highest plateau found in the load-displacement curve). In figure 4 the stiffness and strength of the depicted load-displacement curves are indicated by solid lines.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. The most important mechanical properties (work to straighten the molecule W, stiffness Y and strength σ) obtained as a function of sticky site density, arrangement and coordination. Red and blue symbols denote purely two-fold coordinated structures and three-fold coordinated structures, respectively. Circles and triangles depict a random and an ordered arrangement of sticky sites.
Download figure:
Standard image High-resolution image{kind=link}
The left graph of figure 5 shows the work to straighten the molecule W. For all systems, W shows a linear dependence on the sticky site density (i.e., the number of cross-links). While W does not considerably change between the random and ordered arrangement (as was already observed in [63]), it is consistently higher for the system containing bis-complexes only compared to the system containing trimers. This is remarkable because the complex energies are the same for the dimer and trimer. Thus, at zero temperature it is expected that the area under the curve should be the same for both systems. Partly this effect can be explained by the fact that not all cross-links in the system initially form a trimer (see bottom row of figure 4). Nevertheless, although up to  of the sticky sites are part of a three-fold coordinated cross-link, W is reduced a factor of almost 2 for the system containing trimers compared to the dimer system. Thus, the actual value of W stems from a subtle interplay of bond reformation and entropic effects pointing to the fact that the load-displacement curve corresponds to a derivative of a free energy in nature including entropic effects (see also middle panel of figure 3). In the presented simulations, entropic effects play a more profound role in the system containing trimers than in the system containing only dimers, because the energy per bond is smaller for the first.
of the sticky sites are part of a three-fold coordinated cross-link, W is reduced a factor of almost 2 for the system containing trimers compared to the dimer system. Thus, the actual value of W stems from a subtle interplay of bond reformation and entropic effects pointing to the fact that the load-displacement curve corresponds to a derivative of a free energy in nature including entropic effects (see also middle panel of figure 3). In the presented simulations, entropic effects play a more profound role in the system containing trimers than in the system containing only dimers, because the energy per bond is smaller for the first.
The middle part of figure 5 shows the stiffness Y of the system. For all systems, the stiffness increases with increasing sticky site density. In the case of the ordered arrangement of sticky sites a stiffness cannot be uniquely defined for low sticky site densities due to the presence of many isolated force peaks. This is because in the ordered case the positions of sticky site are the same in all runs. As the position of force peaks in the load displacement curve is highly correlated with the position of sticky sites in the chain, the peaks are not washed out during averaging. This is different for the random arrangement where a stiffness can be defined with reasonable accuracy for all sticky site densities. Here the single peaks are washed out during averaging due to the many different configurations of sticky sites. Unlike W, the arrangement of sticky sites has a large influence on Y. The ordered arrangement shows a higher stiffness than the random arrangement for both the trimer and dimer system. Similar observations were made in [62].
The right part of figure 5 shows the strength of the investigated systems. In the current work, the strength is defined as the highest plateau found in the load-displacement curves. In the case of the highly fluctuating results of the dimer system the plateau is defined as the average of the oscillations (see figure 4). The two-fold coordinated structures show consistently a higher strength than the three-fold coordinated one. This can be partly explained by the fact that the theoretical strength of the two-fold coordinated complex is (Fmax = 1.041 eV R−1) larger than the theoretical strength of the tris-complex (Fmax = 0.47 eV R−1). Furthermore, the figure clearly shows that the theoretical strength is by far not achieved by any of the systems. As worked out in [62], temperature and entropy reduce the effective strength of the systems. As the figure shows, the strength increases with increasing sticky site density for all systems. Furthermore, the distribution of cross-links have no influence on the strength of the system containing tris-complexes, while in the case of dimers the ordered arrangement of sticky sites leads to a lower strength than a random arrangement.
It should be emphasized that although the results presented seem to indicate a (mechanical) inferiority of three-fold coordinated cross-links over two-fold coordinated ones, this first impression is misleading. The mechanical performance depends crucially on the choice of parameters and in the current paper, the complex energies of the trimer and dimer were chosen identical. This means that the energy per bond (as well as the stiffness and strength) is reduced a factor of three for the system containing trimers compared to the system consisting of dimers only. However, figure 5 shows that the mechanical performance of the trimer system is reduced less than a factor of three compared to the dimer system. Thus, relative to the strength of individual bonds the mechanical performance of trimers is even enhanced compared to the dimer system.
4. Summary and conclusions
In this paper, the influence of the coordination of cross-links on the mechanical performance of single polymer chains was investigated. The properties of systems with two-fold coordinated cross-links only were compared to systems containing also tris-complexes. The coordination of cross-links were controlled using the framework provided by potentials of REBO type. To make the systems comparable to previous studies of purely two-fold coordinated structures, the parameters were chosen such that the energy of the bis-complex in the purely two-fold coordinated system matches the energy of the trimer. This means that the energy per bond (and correspondingly the strength) is smaller for the tris- than the bis-complex. It could convincingly be shown that the coordination of cross-links strongly influence the mechanical behavior. For the same sticky site density, all investigated mechanical properties are reduced for the tris-complexes compared to the bis-complex case. This can (at least partly) be understood by the reduction of the strength of the tris-complex for the stiffness and strength, but even the work to straighten the molecule is reduced for the system containing tris-complexes. On the other hand, due to the reduced strength of the individual bonds these are more mobile than in the purely two-fold coordinated system. Future work will consist in investigating the effect of three-fold coordinated cross-links on aligned chain bundles. In this case the loading will be more complicated, because whenever a cross-link connects different chains the loading cannot be simply described by bead 2 being load-free (see figure 1). Furthermore, in reference [65] it was shown that two-fold coordinated cross-links having a quarter of the strength of a covalent bond may reduce the strength of the system. Preliminary simulation results of chain bundle systems indicate that this will probably change in the case of tris-complexes, because the individual strength of bonds is reduced. Another important point is the increased mobility or plasticity of bonds that may allow for bond re-formation and re-ordering during loading and unloading significantly enhancing the mechanics of such systems as is found experimentally [33]. These points need further clarification and will be discussed in a future paper.
In conclusion, a new simulation procedure was presented giving the possibility to investigate cross-links of different coordination, i.e. resembling metal coordination bonds, in polymers. First results show that—with the same complex binding energy—multivalent cross-links show a reduced mechanical performance at the same sticky site density compared to a purely two-fold coordinated structure. Nevertheless, the higher plasticity of bonds as well as the lower strength may also prove beneficial in a system consisting not of single, independent chains, but in fiber networks or fiber bundles.
Acknowledgments
We would like to thank Matthew Harrington, Georg Menzl and Christoph Dellago for helpful discussions. The computational results presented have been achieved using the Vienna Scientific Cluster (VSC). Financial support of the Austrian Science Fund (FWF) in the framework of project P 27882-N27 is gratefully acknowledged.